
Abstract
Gastrointestinal helminth-microbiota associations are shaped by various ecological processes. The effect of the ecological context of the host on the bacterial microbiome and gastrointestinal helminth parasites has been tested in a number of ecosystems and experimentally. This study takes the important step to look at these two groups at the same time and to start to examine how these communities interact in a changing host environment. Fresh faecal samples (N = 335) from eight wild Eulemur populations were collected over 2 years across Madagascar. We used 16S ribosomal RNA gene sequencing to characterise the bacterial microbiota composition, and faecal flotation to isolate and morphologically identify nematode eggs. Infections with nematodes of the genera Callistoura and Lemuricola occurred in all lemur populations. Seasonality significantly contributed to the observed variation in microbiota composition, especially in the dry deciduous forest. Microbial richness and Lemuricola spp. infection prevalence were highest in a previously intensely logged site, whereas Callistoura spp. showed no such pattern. In addition, we observed significant correlations between gastrointestinal parasites and bacterial microbiota composition in these lemurs, with 0.4–0.7% of the variation in faecal bacterial microbiota composition being explained by helminth infections. With this study, we show effects of environmental conditions on gastrointestinal nematodes and bacterial interactions in wild lemurs and believe it is essential to consider the potential role of microbiome-parasite associations on the hosts’ GI stability, health, and survival.
Similar content being viewed by others

Introduction
The gastrointestinal (GI) microbiota plays a vital role in the physiology, health, and nutrition of its host1. Next to the microbiota, GI macroparasites, including protozoa and nematodes, can be present within a host’s digestive tract. Parasitism can impact the host’s health, behaviour, and survival, thereby influencing evolutionary processes and population dynamics2. Besides, parasites are known to affect the host’s reproduction directly through pathologic effects and mate choice as well as indirectly by impaired nutrition and energy deficits3. Researchers suggest a strong connection between specific parasite species and GI bacteria. For example, GI microbiota can prevent gut colonisation by pathogenic microorganisms4 and researchers detected a link between the microbiome and simian immunodeficiency virus in wild primates5. A stable and diverse GI microbiota composition is crucial for mammalian health6, 7, and defining the mechanisms influencing its composition and diversity is considered important8.
Faecal bacterial GI microbiota and macroparasites living at internal body surfaces and the lumen of the GI tract are part of an animals’ microbiome and are involved in key host functions9. The coinfection of a host by multiple microorganisms has important epidemiological and clinical implications, which has been recognised for decades across multiple animal taxa10. As studying wild populations under natural conditions is rather complex, most studies on the determinants of the GI microbiota composition and parasite prevalence either comprise laboratory or clinical studies that focus on a single host species or infection with a single parasite species11, 12. Our work is building on this extensive body of literature on GI bacteria and helminths and their interaction to increase our understanding of ecological processes that shape composition and functionality of GI microbiota and parasites in wild populations12.
The composition of the GI microbiota is known to be shaped by multiple factors, including host genetics, evolutionary history, physiology, sex, and age13, 14. Several recent studies showed that the microbial composition can remain stable over the host’s lifespan15, 16. However, other studies found that extrinsic factors, including diet composition17,18,19, pathogens20, seasonality21, habitat degradation22, and geographical differences23 influence GI microbiota. For example, it has been shown that the microbial composition in black howler monkeys (Alouatta pigra) differs across seasons and is correlated with diet24. Also, the distribution of parasite infections in wild host populations is influenced by a number of factors, including host susceptibility and exposure25. Many helminths can spread through a variety of transmission modes, including the faecal-oral route, which involves ingestion of contaminated soil or food26. The nematodes that are the focus of the present study spend part of their life cycle outside of the host and are therefore exposed to environmental conditions that shape temporal variations in parasite infections. Climatic seasonality has been identified as an important driver of this temporal variation in several wild primate species27, 28. However, studies investigating these links have yielded different outcomes24, 29, 30. It has also been shown that some nematodes have an accelerated development and increased reproduction and survival rates in wetter and warmer conditions28, 31, and desiccate more frequently under dry circumstances27. Several studies found GI parasite richness, prevalence, and abundance to be higher in the warm wet season, compared to the cold dry season, e.g., in lemurs32,33,34, chimpanzees27, as well as howler and spider monkeys (Ateles geoffroyi)35. However, some helminth species (e.g., Enterobius spp.) seem to prefer relatively low temperatures36. Although the underlying processes remain unclear37, these examples show that environmental factors can influence the microbial composition and parasite prevalence14, 21, and require further study in wild mammals.
In addition to environmental factors, the impact of anthropogenic forest disturbance, including logging, on health and pathogens in both wildlife and humans may be far-reaching (Keele et al.38). Anthropogenic forest disturbance may lead to changes in host population densities and interaction patterns of wildlife with humans, domestic animals, and other wildlife species31, 39. Such disturbances can thereby enforce changes in the GI microbiota composition and parasite infections14, 22, 40. Microbiota diversity can be reduced in degraded areas, as has been shown in howler monkeys, red colobus monkeys (Procolobus gordonorum), and other primate species14, 22, 41. Furthermore, increased parasite prevalence, virulence, and transmission rates were found in such disturbed forests40, 42, 43. Although the exact mechanisms influencing the microbial composition and parasite infections in disturbed forests is still unknown, nutritional stress is considered important44. Nutritional stress can alter the microbiome and lower an animal’s immune status, resulting in a higher susceptibility to parasites45. Forest disturbance can also directly influence parasites that spend part of their life cycle outside of the host, as changes in forest structure lead to differences in light exposure, temperature, and humidity46. This integrated study on forest disturbance effects on both the parasites and the microbiome further explores the parasite and microbiome ecology in wild primates living in natural versus human-modified forests.
Microbiota and parasites co-inhabit the GI-tract and have evolved in close association, suggesting that they have the potential to influence each other47. Research on this interplay between host, parasites, and the microbiome has increased over the last decade48, and recent studies in humans showed associations between nematode infections and changes in the GI microbiota structure49,50,51. However, this observation is not consistent across human populations52, 53. Another study experimentally demonstrated that the gut bacterial composition in mice (Mus musculus) can change when exposed to a GI parasite (Trichuris muris)54. Associations between specific bacteria and the abundance of enteric nematodes were also found in wild wood mice (Apodemus sylvaticus)21. Most of these studies mentioned above focussed on mice, pigs (Sus scrofa), or humans. However, recent studies have begun to address the interaction between the microbiome and parasites in primates55, and we aim to contribute with this study more comparative data on the interactive effect of parasite infections and microbiota composition of wild lemurs.
Specifically, we aim to assess the effects of seasonality (i.e., dry versus wet season), and forest disturbance on the interaction between GI parasites and bacterial microbiota composition in two lemur species. Recently, the microbial composition of lemurs has been studied in captive lemurs15, in two sympatric wild lemur species56, and in wild sifakas57. However, the processes leading to the natural variation of faecal microbiota in wild lemurs, and how its variation is influenced by environmental conditions, need further study. Furthermore, only a few studies to date have used a metataxonomic 16S ribosomal RNA (rRNA) gene-targeted approach to address the association and interactive effects between parasites and the microbiome47, 50, 52,53,54, 58,59,60. In the present study, we focus on four congeneric lemur species at eight geographic locations: Eulemur rufifrons, E. fulvus, E. macaco, and E. rubriventer. The substantial heterogeneity in lemur habitats across Madagascar is created by an interaction of the east–west and north–south rainfall gradient61. The four lemur species belong to the genus Eulemur and are morphologically alike62, are present in the distinct geographic regions of Madagascar, and inhabit both large intact forests and forests that have experienced past logging63.
Given the major role of environmental factors in shaping seasonal variation in microbial community structure and parasite infections, we expected that (1) lemurs inhabiting the dry deciduous forests of western Madagascar with strong seasonal variation in rainfall and temperature show larger seasonal contrasts in both parasite infections and microbial composition compared to lemurs in the rainforests of eastern Madagascar with less seasonal variation. We further expected (2) that the microbiota composition is altered and parasite infection prevalence is increased in lemurs whose habitat is restricted to previously logged rainforests compared to lemurs living in less disturbed forests. Lastly, we explored (3) correlations between GI microbiota and natural parasite infections. Hence, in this study, we determine how the GI microbiota and parasite infections vary with their geographic distribution spatially in wild lemurs along with seasonal variation and past logging. In addition, we explore the interactive effects between the parasites and microbiota present.
Results
Seasonality
We found a clear separation of samples by season in the bacterial microbiota composition of multiple lemur populations sampled across Madagascar (Fig. 1, Table 1), using principal coordinate analysis (PCoA) based on the weighted UniFrac distance matrix (early wet season N = 128, early dry season N = 196, R2 = 0.08, Adonis; P = 0.0001, Fig. 2). Distance-based redundancy analysis (dbRDA) identified the area of sample collection as the most influential variable followed by season when considering all samples as a single dataset. We observed an increase in the percentage of explained variance in microbiota composition by seasonality when we focused on samples collected within one area and one lemur species (Fig. 3). Specifically, for E. fulvus populations from Ankarafantsika NP and Andasibe NP, and E. rubriventer and E. rufifrons populations from Ranomafana NP, the percentage of variation in microbiota composition explained by season increased from 5.7% for the entire dataset to 16.9%, 20.2%, 12.5% and 13.5%, respectively (Fig. 3a–d). Therefore, these populations harboured a different microbial composition in the early dry season compared to the early wet season. With regards to alpha diversity, the E. fulvus population in Ankarafantsika showed a significantly higher mean phylogenetic diversity (PD index, P < 0.001) in the early dry season (N = 21) compared to the early wet season (N = 29). No statistically significant differences in alpha diversity were observed for other subsets of samples as defined by the area of habitation and lemur species, as we showed in a previous study64 (Supplementary Material S1, Table S1).

Study sites and the geographic ranges of the different Eulemur species (Google Maps, 2015). Left: Map of Madagascar with the study sites Ranomafana NP (I), Nosy Be, Nosy Komba, and Nosy Tanikely (II), Andasibe NP (III), Ankarafantsika NP (IV), Kirindy Forest Reserve (V), Zombitse NP (VI). Right: the geographic ranges of (a) E. rubriventer, (b) E. rufifrons, (c) E. macaco, d) E. fulvus65. Downloaded from The IUCN Red List of Threatened Species. Version 2016–3. www.iucnredlist.org on 12 February 2017. (a) Eulemur rubriventer—IUCN (International Union for Conservation of Nature) 2014. Eulemur rubriventer. The IUCN Red List of Threatened Species. Version 2016–3; (b) Eulemur rufifrons—IUCN (International Union for Conservation of Nature) 2014. Eulemur rufifrons. The IUCN Red List of Threatened Species. Version 2016–3; (c) Eulemur macaco—IUCN (International Union for Conservation of Nature) 2014. Eulemur macaco. The IUCN Red List of Threatened Species. Version 2016–3; (d) Eulemur fulvus—IUCN (International Union for Conservation of Nature) 2014. Eulemur fulvus. The IUCN Red List of Threatened Species. Version 2016–3.

Lemur faecal microbiota composition across seasons and locations. Ordination of faecal microbial composition in multiple lemur populations across Madagascar samples in different seasons (early dry and early wet) and locations. This figure shows the results of a principal coordinate analysis (PCoA) based on the weighted UniFrac distance matrix, grouping strength of samples by season- R2 = 0.09 (Adonis; P = 0.001).

Lemur faecal microbiota composition across seasons and locations. dbRDA Analyses of the abundance-weighted phylogenetic composition at OTU level of individual lemurs across seasons (early dry and early wet) in different geographic areas visualised in ordination. Faecal microbiota significantly cluster by season. Results are given for the percentage of variation explained by the sum of the first two canonical axes, percentage explained by season with corresponding P-value. (a) Eulemur fulvus in Ankarafantsika National Park, (39.8%, 16.9%, P = 0.001). (b) Eulemur fulvus in Andasibe (46.5%, 20.2%, P = 0.001. (c) Eulemur rufifrons in Ranomafana NP (31.1%, 13.5%, P = 0.001). d) Eulemur rubriventer, Ranomafana NP (31.3%, 12.5%, P = 0.001).
Based on morphological analyses, nematode species of two genera, Callistoura and Lemuricola, were present in the GI tract of nearly all Eulemur individuals from eight geographically distinct populations (Fig. 4, Supplementary Material S1). Of all the sampled lemurs (N = 335), we detected eggs of Callistoura spp. in 188 (56.1%), eggs of Lemuricola spp. in 17 (5.1%), eggs of both parasite species in 34 (10.1%), and no eggs in 96 (28.7%) of the lemurs (Table 2). The observed co-occurrence (10.1%) is very close to the expected co-occurrence for independent infections (67.5% × 15.1% = 9.9%), suggesting that infections with both Callistoura and Lemuricola spp. occur independently, and therefore, coinfection appears to be independent.

Detected parasite species. Callistoura sp. egg (left) and Lemuricola sp. egg (right), isolated from a faecal sample of Eulemur rufifrons, magnification 200x (picture taken by IdW).
In the analysis of Callistoura spp. prevalence across seasons and locations using GLMMs, we did not find alarming problems regarding model diagnostics (Supplementary Material S1). A highly significant full model was found (LRT; P < 0.001, Supplementary Material S1; Table S1; model SC1 vs SC2). This result was solely attributed to the random part of the model (SC1 vs SC3), with a larger part being explained by variation among social groups, representing binomial overdispersion (SC1 vs SC5, P = 0.0004), compared to variation among sites (SC1 vs SC6; P = 0.021). We found no significant effect of species, location, season, and the interaction between location and season (SC1 vs SC4; P = 0.20). The hypothesised interaction of location and season was not significantly different from zero (SC1 vs SC7; P = 0.32).
Also, in the analysis of Lemuricola spp. prevalence across seasons and locations using GLMMs, no alarming problems regarding model diagnostics were found (Supplementary Material S1). The full model explained a significant amount of variation (Supplementary Material S1; Table S1; model SL1 vs SL2; P = 0.012). We did not find significant variation due to random effects for sites or social groups, and hence no binomial overdispersion (SL1 vs SL3; P = 0.50), but the fixed part of the model was significant (SL1 vs SL4; P = 0.026). The hypothesised interaction of location and season was not found (SL1 vs SL6; P = 0.84), but the location main effect was significant (SL1 vs SL7; P = 0.032). The Lemuricola spp. prevalence was estimated as 25% (95% Confidence Interval (CI): 16%-37%) in the dry Western areas compared to 10% (5%-20%) in the wet Eastern areas.
No difference in infection prevalence of Callistoura spp. between animals with and without Lemuricola spp. infection was found (LRT; P = 0.37). Overall, we found that 188 out of 284 lemurs without Lemuricola spp. were infected with Callistoura spp. (66%), and 34 out of 51 Lemuricola spp. infected animals were infected with Callistoura spp. (67%).
Disturbance
A possible association between forest disturbance and parasite infection and faecal bacterial microbiota composition was examined in lemurs from Ranomafana NP (Fig. 5). Bacterial richness was significantly higher in the previously logged site (Talatakely, N = 29), compared to the less disturbed site (Vatoharanana/Valohoaka N = 27) (PD index = 7.3 ± 1.1 vs 5.8 ± 1.7, P = 0.001). The dbRDA also showed that the microbial composition was grouped according to sites with a different disturbance history (P = 0.004, Fig. 5).

Faecal microbiota composition in disturbed and less disturbed sites. Ordination (RDA) of the microbial composition (OTU) across sites with a different disturbance history (disturbed vs. less disturbed) for Eulemur rubriventer and E. rufifrons in Ranomafana National Park, Madagascar. Cumulative variation explained by the first two axes was 26.7% and the sampling location accounted for 3.8% of the total variation (P = 0.002).
In the analysis of prevalence of Callistoura spp. after model checking (Supplementary Material S1), we did not find an overall significant effect of species, disturbance, season or their interaction (Supplementary Material S1; Table S1; model DC1 vs DC2, P = 0.185). When we focus on the specific hypothesis on disturbance, though, a significantly different prevalence between the two subsites was found (model DC1 vs DC3, P = 0.042). The prevalence of Callistoura spp. in the non-disturbed subsite was 85% (95% CI 72–93%) and in the disturbed subsite 53% (36–70%, Fig. 6).

Parasite prevalence with disturbance. (a) Callistoura spp. prevalence, (b) Lemuricola spp. prevalence in Eulemur rufifrons and E. rubriventer populations in a previously disturbed and less disturbed site in Ranomafana NP, Madagascar. Mean with 95% confidence intervals and the letter coding above the bars indicate whether groups are significantly different.
In the analysis of the prevalence of Lemuricola spp., the omnibus test showed highly significant effects of species (Supplementary Material S1; Table S1; model DL1 vs DL2; P < 0.001), which could not be traced back to differences between subsites with different disturbance histories (model DL1 vs DL4; P < 0.001). The prevalence in the less disturbed compared to the previously logged subsite was estimated as 1.2% (95% CI 02–09%) compared to 26% (15–47%) in the less disturbed subsite. The infection rates of Callistoura spp. showed considerable extrabinomial variation, but the infection rates of Lemuricola spp. did not.
Microbiota and parasites
Constrained ordination (dbRDA) showed that the prevalence of Callistoura spp. accounted for 0.4% (P = 0.024) of the variation in microbiota composition found among all samples with available microbial and parasite infection data (N = 324), controlling for host species and habitation64. However, we did not observe significant (P = 0.49) influence of Lemuricola spp. prevalence on microbiota composition. When samples from different seasons were analysed separately, we observed an increase of the relative weight of explained variations to 0.7% with maintained, albeit decreased (due to smaller sample size) significance (P = 0.05) in samples collected during the early-wet season, and no significant influence during the early-dry season.
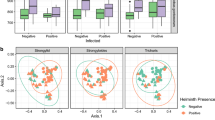
When focussing on lemurs of one species from the same area and season, we could not find statistically significant correlations with Callistoura spp. prevalence. However, among the E. rubriventer population in Ranomafana NP in the early dry season, microbiota composition showed a nearly significant correlation with Lemuricola spp. prevalence (P = 0.055) with 9.2% of variation explained by this factor. Interestingly, a clear separation of samples could be observed in the corresponding dbRDA plots (Figs. S1, S2), albeit without statistical support (all P > 0.05), potentially due to the relatively low and unequal number of samples per group.
Discussion
We assessed the influence of environmental conditions on the faecal bacterial microbiota composition and parasite infections as well as the correlation between GI microbiota and parasites in wild lemurs. The two helminth genera Callistoura66 and Lemuricola67 were the only two nematode genera detected in all Eulemur populations. These microphagous pinworms belong to the family Oxyuridae and are directly transmitted68. They colonise distinct parts of the GI tract of their hosts: Callistoura spp. lives in the ileum and colon and Lemuricola spp. in the caecum and colon68. These parasite species were also found in most other lemur genera67, 68, including other species from the genus Eulemur, i.e. in E. flavifrons69, E. macaco70, E. fulvu71, and E. albifrons72. So, these nematode genera have an extensive distribution throughout Madagascar and do not show apparent specificity to a particular kind of lemur host68.
We hypothesised that lemurs inhabiting dry deciduous forests, with substantial seasonal variation in rainfall and temperature, would show larger seasonal contrasts in both parasite infections and microbial composition compared to lemurs in eastern rainforests with relatively low seasonal variation. Nevertheless, we found a strong seasonal contrast in the microbial composition at the Organisational Taxonomic Unit (OTU) level across all lemur populations. Across Madagascar, lemurs are exposed to seasonality and have been observed to change their diet accordingly63. Diet was found to be an important driver of the GI microbial composition in many human studies (e.g.17). Although humans are assumed to have a stable microbiota over longer periods (> 10 days)73, dietary changes can alter the relative abundance of specific members of the microbiota within 24 h19. With respect to wildlife, e.g., wood mice (Apodemus sylvaticus) were shown to exhibit seasonal shifts in gut microbiota structure that coincide with their annual dietary changes21. Also, in wild Mexican black howler monkeys, temporal changes in the relative abundance of gut bacteria were strongly correlated with dietary variations24. Another study on Eulemurs show that difference in diet in geographically separated population strongly influence intestinal microbiota64. Hence, seasonal diet shifts are likely to explain most of the variation in microbiota in lemurs across seasons.
In addition, the microbial alpha richness from lemurs in Ankarafantsika National Park74 was higher in the early dry season compared to the early wet season. Over the dry season, lemurs experience conditions of relatively low temperatures and food and water restriction, especially in the dry western parts of Madagascar. This nutritional stress may result in a narrower diet, and the microbiota would be more precisely adapted to the food items available. This more restricted diet during the dry season could, therefore, explain the gradual decrease in microbiota richness that we observed. Such dietary change might lead to an altered microbial composition, which potentially facilitates the digestion of specific food items. It is tempting to speculate that this could also lead to an increased caloric intake, which might contribute to increased fitness of both the host and microbiota21.
The presence of different fruit trees results in large dietary differences across populations75, 76. For example, the four most predominant food items consumed by E. fulvus in Ankarafantsika in the early and early wet season, were Buddleja madagascariensis, Psychotria sp., Vitex perrieri and Diospyros tropophylla)75, 77, species that do not occur in Nosy Tanikely or Andasibe76. Furthermore, introduced mango trees (Mangifera indica) are only consumed at Nosy Tanikely. However, there is also some dietary overlap across populations, i.e., Dichapetalum leucosia and Landolphia myrtifolia were consumed by E. fulvus in both Ankarafantsika and Andasibe. Despite the overlap in some fruit species, the geographically separated populations of this lemur species showed major dietary differences, probably leading to major variations in microbiota composition in these populations.
We found a slight, but not significant, indication that parasite infections in the dry regions of Madagascar showed larger seasonal contrasts compared to the eastern rainforest. Another study found a higher parasite richness in areas with a large precipitation range throughout the year78. Many parasites require a certain temperature and humidity to complete their life cycles78 or as microhabitats for their larva79. The drier conditions towards the end of the dry season can prevent egg development and can lead to desiccation of the fragile eggs31. However, some related nematode species are able to survive such short periods of drought by entering a state of hypobiosis, until humidity conditions improve to the point where free-living larval stages can survive80. In addition to these direct seasonal influences on parasites, the lemur host influences these infection patterns as well. The host’s resource use and diet, in general, are considered as major determinants of host exposure to parasites31. It was also experimentally established that host foraging ecology has important consequences for the exposure to and transmission of parasites81. Food scarcity for lemurs is relatively high towards the end of the dry season82, 83 and the associated nutritional stress can have a repressive effect on the hosts’ immune system, which may result in a higher susceptibility to parasite infection40.
Seasonal changes in lemur reproductive status can also lead to changes in parasite infections patterns84. The early dry season coincides with the mating season of Eulemurs85, and more frequent physical contact both within and between lemur groups during this period may enhance parasite infection84. Besides, androgen and glucocorticoid levels of the males and oestrogen levels of the females increase during the mating season, which can lead to a higher susceptibility to parasite infections due to their repressive effect on the immune system86. Furthermore, the early wet season coincides with the weaning season, a season that is energy demanding, especially for lactating females. These behavioural and physiological differences may thus lead to differences in parasite infection status across different seasons. This hypothesis has yet to be explored with data to consider the likelihood of particular explanatory factors.
We also did not find an interactive effect of the two nematode species as coinfection appears to be independent. Lemuricola and Callistoura spp. colonise distinct parts of the gastrointestinal tract of their hosts, the caecum-colon and ileum-colon respectively68, which can explain the lack of interactions between these two species.
We hypothesised the microbiota composition to be altered and parasite infection prevalence to be increased in lemurs whose habitat is restricted to more intensely logged forests. For the microbial composition, we found statistically significant variation between samples taken at a previously logged and at a less disturbed site. Moreover, a higher richness of microbial consortia was observed in the logged area. Other studies that have addressed the impact of anthropogenic disturbance on the gut microbiota of wild primates seem to contradict our findings. For example, habitat disturbance was reported to lead to reductions in Alouatta gut microbial diversity22, and a similar pattern was found in Udzungwa red colobus monkeys14. These results may reflect a general trend of habitat degradation and reduced diversity in the ecological pool of microbial taxa available to colonize hosts22. However, the number of studies in this field is minimal. Besides, the type and intensity of anthropogenic disturbance and the forests’ regeneration time may be important as well87. Logging in our sites occurred nearly thirty years ago, and sites have been regenerating since88, which can explain the deviating patterns that were found in this study. Nevertheless, these forests still differ to a large extent in their structural characteristics, as well as tree species composition89, which may explain the differences in microbiota composition we found.
We found a relatively high abundance of Cyanobacteria in the Eulemur population in the less disturbed compared to the previously logged site. Sequences identified as Cyanobacteria are most probably derived from their non-photosynthetic gut dwelling siblings90. Even though they are part of the normal gut microbiota of mammals, it is not clear which role they play in intestinal ecosystems.
Concerning parasites, the prevalence of Lemuricola spp. was significantly higher in the more intensely disturbed site compared to the less disturbed site, while Callistoura spp. prevalence showed no such pattern. Selective logging results in a suite of alterations that may increase infection risk and susceptibility to certain parasite infections in resident populations39. For example, studies on howler monkeys have reported higher GI parasite diversity and abundance in primates inhabiting degraded areas compared to those in less disturbed areas91. The depletion of the GI microbiota in degraded environments may explain these patterns. However, other studies show only minimal effects of disturbance on patterns of intestinal parasite infection92. As mentioned above, our logged forest site has been regenerating over decades, and it seems that lemurs have been able to adapt to differences in food availability and forest structural differences accordingly89. As eggs of Lemuricola spp. are deposited in the perianal region of their host68, body contact and grooming behaviour may be important factors in explaining the prevalence of this nematode within a population. Interaction rates and local lemur densities may be increased, and home ranges more restricted in the more intensely logged forest, which has been shown to increase parasite infection risks40, 93. This may explain the higher Lemuricola spp. prevalence we found in these forests.
Several other studies observed a relationship between microbiota and GI parasites21, 48,49,50,51, 54. We found a small but significant correlation of microbial composition with the prevalence of Callistoura spp. In addition, the lemur population in Ankarafantsika had a significantly lower infection prevalence of Callistoura spp. compared to lemur populations in other areas and at the same time, this population showed the highest microbiota richness. Despite the statistical significance of the correlations, interpretation of this correlations should be made with care. On the one hand, GI parasites can have a direct influence on intestinal microbiota by damaging the host’s intestinal epithelium, extracting nutrients in the GI tract59, secreting antimicrobial products or inducing an inflammatory response94. On the other hand, observed correlations could not provide direct evidence for these mechanistic aspects. The microbiota is a dynamic ecosystem that has been shown to be affected by a broad range of environmental factors. However, the effect of factors with smaller relative weight is often masked by individual-specific factors like diet and genetic background95. This could incorrectly reflect the true importance of such minor factors, particularly in wildlife studies where individual variation cannot easily be controlled.
Several studies found that the presence of some nematode species is linked to high microbiota diversity, with potential beneficial consequences for host health47, 58, 60, 94, 96. It is assumed that the GI microbiota regulate the immune system, but also that GI nematodes can alter the bacterial composition and structure, thereby creating conditions that can facilitate nematode infestations96. Although it has been shown that some parasites change environmental conditions prevailing in the intestine, and thus affecting also microbial habitats, the exact relations between parasites and the microbiota remain unclear97. Most parasite species, and directly transmitted parasites in particular, co-evolve in association with only a few host species and adapt to the host gut environment and diet, resulting in host-driven diversification98 that allowed to speculate about microbe-parasites evolutionary crosstalk. Understanding underlying mechanisms is critical for improving our knowledge of parasite–microbe interactions in wild primate populations. This can become achievable with a greater longitudinal sampling effort, refined, standardised sample preservation protocols, genetic identification of the nematodes with molecular methods, and if possible in vitro and in vivo model experiments.
In conclusion, this study investigated the impact of seasonality and past logging on host-associated parasite infections, faecal bacterial communities, and correlative patterns between these GI inhabitants in geographically separated Eulemur populations. Our results show that seasonal differences and past logging events significantly contributed to explaining the observed temporal variations in parasite infections and microbial diversity. The variation in microbiota composition at the genus level showed a significant correlation with the presence of parasites, suggesting a relationship between gastrointestinal parasites and microbiota composition under natural conditions. The factors that influence microbiota composition and presence of parasites may, in turn, affect host nutrition, behaviour, and health. These findings likely apply to other wild mammal communities as well. We believe it is crucial to consider the potential role of microbiome-parasite associations on the hosts’ GI stability, health, and survival.
Methods
Study site
Our research was performed in eight geographically distinct sites (Fig. 1, Table 1). Kirindy Forest, Ankarafantsika National Park (NP), and Zombitse NP are located on the western, north-western, and south-western side of Madagascar, respectively. They consist of dry deciduous forest with pronounced seasonality 99. These western regions have a higher annual mean temperature than the eastern rainforests but receive less rainfall.
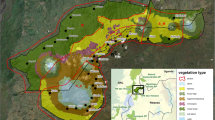
In contrast, Andasibe Mantadia NP and Ranomafana NP are located on the eastern side of Madagascar. They are relatively wet rain forests with a less distinct dry season compared to the western areas100. Within Ranomafana NP, we distinguished two research sites, Talatakely (TALA) and Vatoharanana-Valohoaka (VATO-VALO) with different degrees of anthropogenic disturbance (Fig. 7)89, 101. Before the establishment of the national park in 1991, the forests in this area were used by local inhabitants, amongst others for slash-and-burn agriculture100. Now, more than 25 years after the last logging activities, Ranomafana NP shows a high heterogeneity in forest structure.

Map of Ranomafana National Park and the two forest sites that were surveyed in this study. Talatakely (white dot) experienced relatively intense logging in the past, while Vatoharanana- Valohoaka (black dot) experienced no such disturbances. This map was generated via ArcGIS version 10.5. Data was downloaded from UNEP-WCMC and IUCN (2016), Protected Planet: [National Parks of Madagascar; The World Database on Protected Areas (WDPA) [On-line], [May 2016], Cambridge, UK: UNEP-WCMC and IUCN.
The islands Nosy Be, Nosy Komba, and Nosy Tanikely, are located in the north-west of Madagascar. The forests of Nosy Be (~ 320 km2) are largely replaced by coffee, fruit, and ylang-ylang plantations, and by rice and sugar cane fields. Only Lokobe NP (~ 7 km2) at the south-eastern part of the island still contains the island’s original forest vegetation. Nosy Komba and Nosy Tanikely are located in between Nosy Be and the mainland. The vegetation on Nosy Komba (~ 25 km2) is similar to Nosy Be. The vegetation at Nosy Tanikely (~ 0.06 km2) mainly consists of low forest and bushy vegetation, including palm trees and planted banana and mango trees, surrounded by a sandy shore with large rock formations (de Winter, pers. obs.102).
Study species
True lemurs (genus Eulemur, family Lemuridae) are medium-sized (body and tail length 30–50 cm, 2–4 kg) arboreal primates that occasionally move on four legs on the ground. They live in social groups ranging from two to fifteen individuals, and their diet primarily consists of fruits, flowers, and leaves62. We studied four Eulemur species: Eulemur rufifrons, E. fulvus, E. macaco, and E. rubriventer. The main difference between the Eulemur species is their group size E. rufifrons, E. macaco, and E. fulvus live in multi-male, multi-female groups from four to 18 individuals103,104,105, whereas E. rubriventer lives in small monogamous groups from two up to five individuals106. Eulemur rufifrons lives in the southwest and east, and the native range of Eulemur fulvus is in the north of Madagascar, on both the east and west side107. This species has also been introduced to the northern island Nosy Tanikely. Eulemur macaco is found on the mainland and several islands in the north-west, while Eulemur rubriventer inhabits forests in eastern Madagascar (Fig. 1, Table 1). Eulemur rubriventer and E. rufifrons live sympatrically in Ranomafana NP108.
Faecal sample collection
We collected 338 faecal samples between October 2013 and February 2015 (Table 1), of which 133 were also used in a previous study64. Within Ranomafana NP, we collected 103 samples; 38 samples from a previously logged site (Talatakely) and 65 from a site that was considered a less disturbed site in terms of its logging history (Vatoharanana-Valohoaka). Immediately after defecation, fresh faecal samples (3–4 g) were collected non-invasively. We noted visual characteristics, i.e., consistency, colour, presence of blood, mucus, or tapeworm proglottids. We also reported GPS coordinates, time, group size, group composition, age (sub-adult if ≤ 2 years old or adult if ≥ 3 years old), and sex. We allocated a body fur condition score to the individuals whose faeces were collected109. We aimed at sampling all adults within a social group, and we did not resample the same individuals. When encountering a new social group, we first recorded as many distinguishable features as possible of all lemur individuals. As soon as we were not sure whether the faeces were from a new individual or whether we already sampled the animal, we moved on to another group. As we worked mostly within National Parks or Reserves, the lemurs were all habituated to human observers, mainly due to the frequent visits by tourists or researchers, which facilitated the faecal collection. We found no abnormalities in the consistency and colour of the faeces and we did not find blood, mucus or tapeworm proglottids in any of the faecal samples. Within 12 h after collection, each faecal sample was divided over two sterile tubes: one gram of faeces was stored in a tube filled with 5 ml of 70% ethanol (Supplementary Material S2) and two grams of faeces was placed in a tube filled with 15 ml SAF fixative44, 110. Samples were analysed at the Laboratory of Microbiology, Wageningen University & Research, and the Department of Infectious Diseases and Immunology, Utrecht University. All described methods were performed in accordance with the relevant guidelines and regulations and were approved by the trilateral commission (CAFF/CORE) in Madagascar (permits 297/13 and 143/14/MEF/SG/DGF/DCB.SAP/SCBSE).
DNA-based bacterial composition analyses
Faecal bacterial microbiota composition, determined by next-generation sequencing of 16S rRNA gene fragments, was used as a proxy for the intestinal microbial community. We extracted microbial DNA from the faecal samples collected in Ranomafana NP following a modified double bead-beating procedure using the QIAamp® DNA Stool Mini Kit (Qiagen, Valencia, CA, USA) (Based on111). For the sample processing, we used the protocol proposed by Yu and Morrison (2004), modified by112. Prior to DNA extraction, faecal material was air-dried during 15–20 min in a fume hood to remove ethanol from samples. We extracted DNA from samples collected at the other sites using the Maxwell® 16 Research Instrument (Promega, Madison, USA) in combination with the corresponding RNA extraction kit customised for faecal DNA extraction according to manufacturer’s instructions. Prior to DNA extraction, samples were rehydrated through a series of ethanol solutions with decreasing proportions of ethanol in steps of 10%. For rehydration, 1.5 ml of 70% ethanol with faecal particles was transferred into a fresh 2 ml tube and centrifuged at 13,000 rpm for 5 min. After centrifugation, part of the supernatant was replaced with the same amount of distilled water to decrease ethanol concentration by 10%, vortexed, and incubated for 10 min at RT. These steps were repeated until the ethanol was entirely replaced by distilled water. Cell disruption and lysis were performed as described above, but instead of lysis buffer, we used S.T.A.R buffer (Roche Molecular Systems, USA). DNA quality and concentration were spectrophotometrically verified (Nanodrop Technologies, Wilmington, USA. For each sample, barcoded amplicons were generated from 40 ng of extracted DNA using a two-step PCR method in a LabCycler Gradient (SensoQuest, Germany) and pooled afterwards as described previously113. Briefly, the V1–V2 region of the 16S rRNA was first amplified by PCR (25 cycles of 95 °C (30 s), 52 °C (40 s), and 72 °C (90 s)), followed by post-elongation (72 °C, 7 min) using primer pair 27F–DegS: 5′-GTTYGATYMTGGCTCAG-3′114 and 338R–I: 5′-GCWGCCTCCCGTAGGAGT-3′/338R–II: 5′-GCWGCCACCCGTAGGTGT-3′115 that contained forward and reverse linkers UniTag I (5′-GAGCCGTAGCCAGTCTGC-3′) and UniTag2 (5′-GCCGTGACCGTGACATCG-3′), respectively. Amplicons were then used as a template for a second PCR in order to introduce sample-specific barcodes, using individual barcode primers targeting Unitag1 and UniTag2 sequences. The amount and size of the amplicons were checked visually by agarose gel electrophoresis. The PCR products were purified using the HighPrepTM PCR kit (MagBio Genomics), concentrated using magnetic beads (MagBio, Switzerland) according to the HighPrep protocol, quantified using the Qubit dsDNA BR Assay Kit (Life Technologies, USA), and pooled in equimolar amounts into libraries of 48 samples, including two mock communities of defined composition, for paired-end sequencing (300 bp) on the Illumina Miseq platform at GATC Biotech (Konstanz, Germany; now part of Eurofins Genomics Germany GmbH). Mock communities i.e. mixes of quantified and purified copies of bacterial 16 s rRNA genes in known proportions, are routinely used in our laboratory to assess quality and reliability of a sequencing run, amplicon preparations, and quality of data processing, as was described previously116.
The amplicon sequences were demultiplexed, and the subsequent analysis of raw rRNA gene sequence data was performed using NG-Tax116. Reads assigned to OTUs of plant origin such as chloroplast and plant mitochondrial DNA were removed from the dataset used for downstream analysis. The raw data was ranked per individual sample based on the matching of reads to OTUs, allowing an error of one nucleotide.
Parasite isolation
The collected faecal samples were examined for the presence of GI nematodes with the use of the Centrifugation-Sedimentation-Flotation (CSF) method117. GI nematode species identification was based on morphological traits such as colour, shape, size, and content of eggs68, 84, 118. A rough estimation of the number of parasite eggs per gram of faeces (EGP) was obtained by simple counts. Since the number of eggs that end up in the faeces is not a reliable index of adult worm burden119, the egg count cannot be regarded as a measurement of infection intensity, but rather as a measurement of infectivity.
Statistical analysis
After initial sequence data processing with NG-tax, we combined the OTU table, metadata, and phylogenetic tree into a “phyloseq” object, as implemented in the “phyloseq” R package (v.1.22.3). Further analyses were carried out in R (v 3.4.1) (Supplementary Material S2). OTUs that were encountered in less than three samples, OTUs not assigned to any taxonomic level (NA) and OTUs identified as chloroplast and mitochondria were removed. In addition, samples with a low number of the reads (less than 1000 reads), missing metadata of interest, and one sample (i.e., ‘NT9F’, due to the low quality of the starting material) were removed from the data set (Supplementary Material S1, Table S2, and Supplementary Material S2). For beta diversity analysis, the weighted UniFrac distance matrix was calculated from the OTU table and phylogenetic tree as implemented in the “phyloseq” package, with the phylogenetic tree rooted at the midpoint (package “phangorn”). Multidimensional scaling with weighted UniFrac as a distance matrix (PCoA) was applied (package “phyloseq”) to obtain a first insight into the beta diversity of faecal microbial communities in the investigated lemur populations. We used dbRDA (dist.wu ~ Site + Species + Fieldseason + CallPrev + Sex + Age + LemPrev) and an ANOVA like permutation test (anova.cca; permutations = 9999) to identify variables that significantly contribute to explaining the observed variation in microbial composition (package “vegan”). To this end, we employed dbRDA to enhance visualization of the results when the focus shifted from providing an overall picture to investigating individual factors. Constrained ordination techniques, such as dbRDA, allow researchers and readers to zoom in on a two-dimensional representation with the best separation of samples. Variable “Social Group” was excluded from the analysis due to extremely uneven sample distribution (Supplementary Material S2, Tables S1, S3), with 28 out of a total of 92 social groups including only one sample. The degree to which individual factors could explain microbiota composition was estimated by partial dbRDA with control for variables that were not used as a constraint. R2 values were used as an estimator of variation explained by a constraint (package “vegan”). Phylogenetic diversity was used as a primary alpha diversity measure. It was calculated from the phyloseq object with the OTU table rarefied at a read depth of 1051, using a custom function (author Thomas W. Battaglia, https://github.com/twbattaglia). Statistical differences between alpha diversity of pre-defined sample groups were assessed by posthoc Kruskal Nemenyi-tests (package “PMCMR”). The datasets generated during this study are available in the Supplementary Material S1 and the public read archive EBI, study name ‘ena-STUDY-WAGENINGEN UNIVERSIT-03-04-2017–14:57’, with accession number ‘PRJEB20227’ (link: https://www.ebi.ac.uk/ena/data/view/PRJEB20227).
To analyse the effect of seasonality (early dry vs early wet) and location (western dry deciduous forests vs eastern rainforests) on the infection prevalence of Callistoura and Lemuricola spp. in Eulemur species, GLMMs were used, assuming a Binomial distribution and logit link function for data aggregated per social group. We included random effects for sites within a location and observation-level random effects for social groups, and fixed effects for species, season, location, and the interaction between season and location. The observation-level random effects handle possible binomial overdispersion. The factor species entered the model as a control variable to avoid confounding of location effects with species effects. We focused specifically on the interaction between location and season to test the seasonality hypothesis as formulated in the Introduction. To present estimated infection prevalence with 95% confidence intervals (CI) on the probability scale, we back-transformed the results (on the logit-scale) from the GLMMs first. Next, we applied a shrinkage factor120, which is needed for GLMMs, to obtain predicted population means instead of medians. To test whether infections by the two nematode genera occurred independently, we modified the GLMM for Callistoura spp., using unaggregated data, by adding an indicator variable for Lemuricola spp. as a regressor to the model. In this way, we allowed the infection prevalence for Callistoura spp. could be different among lemurs with or without Lemuricola spp. infections.
In a subset of the data (Ranomafana NP; N = 103 individuals of E. rubriventer and E. rufifrons only), we analysed Callistoura and Lemuricola spp. infection prevalence comparing disturbed and less disturbed subsites. Again, we aggregated infection scores per social group and used ordinary Generalised Linear Models (GLMs) assuming a binomial distribution for the number of infected animals per social group and logit link function. We entered effects for the control factor species and for the main factors of interest: disturbance (less vs more disturbed subsites), season (early dry vs early wet), and their interaction into the model. In the analysis of Callistoura spp. prevalence, a smaller model was fitted due to the low numbers of cases (14 cases, with just 1 in the less disturbed site). Extra-binomial variation could not be ruled out, because individuals within social groups may have correlated responses. Because of different group sizes (range 1–7), we used Williams’ method as available in the dispmod package of R121. If the overdispersion was not present, when judging the residual deviance, we used an ordinary binomial GLM. We calculated back-transformed predicted means presented with 95% CI for the previously disturbed and less disturbed sites.
Model assumptions were checked by inspection of residuals, leverages, and collinearity statistics, and model stability (for GLMM) and dfbetas (for GLM) were assessed. After model checking, comparisons of the full model (separately for the analyses of Callistoura and Lemuricola spp. infections, seasonality, and disturbance analyses) with reduced models were made using likelihood ratio tests (LRT), followed by tests for individual factors in case of significant results. Regardless of the results from omnibus tests, we tested the specifically formulated hypotheses regarding seasonality and forest disturbance (see “Introduction”). Pseudo R2122 for the full models were calculated (results are shown in Supplementary Material S1, Table S1).
We performed the statistical analyses in base R123, using the R packages lme4 for the GLMMs124 and emmeans125 for prediction of group means (Supplementary Material S2), with car126 for variance inflation factors, DHARMAa127 for residual checking in GLMMs, and MuMIn128 for creating pseudo R2 values (elaborate results of statistical analyses are presented in the Supplementary Material S2).
References
Pfeiffer, J. K. & Virgin, H. W. Transkingdom control of viral infection and immunity in the mammalian intestine. Science 351, 5872 (2016).
Ramanan, D. et al. Helminth infection promotes colonization resistance via type 2 immunity. Science 352, 608–612 (2016).
Leclaire, S. & Faulkner, C. T. Gastrointestinal parasites in relation to host traits and group factors in wild meerkats Suricata suricatta. Parasitology 141, 925–933 (2014).
Kabat, A. M., Srinivasan, N. & Maloy, K. J. Modulation of immune development and function by intestinal microbiota. Trends Immunol. 35, 507–517 (2014).
McHardy, I. L. X. T. M. et al. HIV Infection is associated with compositional and functional shifts in the rectal mucosal microbiota. Microbiome 1, 1 (2013).
Sekirov, I., Russell, S. & Antunes, L. Gut microbiota in health and disease. Physiol. Rev. 90, 859–904 (2010).
de Vos, W. M. & de Vos, E. A. Role of the intestinal microbiome in health and disease: from correlation to causation. Nutr. Rev. 70, 45–56 (2012).
Patterson, E. et al. Gut microbiota, the pharmabiotics they produce and host health. Proc. Nutr. Soc. 73, 477–489 (2014).
Bennett, G. et al. Host age, social group, and habitat type influence the gut microbiota of wild ring-tailed lemurs (Lemur catta). Am. J. Primatol. 1, 1–10 (2016).
Belongia, E. A. Epidemiology and impact of coinfections acquired from Ixodes ticks. Vector-Borne Zoonotic Dis. 2, 265–273 (2002).
Eckburg, P. B. et al. Diversity of the human intestinal microbial flora. Science 308, 1635–1638 (2015).
Tompkins, D. M., Dunn, A. M., Smith, M. J. & Telfer, S. Wildlife diseases: from individuals to ecosystems. J. Anim. Ecol. 80, 19–38 (2011).
Hansen, J., Gulati, A. & Sartor, R. B. The role of mucosal immunity and host genetics in defining intestinal commensal bacteria. Curr. Opin. Gastroenterol. 26, 564–571 (2010).
Barelli, C. et al. Habitat fragmentation is associated to gut microbiota diversity of an endangered primate: implications for conservation. Sci. Rep. 5, 14862 (2015).
McKenney, E. A., Rodrigo, A. & Yoder, A. D. Patterns of gut bacterial colonization in three primate species. PLoS ONE 10, 1–18 (2015).
Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansson, J. K. & Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230 (2012).
De Filippo, C. et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. 107, 14691–14696 (2010).
Muegge, B. D. et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Proc. Natl. Acad. Sci. 332, 970–974 (2012).
Wu, G. D. et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108 (2011).
Boutin, S., Bernatchez, L., Audet, C. & Derom̂e, N. Network analysis highlights complex interactions between pathogen, host and commensal microbiota. PLoS ONE 8, 1–16 (2013).
Maurice, C. F. et al. Marked seasonal variation in the wild mouse gut microbiota. ISME J. 9, 2423–2434 (2015).
Amato, K. R. et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 716, 1344–1353 (2013).
Dishaw, L. J. et al. The gut of geographically disparate Ciona intestinalis harbors a core microbiota. PLoS ONE 9, e93386 (2014).
Amato, K. R. et al. The gut microbiota appears to compensate for seasonal diet variation in the wild black howler monkey (Alouatta pigra). Microb. Ecol. 69, 434–443 (2015).
Moore, S. L. & Wilson, K. Parasites as a viability cost of sexual selection in natural populations of mammals. Science 297, 2015–2018 (2002).
Nunn, C. L., Thrall, P. H., Leendertz, F. H. & Boesch, C. The spread of fecally transmitted parasites in socially-structured populations. PLoS ONE 6, e21677 (2011).
Huffman, M. A., Gotoh, S. & Turner, L. A. Seasonal trends in intestinal nematode infection and medicinal plant use among chimpanzees in the mahale mountains. Tanzania. 38, 111–125 (1997).
Benavides, J. A. et al. From parasite encounter to infection: multiple-scale drivers of parasite richness in a wild social primate population. Am. J. Phys. Anthropol. 147, 52–63 (2012).
Barrett, M. A., Brown, J. L., Junge, R. E. & Yoder, A. D. Climate change, predictive modeling and lemur health: assessing impacts of changing climate on health and conservation in Madagascar. Biol. Conserv. 157, 409–422 (2013).
Aivelo, T., Laakkonen, J. & Jernvall, J. Population and individual level dynamics of intestinal microbiota of a small primate. Appl. Environ. Microbiol. 82, 00559–16 (2016).
Nunn, C. C. & Altizer, S. S. Infectious Diseases in Primates: Behavior, Ecology and Evolution (Oxford University, Press, 2006).
Raharivololona, B. & Ganzhorn, J. Seasonal variations in gastrointestinal parasites excreted by the gray mouse lemur Microcebus murinus in Madagascar. Endanger. Species Res. 11, 113–122 (2010).
Huffman, M. & Chapman, C. Primate Parasite Ecology: the Dynamics and Study of Host–Parasite Relationships (Cambridge University, Press, 2009).
Setchell, J. M. et al. Parasite prevalence, abundance, and diversity in a semi-free-ranging colony of Mandrillus sphinx. Int. J. Primatol. 28, 1345–1362 (2007).
Maldonado-López, S., Maldonado-López, Y., Gómez-Tagle, C. A., Cuevas-Reyes, P. & Stoner, K. E. Patterns of infection by intestinal parasites in sympatric howler monkey (Alouatta palliata) and spider monkey (Ateles geoffroyi) populations in a tropical dry forest in Costa Rica. Primates 55, 383–392 (2014).
Caldwell, J. P. Pinworms (enterobius vermicularis). Can. Fam. Physician 28, 306–309 (1986).
Hanson, C. A., Fuhrman, J. A., Horner-Devine, M. C. & Martiny, J. B. H. Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Microbiol. 10, 1–10 (2012).
Keele, B. et al. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science 313, 523–526 (2006).
Gillespie, T. R., Chapman, C. A. & Greiner, E. C. Effects of logging on gastrointestinal parasite infections and infection risk in African primates. J. Appl. Ecol. 42, 699–707 (2005).
Chapman, C. A., Gillespie, T. R. & Goldberg, T. L. Primates and the ecology of their infectious diseases: how will anthropogenic change affect host-parasite interactions?. Evol. Anthropol. 14, 134–144 (2005).
McCord, A. I. et al. Fecal microbiomes of non-human primates in Western Uganda reveal species-specific communities largely resistant to habitat perturbation. Am. J. Primatol. 76, 347–354 (2014).
Chapman, C., Speirs, M. & Gillespie, T. Life on the edge: gastrointestinal parasites from the forest edge and interior primate groups. Am. J. Primatol. 409, 397–409 (2006).
Kowalewski, M. M. et al. Black and gold howler monkeys (Alouatta caraya) as sentinels of ecosystem health: patterns of zoonotic protozoa infection relative to degree of human-primate contact. Am. J. Primatol. 73, 75–83 (2011).
Chapman, C. A. et al. do food availability, parasitism, and stress have synergistic effects on red colobus populations living in forest fragments?. Am. J. Phys. Anthropol. 131, 525–534 (2006).
Hughes, S. & Kelly, P. Interactions of malnutrition and immune impairment, with specific reference to immunity against parasites. Parasite Immunol. 28, 577–588 (2006).
Angelstam, P. et al. Habitat modelling as a tool for landscape-scale conservation : a review of parameters for focal forest birds source. Ecol. Bull. 51, 427–453 (2004).
Kreisinger, J., Bastien, G., Hauffe, H. C., Marchesi, J. & Perkins, S. E. (2015) Interactions between multiple helminths and the gut microbiota in wild rodents. Philos. Trans. R. Soc. B Biol. Sci.370, 20140295.
Mutapi, F. The gut microbiome in the helminth infected host. Trends Parasitol. 31, 405–406 (2015).
Kay, G. L. et al. Differences in the faecal microbiome in schistosoma haematobium infected children vs. uninfected children. PLoS Negl. Trop. Dis. 9, e0003861 (2015).
Lee, S. C. et al. Helminth colonization is associated with increased diversity of the gut microbiota. PLoS Negl. Trop. Dis. 8, 1–10 (2014).
Morton, E. R. et al. Variation in rural African gut microbiomes is strongly shaped by parasitism and diet. bioRxiv (2015).
Cooper, P. et al. Patent human infections with the whipworm, Trichuris trichiura, are not associated with alterations in the faecal microbiota. PLoS ONE 8, 1–10 (2013).
Cantacessi, C. et al. Impact of experimental hookworm infection on the human gut microbiota. J. Infect. Dis. 210, 1–4 (2014).
Houlden, A. et al. Chronic Trichuris muris infection in C57BL/6 mice causes significant changes in host microbiota and metabolome: effects reversed by pathogen clearance. PLoS ONE 10, e0125945 (2015).
McKenney, E. A., Greene, L. K., Drea, C. M. & Yoder, A. D. Down for the count: cryptosporidium infection depletes the gut microbiome in Coquerel’s sifakas. Microb. Ecol. Health Dis. 28, 1335165 (2017).
Fogel, A. T. The gut microbiome of wild lemurs: a comparison of sympatric lemur catta and propithecus verreauxi. Folia Primatol. 86, 85–95 (2015).
Springer, A. et al. Patterns of seasonality and group membership characterize the gut microbiota in a longitudinal study of wild Verreaux’s sifakas (Propithecus verreauxi). Ecol. Evol. 7, 5732–5745 (2017).
Walk, S. T., Blum, A. M., Ewing, S. A. S., Weinstock, J. V. & Young, V. B. Alteration of the murine gut microbiota during infection with the parasitic helminth Heligmosomoides polygyrus. Inflamm. Bowel Dis. 16, 1841–1849 (2010).
Li, R. W. et al. Alterations in the porcine colon microbiota induced by the gastrointestinal nematode Trichuris suis. Infect. Immun. 80, 2150–2157 (2012).
Rausch, S. et al. Small intestinal nematode infection of mice is associated with increased enterobacterial loads alongside the intestinal tract. PLoS ONE 8, 1–13 (2013).
Irwin, M. T., Johnson, S. E. & Wright, P. C. The state of lemur conservation in south-eastern Madagascar: population and habitat assessments for diurnal and cathemeral lemurs using surveys, satellite imagery and GIS. Oryx 39, 204–218 (2005).
Markolf, M. et al. True lemurs…true species: species delimitation using multiple data sources in the brown lemur complex. BMC Evol. Biol. 13, 233 (2013).
Wright, P. et al. Long-term lemur research at Centre Valbio, Ranomafana National Park, Madagascar. in Long-term field studies of primates (eds. Kappeler, P. M. & Watts, D. P.) 67–100 (Springer, Berlin Heidelberg, 2012). https://doi.org/10.1007/978-3-642-22514-7
de Winter, I. et al. Occupancy strongly influences faecal microbial composition of wild lemurs. Microbiol. Ecol. 94, 1–13 (2018).
IUCN. The IUCN Red List of Threatened Species. Version 2016–1. (2016).
Chabaud, A. G. & Petter, A. J. Les nématodes parasites de Lémuriens malgaches, II Un nouvel oxyure: Lemuricola contagiosus. Mém. Inst. Sci. MadagascarA, 127–158 (1959).
Chabaud, A. G., Brygoo, E.-R. & Petter, A.-J. Les Nématodes parasites de Lémuriens malgaches VI. Description de six espèces nouvelles et conclusions générales. Ann. Parasitol. Hum. Comparée 181–214 (1965).
Irwin, M. T. & Raharison, J. A review of the endoparasites of the lemurs of Madagascar. 66–93 (2009).
Schwitzer, N. et al. Parasite prevalence in blue-eyed black lemurs Eulemur flavifrons in differently degraded forest fragments. Endanger. Species Res. 12, 215–225 (2010).
Junge, R. E. & Louis, E. E. Biomedical evaluation of black lemurs (Eulemur macaco macaco) in Lokobe Reserve, Madagascar. J. Zoo Wildl. Med. 38, 67–76 (2007).
Nègre, A., Tarnaud, L., Roblot, J. F., Gantier, J. C. & Guillot, J. Plants consumed by Eulemur fulvus in Comoros Islands (Mayotte) and potential effects on intestinal parasites. Int. J. Primatol. 27, 1495–1517 (2006).
Junge, R. E. et al. Comparison of biomedical evaluation for white-fronted brown lemurs (Eulemur fulvus albifrons) from four sites in Madagascar. J. Zoo Wildl. Med. 39, 567–575 (2008).
Rajilić-Stojanović, M., Heilig, H. G. H. J., Tims, S., Zoetendal, E. G. & De Vos, W. M. Long-term monitoring of the human intestinal microbiota composition. Environ. Microbiol. 15, 1146–1159 (2013).
Crowley, B. E., McGoogan, K. C. & Lehman, S. M. Edge effects on foliar stable isotope values in a Madagascan tropical dry forest. PLoS ONE 7, e44538 (2012).
Sato, H., Ichino, S. & Hanya, G. Dietary modification by common brown lemurs (Eulemur fulvus) during seasonal drought conditions in western Madagascar. Primates 55, 219–230 (2014).
Styger, E., Rakotoarimanana, J. E. M., Rabevohitra, R. & Fernandes, E. C. M. Indigenous fruit trees of Madagascar: potential components of agroforestry systems to improve human nutrition and restore biological diversity. Agrofor. Syst. 46, 289–310 (1999).
Sato, H. Habitat shifting by the common brown lemur (Eulemur fulvus fulvus): a response to food scarcity. Primates 54, 229–235 (2013).
Guernier, V., Hochberg, M. E. & Guégan, J. F. Ecology drives the worldwide distribution of human diseases. PLoS Biol. 2, e141 (2004).
Froeschke, G., Harf, R., Sommer, S. & Matthee, S. Effects of precipitation on parasite burden along a natural climatic gradient in southern Africa: implications for possible shifts in infestation patterns due to global changes. Oikos 119, 1029–1039 (2010).
Brooker, S., Clements, A. C. A. & Bundy, D. A. P. Global epidemiology, ecology and control of soil-transmitted helminth infections. Adv. Parasitol. 62, 221–261 (2006).
Luong, L. T., Grear, D. A. & Hudson, P. J. Manipulation of host-resource dynamics impacts transmission of trophic parasites. Int. J. Parasitol. 44, 737–742 (2014).
Wright, P. C., Vololontiana, R. & Pochron, S. T. The key to Madagascar frugivores. in Tropical fruits and frugivores 121–138 (Springer, Dordrecht, 2005).
Tecot, S. R. Seasonality and predictability: The hormonal and behavioral responses of the redbellied lemur (Eulemur rubriventer) in Ranomafana National Park, southeastern Madagascar (University of Texas, Austin, 2008).
Clough, D. Gastro-intestinal parasites of red-fronted lemurs in Kirindy Forest, western Madagascar. J. Parasitol. 96, 245–251 (2010).
Overdorff, D. J. & Johnson, S. E. Eulemur, true lemurs. in The Natural History of Madagascar (eds. Goodman, S. M. & Benstead, J.) 1320–1324 (University of Chicago Press, Chiago, 2003).
Ostner, J., Kappeler, P. M. & Heistermann, M. Androgen and glucocorticoid levels reflect seasonally occurring social challenges in male redfronted lemurs (Eulemur fulvus rufus). Behav. Ecol. Sociobiol. 62, 627–638 (2008).
Johns, A. D. & Skorupa, J. P. Responses of rain-forest primates to habitat disturbance: a review. Int. J. Primatol. 8, 157–191 (1987).
Wright, P. & Andriamihaja, B. Making a rain forest national park work in Madagascar: Ranomafana National Park and its long-term research commitment. in Making parks work: Strategies for preserving tropical nature 112–136 (2002).
de Winter, I. I. et al. Past disturbance effects on forest structure and lemur abundances (Biol, Conserv, 2018).
Di Rienzi, S. C. et al. The human gut and groundwater harbor non-photosynthetic bacteria belonging to a new candidate phylum sibling to cyanobacteria. Elife 2, 1 (2013).
Vitazkova, S. & Wade, S. Effects of ecology on the gastrointestinal parasites of Alouatta pigra. Int. J. Primatol. 28(28), 1327–1343 (2007).
Martinez-Mota, R. The effects of habitat disturbance, host traits, and host physiology on patterns of gastrointestinal parasite infection in black howler monkeys (Alouatta pigra). PhD dissertation, Department ofAnthropology, University of Illinois (2015).
Arneberg, P. Host population density and body mass as determinants of species richness in parasite communities: comparative analyses of directly transmitted nematodes of mammals. Ecography 25, 88–94 (2002).
Reynolds, L. A. et al. Commensal-pathogen interactions in the intestinal tract: lactobacilli promote infection with, and are promoted by, helminth parasites. Gut Microbes 5, 522–532 (2014).
Zhernakova, A. et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–569 (2016).
Hayes, K. S. et al. Exploitation of the intestinal microflora by the parasitic nematode Trichuris muris. Science 328, 1391–1394 (2010).
Reynolds, L. A., Finlay, B. B. & Maizels, R. M. Cohabitation in the intestine: interactions among helminth parasites, bacterial microbiota, and host immunity. J. Immunol. 195, 4059–4066 (2015).
Pedersen, A. B., Altizer, S., Poss, M., Cunningham, A. & Nunn, C. L. Patterns of host specificity and transmission among parasites of wild primates. Int. J. Parasitol. 35, 647–657 (2005).
Goodman, S. M. et al. The distribution and conservation of bats in the dry regions of Madagascar. Anim. Conserv. 8, 153–165 (2005).
Irwin, M. T. et al. Patterns of species change in anthropogenically disturbed forests of Madagascar. Biol. Conserv. 143, 2351–2362 (2010).
Balko, E. A. & Underwood, H. B. Effects of forest structure and composition on food availability for Varecia variegata at Ranomafana National Park, Madagascar. Am. J. Primatol. 66, 45–70 (2005).
Köhler, J., Glaw, F. & Vences, M. First record of Mabuya comorensis (Reptilia: Scincidae) for Madagascar, with notes on the herpetofauna of Nosy Tanikely. Boll. Mus. Reg. Sci. Nat. Torino 15, 75–82 (1998).
Erhart, E. M. & Overdorff, D. J. Population demography and social structure changes in Eulemur fulvus rufus from 1988 to 2003. Am. J. Phys. Anthropol. 136, 183–193 (2008).
Johnson, S. E., Gordon, A. D., Stumpf, R. M., Overdorff, D. J. & Wright, P. C. Morphological variation in populations of Eulemur albocollaris and E. fulvus rufus. Int. J. Primatol. 26, 1399–1416 (2005).
Pyritz, L. W., Kappeler, P. M. & Fichtel, C. Coordination of group movements in wild red-fronted lemurs (Eulemur rufifrons): processes and influence of ecological and reproductive seasonality. Int. J. Primatol. 32, 1325–1347 (2011).
Tecot, S. R. It’s all in the timing: birth seasonality and infant survival in Eulemur rubriventer. Int. J. Primatol. 31, 715–735 (2010).
Mittermeier, R. A. et al. Lemur diversity in Madagascar. Int. J. Primatol. 29, 1607–1656 (2008).
Overdorff, D. Similarities, differences, and seasonal patterns in the diets of Eulemur rubriventer and Eulemur rufifrons in the Ranomafana National Park, Madagascar. Int. J. Primatol. 14, 721–753 (1993).
Berg, W., Jolly, A., Rambeloarivony, H., Andrianome, V. & Rasamimanana, H. A scoring system for coat and tail condition in ringtailed lemurs, Lemur catta. Am. J. Primatol. 71, 183–190 (2009).
Van Gool, T., Weijts, R., Lommerse, E. & Mank, T. G. Triple faeces test: an effective tool for detection of intestinal parasites in routine clinical practice. Eur. J. Clin. Microbiol. Infect. Dis. 22, 284–290 (2003).
Yu, Z. & Morrison, M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 36, 808–812 (2004).
Salonen, A. et al. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiol. Methods 81, 127–134 (2010).
Tian, L. et al. Effects of pectin supplementation on the fermentation patterns of different structural carbohydrates in rats. Mol. Nutr. Food Res. 1–11 (2016).
van den Bogert, B., de Vos, W. M., Zoetendal, E. G. & Kleerebezem, M. Microarray analysis and barcoded pyrosequencing provide consistent microbial profiles depending on the source of human intestinal samples. Appl. Environ. Microbiol. 77, 2071–2080 (2011).
Daims, H., Brühl, A., Amann, R., Schleifer, K. H. & Wagner, M. The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a more comprehensive probe set. Syst. Appl. Microbiol. 22, 434–444 (1999).
Ramiro-Garcia, J. et al. NG-Tax, a highly accurate and validated pipeline for analysis of 16S rRNA amplicons from complex biomes. NF1000 Res. 5, 5 (2016).
Dryden, M. W., Payne, P. A., Ridley, R. & Smith, V. Comparison of common fecal flotation techniques for the recovery of parasite eggs and oocysts. Vet. Ther. 6, 15–28 (2005).
Gillespie, T. R. Noninvasive assessment of gastrointestinal parasite infections in free-ranging primates. Int. J. Primatol. 27, 1129–1143 (2006).
Gillespie, T. R. & Chapman, C. A. Prediction of parasite infection dynamics in primate metapopulations based on attributes of forest fragmentation. Conserv. Biol. 20, 441–448 (2006).
Zeger, S. L., Liang, K.-Y. & Albert, P. S. Models for longitudinal data: a generalized estimating equation approach. Biometrics 44, 1049–1060 (1988).
Scrucca, L. Dispmod: Dispersion Models. R package version 1.1. (2012). https://cran.r-project.org/package=dispmod.
Nakagawa, S. & Schielzeth, H. A general and simple method for obtaining R2 from generalized linear mixed-effects models. Methods Ecol. Evol. 4, 133–142 (2013).
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria (2018). https://www.r-project.org/.
Bates, D., Maechler, M., Bolker, B. & Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48 (2015).
Lenth, R. V. Least-squares means: the R package lsmeans. J. Stat. Softw.69, (2016).
Fox, J., Friendly, M. & Weisberg, S. Hypothesis tests for multivariate linear models using the car package. R J. 5, 39–52 (2013).
Hartig, F. DHARMa: Residual Diagnostics for Hierarchical (multi-level/mixed) Regression Models. R package version 0.1. 5. (2017).
Barton, K. MuMIn: Multi-model Inference, R package version 0.12. 0. (2009).
Acknowledgements
For their advice in the field, the authors want to thank Dr. P. Wright, Dr. E. Larney, and John E. Cadle, and for their logistical support we want to thank the Centre Valbio staff (CVB), the Institute for the Conservation of Tropical Environments (ICTE) and the Madagascar Institute pour la Conservation des Ecosystèmes Tropicaux (MICET), especially B. Andriamihaja, who facilitated arrangements on necessary permits. Thanks go to the Malagasy government and Madagascar National Parks (MNP), Centre de Formation Professionelle Forestière (CFPF), and Mitsinjo Association for their formal permission. For their fieldwork assistance and indispensable knowledge, we want to thank all field technicians and guides that helped as at the various national park, as well as the following MSc students: Anna Luijten, Jurian van Duursen, Nathalie Houtman, Jeroen Schütt, Jeroen Bentinck, Hilde de Laat, Elke van Gils, Freek IJdema, Maartje Papegaaij, and Rakotosolofo Andrianadrasanarivelo. We thank Harm Ploeger, Rolf Nijsse, and Herman Cremers (Utrecht University) for their aid with parasite identification and supervising students in the laboratory. Furthermore, we would like to thank J. Ramiro Garcia for assistance with statistical analysis. We would also like to thank the anonymous reviewers for their helpful and constructive comments, as well as Dr. R. Mundry (Max Planck Institute of Evolutionary Anthropology) for making available his R script for stability analyses in GLMM. This work is part of the PhD thesis of IdW and AU and was supported by an NWO/ALW and PE&RC grant (Grant nr. 1208, project nr. 5120873001). AU was supported by a PhD fellowship from the European Union Erasmus-Mundus programme (EM Action 2- Partnership IAMONET- 20132520). Other necessary funds to conduct the fieldwork of this study were provided by: the KNAW Academy Ecology Fund, the Dr. J.L. Dobberke Foundation, the Treub Foundation, the Dutch Fund for Research on Nature Conservation, the Dutch Royal Botanical Society, and the Dutch Royal Zoological Society.
Author information
Authors and Affiliations
Contributions
As corresponding author, I.d.W. was involved in all aspects that resulted in the final manuscript, including conception and design, acquisition of data, and analysis and interpretation of data; I.d.W. and A.U. wrote the main manuscript text and prepared all figures and tables. A.U., W.N., P.v.H., I.H., H.P. and H.S. made substantial contributions to conception and design of the work and A.U., G.G., W.N., P.v.H., I.H., P.K. and H.S. in the analysis and interpretation of data. WN and AU were involved in data collection and A.U., G.G., P.v.H., I.H. and H.S. contributed significantly to the data analysis and interpretation. All authors participated in drafting the article, reviewed the manuscript, and revised it critically for important intellectual content and they will all give final approval of the version to be submitted and any revised version to be published.
Corresponding author
Ethics declarations
Competing interests
On behalf of all authors involved, I certify that all authors are accountable for the articles submitted to Scientific Reports and certify that they have no competing interests that might be construed as influencing the results or interpretation of a reported study.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Winter, I.I., Umanets, A., Gort, G. et al. Effects of seasonality and previous logging on faecal helminth-microbiota associations in wild lemurs. Sci Rep 10, 16818 (2020). https://doi.org/10.1038/s41598-020-73827-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-73827-1
This article is cited by
-
Influence of habitat alteration on the structure of helminth communities in small mammals: a systematic review and critical appraisal of theory and current evidence
Parasitology Research (2023)
-
Metabarcoding of bacteria and parasites in the gut of Apodemus agrarius
Parasites & Vectors (2022)
-
Significant effects of host dietary guild and phylogeny in wild lemur gut microbiomes
ISME Communications (2022)
-
Interactions between parasitic helminths and gut microbiota in wild tropical primates from intact and fragmented habitats
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
