
Abstract
The ability to inhibit host cell apoptosis is important for the intracellular replication of the obligate intracellular pathogen Coxiella burnetii, as it allows the completion of the lengthy bacterial replication cycle. Effector proteins injected into the host cell by the C. burnetii type IVB secretion system (T4BSS) are required for the inhibition of host cell apoptosis. AnkG is one of these anti-apoptotic effector proteins. The inhibitory effect of AnkG requires its nuclear localization, which depends on p32-dependent intracellular trafficking and importin-α1-mediated nuclear entry of AnkG. Here, we compared the sequences of ankG from 37 C. burnetii isolates and classified them in three groups based on the predicted protein size. The comparison of the three different groups allowed us to identify the first 28 amino acids as essential and sufficient for the anti-apoptotic activity of AnkG. Importantly, only the full-length protein from the first group is a bona fide effector protein injected into host cells during infection and has anti-apoptotic activity. Finally, using the Galleria mellonella infection model, we observed that AnkG from the first group has the ability to attenuate pathology during in vivo infection, as it allows survival of the larvae despite bacterial replication.
Similar content being viewed by others

Introduction
Coxiella burnetii is a zoonotic Gram-negative pathogen with an obligate intracellular lifestyle. The bacteria cause Q fever in humans. The acute form of Q fever is mainly characterized by a self-limiting flu-like illness and rarely by an interstitial pneumonia or hepatitis1. Acute Q fever is treatable with doxycycline, but many patients might also develop the post-Q fever fatigue syndrome2, which can last for up to 10 years3. At present, there is no evidence-based recommendation for treatment of post-Q fever fatigue syndrome. In addition, the infection may progress to chronic Q fever, which mainly manifests as a potentially fatal endocarditis1. Chronic Q fever develops months or years after infection, suggesting that C. burnetii persists silently within the host before massive bacterial replication leads to chronic Q fever. To control the infection, patients have to be treated for at least 18 months with doxycycline in combination with chloroquine4. Thus, a much more effective treatment for chronic Q fever would be highly desirable.
In general, humans become infected by inhaling C. burnetii-containing aerosols. Monocytes and macrophages are the primary target cells that take up C. burnetii by αvβ3 integrin-mediated phagocytosis5,6. The invasion into non-phagocytic cells seems to be at least partially dependent on the outer membrane protein A (OmpA) from C. burnetii7. After uptake, the C. burnetii-containing vacuole (CCV) matures by consecutive fusion events to a phagolysosome-like compartment8,9,10. Usually, phagocytosis of bacterial microorganisms leads to their destruction in the phagolysosome11. However, C. burnetii survives within the phagolysosome-like environment and requires these conditions for the generation of its progeny12. Cellular apoptosis is also important in the defense against intracellular pathogens13. As this is an anti-inflammatory mechanism it eliminates infected cells without inducing inflammation and tissue destruction14. Yet, C. burnetii efficiently inhibits host cell apoptosis15,16. Thus, inhibition of apoptosis and withstanding phagosomal maturation are thought to be important evasion strategies of C. burnetii17. These processes are dependent on the T4BSS18,19, which injects effector proteins into the host cell cytosol to manipulate the host cell for the benefit of the pathogen20. Roughly 150 putative C. burnetii effector proteins have been identified; however, functions have been assigned to only a few of them21,22,23,24,25,26,27,28,29. Anti-apoptotic activity was shown for the three effector proteins AnkG, CaeA and CaeB23,26,30,31,32,33. Their exact mode(s) of interference with the host cell apoptotic machinery has not been determined. AnkG is the best-characterized C. burnetii effector protein with anti-apoptotic activity32,33. It is a 38.6 kDa protein and contains five predicted ankyrin-repeats, spanning from amino acids 15 to 80 and from 90 to 194. Translocation of AnkG into the host cell was suggested to be independent of the chaperone IcmS34. Inside the host cell, AnkG associates mainly with mitochondria and, after stress induction, localizes in the host cell nucleus32. Only nuclear-localized AnkG is able to prevent pathogen-induced host cell death26,32. Migration to and uptake into the nucleus depend on binding to the host cell proteins p32 and importin-α1, respectively32,33. Here, we aimed to increase our knowledge of the molecular requirements of AnkG activity, using dissimilarities found in the AnkG sequence from 37 C. burnetii strains.
Results
C. burnetii isolates reveal genetic variability of AnkG
Recently, the sequences of the C. burnetii anti-apoptotic effector protein CaeA from 25 different isolates were compared, which allowed the identification of a functional domain with anti-apoptotic activity31. In this study, we analyzed the ankG sequences from 37 C. burnetii isolates, including the 25 isolates from the CaeA study (Table 1) in order to learn more about the link between the ankG genetic background and protein function. Importantly, most of the isolates were collected from ruminants with an abnormal birthing situation, suggesting that these isolates were associated with pathology. However, if C. burnetii either caused or was involved with the pathology is unknown.
By comparing the 37 different ankG sequences, we observed a high degree of similarity at the nucleotide level (Fig. 1a). However, some strains contained base pair deletions or insertions, leading to frame shifts and, consequently, the introduction of a premature stop codon. Based on this in silico analysis the strains were classified into three groups (Fig. 1b).

Genomic diversity of ankG. (a) The ankG alignment defines three groups, which are highlighted by colors (blue, red and green). Single base insertions, deletions and nucleotide substitutions are indicated either by dashes or highlighted in yellow. (b) AnkG amino acid sequences of three C. burnetii isolates –each representatives for its group. The first group contains the reference strain Nine Mile II (AnkGNM) and nineteen additional strains expressing a 338 amino acid protein (blue). The second group includes thirteen isolates and is represented by C. burnetii Soyta (AnkGSoyta) expressing a 92 amino acid protein. AnkGSoyta is identical in the first 83 N-terminal amino acids (red), but harbors 9 different amino acids at the C-terminus (yellow) compared to AnkGNM. The third group contains four isolates and is represented by C. burnetii strain F3 (AnkGF3) expressing a 51 amino acid protein. AnkGF3 has an amino acid exchange at position 11 (isoleucine to leucine) and is otherwise identical to AnkGNM in the first 28 N-terminal amino acids (green), but contains 23 different amino acids at the C-terminus (yellow). The asterisk (*) at the C-terminus of the amino acid sequence representing the second and third group indicates premature truncation.
The reference strain Nine Mile (NM) and nineteen further isolates express the full-length 338 amino acid protein of 38.6 kDa and belong to the first group. Importantly, the sequence of ankG is identical in NM phase I and NM phase II (Fig. 1a). The thirteen isolates of the second group contain a two base pair deletion directly after amino acid 83, leading to a 92 amino acid protein of 10.4 kDa. Thus, AnkG from this group (AnkGSoyta) is identical to the first group in amino acids 1 to 83, but substantially differs afterwards in both size and protein sequence.
Only four isolates belong to the third group, which differs from the groups one and two in an exchange of isoleucine into leucine at position 11. This mutation is also present in the Dugway strain. It leads to a slightly, but significantly, better protection from staurosporine-induced apoptosis33. In addition, AnkG from the third group contains a base pair insertion at the codon for amino acid 29, leading to a premature stop at amino acid codon 51 and a protein of 6 kDa. AnkG from this group (AnkGF3) is identical to AnkGNM in the amino acids 1–28, but different in the amino acids 29–51.
The AnkG variants from the three groups respond differently to apoptosis induction
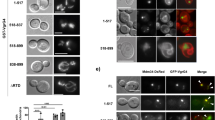
We previously demonstrated that the amino acids 1–69 of AnkGNM are required for anti-apoptotic activity26. Therefore, we analyzed whether AnkGSoyta (member of the second group), which contains all 69 amino acids of the first group, or AnkGF3 (member of the third group), where only the first 28 amino acids, except for amino acid 11, are identical to AnkGNM, would affect host cell survival (Fig. 2a). We ectopically expressed GFP or GFP-AnkG variants transiently in CHO cells. First, we demonstrated stable expression by immunoblot analysis (Fig. 2b). Next, we quantified the percentage of fragmented nuclei as readout for apoptosis after treatment with the apoptosis-inducer staurosporine. Our results demonstrated that AnkG of the first group (AnkGNM) inhibited apoptosis, AnkG of the second group (AnkGSoyta) did not affect apoptosis induction, and AnkG from the third group (AnkGF3) displayed apoptosis-promoting activity (Fig. 2c). Importantly, without staurosporine-induction, AnkGF3 did not display any pro-apoptotic activity (data not shown). As the anti-apoptotic activity of AnkGNM requires nuclear localization, which is mediated by binding to p32- and importin-α132,33, we analyzed the interaction of the AnkG variants with the host cell proteins p32 and importin-α1. We hypothesized that AnkGSoyta and AnkGF3 bind both host cell proteins, as the binding of AnkGNM to importin-α1 was mapped to the amino acids surrounding residue 11, while the amino acids neighbouring residues 22 and 23 had been identified as the binding region for the interaction with p3232,33. Indeed, as shown in Figs. 2d and 2e, all three AnkG variants were able to bind to p32 and importin-α1. The interaction of the HA-AnkG variant with Flag-tagged importin-α1 is not mediated indirectly by the anti-FLAG beads, as shown by us before33. Next, we analyzed the subcellular localization of the AnkG variants, as nuclear localization of AnkGNM was demonstrated to be essential for its anti-apoptotic activity32. AnkGNM displays vesicular staining with close association with host cell mitochondria in healthy cells. After cellular stress, AnkGNM migrates to the host cell nucleus. In contrast, AnkGNM 1–69 was present within the host cell nucleus under all conditions32. Thus, we hypothesized that without cell stress induction the AnkG variants from the second and third groups might also localize to the nucleus, which was, in fact, the case (Fig. 2f). Taken together, although AnkGSoyta bound to p32 and importin-α1 and was translocated to the nucleus, it was unable to inhibit apoptosis. AnkGF3 was also found in the nucleus and bound to p32 and importin-α1, but displayed staurosporine-dependent pro-apoptotic activity in contrast to the anti-apoptotic activity of AnkGNM.

The three AnkG groups display different anti-apoptotic activities. (a) Schematic representation of the AnkG variants used in this study. Amino acids are shown above each of the schematic diagrams. Striped area represents altered amino acids as compared to the reference AnkGNM. (b) CHO-FcR cells were transiently transfected with GFP, GFP-AnkGNM, GFP-AnkGSoyta or GFP-AnkGF3. Proteins were separated by SDS-PAGE, transferred to a PVDF membrane and probed with an antibody against GFP. Depicted is one representative immunoblot of three independently performed experiments with similar results. (c) CHO-FcR cells expressing GFP, GFP-AnkGNM, GFP-AnkGSoyta or GFP-AnkGF3 were treated with staurosporine for 4 h. The cells were fixed, permeabilized and the nuclei were stained with DAPI. The nuclear morphology of at least 100 GFP-expressing cells was scored in three independent experiments. Error bars indicate ± SD. **p < 0.01; ***p < 0.001, n.s. not significant. (d) HEK293 cells were co-transfected with plasmids encoding HA-tagged p32, GFP-tagged AnkGNM, AnkGSoyta and AnkGF3. The proteins were precipitated from the cell lysates using a GFP-trap. Immunoblot analysis was used to detect p32 (anti-HA) and AnkG-protein variants (anti-GFP) in the lysates (pre-IP) and in the precipitates (IP). Shown is a representative experiment out of three independent experiments with similar results. (e) Cell lysates from HEK293 cells transiently expressing HA-tagged AnkGNM, AnkGSoyta and AnkGF3 were incubated with Flag-tagged importin-α1 bound to an anti-Flag M2 affinity gel. Cell lysates and pull-down fractions were subjected to immunoblot analysis using anti-HA and anti-Flag antibodies. Shown is a representative experiment from three independent experiments with similar results. *Unspecific band. (f) Representative immunofluorescence micrographs show CHO-FcR cells transiently expressing GFP-AnkGNM, GFP-AnkGSoyta or GFP-AnkGF3 (all green) stained with DAPI to visualize the nuclei (blue). Scale bars, 10 µm.
Characterization of AnkGSoyta activity
AnkGSoyta represents the second group and forms a 10.4 kDa protein. While the first 83 amino acids are identical to AnkGNM, the residues 84 – 92 differ. Although AnkGSoyta bound to p32 and importin-α1 and displayed nuclear localization, it was unable to inhibit apoptosis. To investigate the underlying reason(s), we first constructed truncations of AnkGSoyta by stepwise removal of amino acids from its C-terminus (Fig. 3a). We ectopically expressed GFP or GFP-AnkGSoyta variants transiently in CHO cells and demonstrated stable expression by immunoblot analysis (Fig. 3b). Next, we measured apoptosis after stimulation with staurosporine by quantifying nuclear fragmentation, which was visualized by DAPI staining. As shown in Fig. 3c, stepwise removal of amino acids from the C-terminal region in AnkGSoyta restored its anti-apoptotic activity. This suggests that the C-terminal region in AnkGSoyta disturbs the activity of the protein. We hypothesized that this might have been caused by the length of the protein. To address this experimentally, we constructed a truncated AnkGNM 1–92 to clarify whether the length of the protein might be the cause for the lost activity of AnkGSoyta, which also contains 92 amino acids. As shown in Fig. 3d, all constructs were ectopically expressed following transient transfection of CHO cells. AnkGNM 1–92 inhibited staurosporine-induced apoptosis as efficiently as full-length AnkG (Fig. 3e), indicating that the reduced length is not the cause for the missing anti-apoptotic activity of AnkGSoyta. These results suggest that the amino acids 84–92 of AnkGSoyta might influence the folding or intra- or intermolecular binding capacity of the protein.

Characterization of AnkGSoyta-mediated apoptosis modification. (a) Schematic representation of the AnkGNM and AnkGSoyta truncations used. Amino acids are shown above each of the schematic diagrams. The striped area represents altered amino acids as compared to the reference sequence AnkGNM. (b–e) CHO-FcR cells were transiently transfected with GFP or with the GFP-tagged AnkG variants indicated. (b,d) Proteins were separated by SDS-PAGE, transferred to a PVDF membrane and probed with an antibody against GFP. Depicted is one representative immunoblot of three independently performed experiments with similar results. (c,e) CHO-FcR cells expressing either GFP or the indicated GFP-tagged AnkG variants were treated with staurosporine for 4 h. The cells were fixed, permeabilized and the nuclei stained with DAPI. The nuclear morphology of at least 100 GFP-expressing cells was scored in three independent experiments. Error bars indicate ± SD. **p < 0.01; ***p < 0.001, n.s. not significant.
Characterization of AnkGF3 activity
AnkGF3 represents the third group and forms a 6 kDa protein. While the first 28 amino acids, except for amino acid 11, are identical to AnkGNM, the amino acids 29 – 51 are different. AnkGF3 bound to p32 and importin-α1 and translocated into the nucleus, but it showed pro-apoptotic activity after apoptosis-induction. To shed light on the underlying reason, we created different AnkG truncations (Fig. 4a). We constructed a truncated variant AnkGF3 1–28 to clarify whether the first 28 amino acids, which are identical between AnkGF3 and AnkGNM, are sufficient for anti-apoptotic activity. Additionally, we constructed AnkGNM 1–51 to clarify whether the length of the protein might result in a change of its activity. First, we ectopically expressed GFP or the GFP-AnkG variants transiently in CHO cells and demonstrated stable expression by immunoblot analysis (Fig. 4b). As shown in Fig. 4c, while GFP-AnkGF3 significantly increases staurosporine-induced apoptosis, GFP-AnkGF3 1–28 inhibited apoptosis similarly to GFP-AnkGNM. This demonstrates that AnkGF3 contains the anti-apoptotic domain, which comprises the first 28 amino acids. Expression of GFP-AnkGNM 1–51 also resulted in inhibition of apoptosis, indicating that the length of AnkGF3, which contains 51 amino acids, cannot explain its pro-apoptotic activity. These findings indicate that the amino acids 29–51 of AnkGF3 negatively affect the anti-apoptotic activity of the N-terminal region.

Characterization of AnkGF3-mediated apoptosis induction. (a) Schematic representation of the AnkG variants used. Amino acids are shown above each of the schematic diagrams. The striped area represents altered amino acids as compared to the reference AnkGNM sequence. (b) CHO-FcR cells were transiently transfected with GFP or with the GFP-tagged AnkG variants indicated. Proteins were separated by SDS-PAGE, transferred to a PVDF membrane and probed with an antibody against GFP. Depicted is one representative immunoblot of three independently performed experiments with similar results. (c, d) CHO-FcR cells expressing GFP or the indicated GFP-tagged AnkG variants or truncations were treated with staurosporine for 4 h. The cells were fixed, permeabilized and the nuclei were stained with DAPI. The nuclear morphology of at least 100 GFP-expressing cells was scored in three independent experiments. Error bars indicate ± SD. *p < 0.05; **p < 0.01, ***p < 0.001.
To determine whether the amino acids 1–28 are necessary and sufficient for the anti-apoptotic activity, we ectopically expressed GFP, GFP-AnkGNM, GFP-AnkGNM 1–28 or GFP-AnkGNM 29–338 transiently in CHO cells and determined cell death after staurosporine treatment. The expression of GFP-AnkGNM 1–28 significantly inhibited staurosporine-induced apoptosis. In contrast, the expression of GFP-AnkGNM 29–338 did not protect the cell from apoptosis (Fig. 4d). These data suggest that the amino acids 1–28 are necessary and sufficient for the anti-apoptotic activity of AnkG.
Analyzing transcription of the ankG gene from the three different groups
The ankG gene from the first group is transcribed and translated as a full-length protein. The ankG alleles from the second and third groups contain a premature stop codon leading to the translation of a 10 and 6 kDa protein, respectively. To determine whether the ankG variants from the second and third groups encode additional proteins containing the C-terminal translocation signal, we inspected the ankG sequences for the presence of additional CDS within the original full-length transcript. Indeed, ankG contains three further start codons, which are in-frame with the stop codon at the C-terminus. These start codons are located at slightly varying base pair positions in the three ankG alleles near bp 337, 532 and 598 in the respective alleles (Fig. 5a). In order to determine whether the C-terminal fragments are transcribed, we analyzed the mRNA from C. burnetii isolates belonging to the three different groups. As representative for the first group, we chose Nine Mile I (NMI) and for the second and third groups 19/34 and Z3574-1/92, respectively. First, we had to establish axenic growth of these isolates, as axenic media does not support growth of all C. burnetii strains35. While growth of NMI in ACCM-2 and ACCM-D has been shown, growing 19/34 and Z3574-1/92 in axenic media has never been attempted. The strain 19/34 grew similarly well in ACCM-2 as NMI. However, Z3574-1/92 was only able to grow in ACCM-D media. As C. burnetii has different growth kinetics in ACCM-2 and ACCM-D media36, which might influence transcriptional activity, we always used NMI grown in the respective media as control. Thus, we isolated RNA from 19/34 and NMI grown in ACCM-2 and RNA from Z3574-1/92 and NMI grown in ACCM-D. After treatment with DNase, we confirmed the lack of DNA contamination by performing a no reverse transcriptase control using specific primers for the dotA gene (Fig. 5b). Next, we reverse transcribed the mRNA into cDNA and determined whether the RNA fragment encoding the N-terminal residues of the three different ankG groups was transcribed. As shown in Fig. 5c, the base pairs 3–150 were transcribed in all three groups, suggesting that the 5´part of the mRNA can be transcribed and translated into the respective proteins. Next, we analyzed if the full-length ankG open reading frame of all three groups is transcribed into the corresponding mRNA. This is indeed the case for all three groups (Fig. 5d). Thus, the 3´ part of the ankG mRNA from the second and third groups is transcribed and might be translated into a protein containing the C-terminal translocation signal. Still, although these proteins might have the potential to be injected into the host cell via the T4BSS, they most likely lack anti-apoptotic activity due to missing amino acids 1–28.

Determination of ankG variant transcripts. (a) Schematic overview of possible transcripts of the three different AnkG groups is shown. The first group is represented by NMI, the second group by 19/34, and the third group by Z3574-1/92. The numbers of the first base pair of potential internal start codons are given, which are in frame with the stop codon at base pair position 1,015–1,017 (group 1), 1,013–1,015 (group 2) or 1,004–1,006 (group 3). (b) DNase treated RNA was analyzed for DNA contamination using dotA specific primers. As positive control C. burnetii NMII gDNA was used in different amounts. Gel electrophoretic analysis was performed using a 2% agarose gel. (c, d) Gel electrophoresis image of the different ankG mRNA sections probed in the distinct C. burnetii strains. gDNA served as positive control, while the non-template control (NTC) contains only water. RNA was isolated from all axenically grown strains (2—grown in ACCM-2; D—grown in ACCM-D), reverse-transcribed in cDNA, and amplified by PCR. Gel electrophoretic analysis was performed using a 2% agarose gel. The cDNA was analyzed for the presence of (c) the mRNA region encoding the N-terminal part of the protein (base pairs 3–150) and (d) the full-length transcript (base pairs 3–1,014).
Role of AnkGSoyta and AnkGF3 during a C. burnetii infection
During a C. burnetii infection AnkGNM is transported into the host cell nucleus, the site of its anti-apoptotic activity33. While we do not fully understand the mechanism by which an effector protein is recognized for delivery through the T4BSS, we know that the C-terminal end of the effector serves as the translocation signal37. In contrast to AnkGNM, AnkGF3 and AnkGSoyta lack the C-terminal end and should therefore not be translocated through the T4BSS. However, it was demonstrated that AnkGNM, AnkGNM 1–69 as well as AnkGNM 70–338 are translocated by the Legionella pneumophila T4BSS26. This indicates that AnkGNM might contain two translocation signals, one in its C-terminal part and another one within the first 69 amino acids. Therefore, AnkGSoyta and/or AnkGF3 might also be recognized as an effector protein and translocated into the host cell by the T4BSS. To determine if this is the case, we transformed C. burnetii with plasmids containing 3xFlag-tagged AnkGF3 or AnkGSoyta. Single C. burnetii pFlag-AnkGF3 or pFlag-AnkGSoyta clones were isolated and transgene expression was analyzed (data not shown). Mouse embryonic fibroblasts (MEFs) were infected with C. burnetii pFlag-AnkGNM, C. burnetii pFlag-AnkGF3 and C. burnetii pFlag-AnkGSoyta under inducing conditions and the localization of Flag-AnkG was analyzed by confocal imaging. As shown in Fig. 6a, Flag-AnkGNM was associated with the bacteria and localized within the host cell nucleus, confirming previous results33. This indicates that Flag-tagged AnkGNM can be translocated by C. burnetii into the host cell. In contrast, Flag-AnkGSoyta and Flag-AnkGF3 were only associated with the bacteria and not present in the host cell. This indicates that neither AnkGSoyta nor AnkGF3 are T4BSS effector proteins. To support this assumption, we fused AnkGNM, AnkGSoyta and AnkGF3 to the calmodulin-dependent adenylate cyclase toxin (Cya). CHO cells were infected with C. burnetii producing either Cya-AnkGNM, Cya-AnkGSoyta or Cya-AnkGF3 and the amount of cyclic AMP (cAMP) was measured. As Cya catalyzes the production of cAMP its amount correlates with the translocation of Cya-tagged AnkG variants into the cytoplasm. A concentration of 2.5-fold more cAMP than the negative control (Cya alone) was regarded as proof for translocation of the protein. Cya-AnkGNM was translocated into the host cell cytosol (Fig. 6b), supporting previous results26. In contrast, Cya-AnkGSoyta and Cya-AnkGF3 were not translocated into the host cell cytoplasm. This data supports the hypothesis that neither AnkGSoyta nor AnkGF3 are T4BSS effector proteins.

Only AnkGNM is a bona fide T4BSS effector protein. (a) Representative confocal micrographs of mouse embryonic fibroblasts (MEFs) infected with C. burnetii expressing Flag-AnkGNM, Flag-AnkGSoyta or Flag-AnkGF3. MEFs were fixed 48 h post-infection and were stained with an antibody specific for Flag (green) and Coxiella (red). DNA was stained with DAPI (blue). Scale bars, 10 μm. (b) The amount of cAMP is given in fmol/well for CHO cells either uninfected or infected for 72 h with C. burnetii producing Cya, Cya-AnkGNM, Cya-AnkGSoyta or Cya-AnkGF3. The threshold for secretion was set to 2.5-fold the cAMP-level of the controls and is depicted as a dotted line. Shown is the mean of at least three experiments ± SD. ***p < 0.001.
AnkGNM attenuates pathology during C. burnetii infection in Galleria mellonella
AnkGNM is translocated into the host cell during infection and has anti-apoptotic activity. Whether AnkG is important for the C. burnetii infection in vitro and more importantly in vivo is unknown. Recently, the Galleria mellonella infection model was used to analyze C. burnetii infection in vivo Galleria mellonella is susceptible to infection by C. burnetii NMI and NMII24,27,38,39,40, suggesting that the pathogenicity of C. burnetii is independent of the phase variation in this infection model. Death of the larvae occurred in a dose-dependent manner and depended on a functional T4BSS38. Furthermore, this in vivo infection model was successfully used to assess the contribution of the T4BSS effector CvpB/Cig2 during infection24,27. Therefore, we used the Galleria mellonella model to determine the role of AnkG during an in vivo infection. To assess virulence, a lethal dose of 106 C. burnetii NMII wildtype (WT), C. burnetii NMII ΔdotA (ΔdotA), C. burnetii pFlag-AnkF (AnkF) or C. burnetii pFlag-AnkG (AnkG) per larvae was injected in the upper right proleg and larval survival was monitored for 7 days. Larvae injected with PBS solution served as a control for the injection stress, the ΔdotA mutant for the activity of the T4BSS and AnkF-expressing C. burnetii for the overexpression of an effector protein. 90% of the larvae receiving PBS solution survived, whereas 100% of the larvae infected with wildtype bacteria had died by day 7 post-injection. In contrast, the majority (~ 95%) of larvae infected with C. burnetii ΔdotA survived (Fig. 7a). This result confirms the importance of the T4BSS for C. burnetii virulence. All larvae infected with C. burnetii pFlag-AnkG survived till day 6 post-injection while all larvae infected with C. burnetii pFlag-AnkF had died by this time point (Fig. 7a). To verify that the observed pro-survival phenotype of larvae infected with C. burnetii pFlag-AnkG is due to increased expression of AnkG, we performed qRT-PCR to determine the expression level of AnkG in C. burnetii pFlag-AnkG either induced with IPTG or not induced. As shown in Fig. 7b, induction with IPTG resulted in increased expression of AnkG. The expression level decreased over time, but at seven days post-induction it was still significantly higher than in non-induced bacteria. This supports the assumption that the increased translocation of overexpressed AnkG into the host cells mediates improved vitality of the C. burnetii infected larvae. To exclude that this phenotype is due to a reduced infection or/and replication ability of the bacteria expressing pFlag-AnkG, we determined the ability of C. burnetii to infect and replicate in the larvae by qRT-PCR and immunofluorescence. Only C. burnetii ΔdotA was unable to replicate in the larvae. In contrast C. burnetii WT, Flag-AnkG and Flag-AnkF replicated in the larvae around 100-fold from day 1 to 5 (Fig. 7c). In agreement with this result, we observed that at day 5 post-infection, the majority of hemocytes infected with WT, Flag-AnkG- or Flag-AnkF-expressing C. burnetii contained replicative CCVs. This was not the case in hemocytes infected with C. burnetii ΔdotA (Fig. 7d). These results demonstrate that a functional T4BSS is essential for intracellular replication, as demonstrated before38. In addition, these data showed that C. burnetii expressing Flag-AnkG replicates in hemocytes. Next, the infection rate of hemocytes was quantified (Fig. 7e). All bacteria exhibited a similar percentage of infection at day 1 post-infection. At day 5 post-infection, the infection rate had increased when infected with WT, Flag-AnkG- or Flag-AnkF-expressing C. burnetii, but not when infected with the C. burnetii ΔdotA mutant. In the latter case, the rate of infection stayed nearly constant. Thus, C. burnetii expressing Flag-AnkG have no defect in the ability to infect hemocytes, to establish a replicative CCV and to spread. We concluded from these data that exogenous AnkG mediates increased survival of C. burnetii infected larvae. How AnkG boosts survival of the infected larvae is currently unknown, but it cannot be explained by attenuated bacterial fitness of Flag-AnkG-expressing C. burnetii. These data suggest that AnkG might be an important virulence factor. In order to start investigating the role of AnkG for pathogenesis in vivo, we infected larvae with either wild type C. burnetii or a ΔankG mutant and monitored their survival over 8 days. Larvae infected with the ΔankG mutant were significantly attenuated in virulence, as demonstrated by improved survival rates (Fig. 7f). However, the underlying reason for this attenuation in virulence will be investigated in future studies. Thus, whether there is a direct or indirect link between the anti-apoptotic activity of AnkG, which might be responsible for the survival of larvae infected with Flag-AnkG-expressing C. burnetii, and the attenuation of the ΔankG mutant has to be unraveled.

AnkGNM mediates host tolerance to C. burnetii infection. (a) Survival of infected Galleria mellonella larvae was analyzed over a 7 day period. 10 larvae each were infected with 106 bacteria (C. burnetii wildtype (WT), C. burnetii ΔdotA (ΔdotA), C. burnetii pFlag-AnkG (AnkG) or C. burnetii pFlag-AnkF (AnkF)) per larvae in 20 µl PBS or with a PBS control. Survival was controlled every 24 h. Shown is the mean of three experiments ± SD. **p < 0.01; ***p < 0.001. (b) AnkG level was measured by RT-PCR 1, 3 and 7 days post-induction with 2 mM IPTG and compared to the AnkG level of respective the untreated control. RNA was isolated and reverse transcribed in cDNA. Fold replication was calculated using the ΔΔCT method. Shown is the mean of three independent experiments. *p < 0.05. (c) C. burnetii replication between day 1 and day 5 post-infection was measured via RT-PCR. At days 1 and 5 post-infection, bacterial DNA was isolated from two larvae and genomic equivalents (GE) were measured by RT-PCR. The fold replication was determined by dividing the GE from day 5 post-infection by the GE from day 1 post-infection. Shown is the mean of three independent experiments. *p < 0.05. (d, e) The infection rate of Galleria mellonella hemocytes on days 1 and 5 post-infection was determined. Hemocytes from three pooled larvae per infected strain were isolated on days 1 and 5 post-infection and stained for immunofluorescence. (d) Representative immunofluorescence micrographs of infected hemocytes are shown. The cytoskeleton was stained with phalloidin (red), C. burnetii was stained with an anti-Coxiella antibody (green) and the nuclei were stained with DAPI (blue). Scale bars, 10 µm. (e) The infection rate of at least 100 hemocytes were scored in three independent experiments. Shown is the mean of three experiments. *p < 0.05; **p < 0.01; ***p < 0.001, n.s. not significant. (f) Survival of infected Galleria mellonella larvae was analyzed over a period of 8 days. Ten larvae each were infected with 106 bacteria (C. burnetii wildtype (WT) or C. burnetii ΔankG (ΔAnkG)) per larvae in 20 µl PBS or with a PBS control. Survival was controlled every 24 h. Shown is the mean of three experiments ± SD. ***p < 0.001.
Discussion
Comparison of genomes from different C. burnetii isolates pointed to a considerable heterogeneity in the repertoire of T4BSS effector proteins41. Noticeable variation was detected in the sequences of the C. burnetii T4BSS effector CaeA derived from 25 different C. burnetii isolates31. CaeA was classified in seven different genotypic groups. In the current study, we focused on another effector (AnkG) and analyzed the AnkG nucleotide and amino acid sequences from the same 25 C. burnetii isolates in addition to 12 further isolates. These 37 different isolates were divided into three groups according to their respective ankG sequences (Fig. 1a,b). To get a better impression of the heterogeneity of the ankG sequence, we compared the ankG sequences from 57 complete, scaffold and draft genomes of C. burnetii strains (Tab. S1). These strains encode nine alleles of the ankG gene. Genome groups I, IIb and III42 express the wild type sequence of the Nine Mile reference strain (group 1 in this study). Genome group IIa encodes a truncated protein of 92 residues due to a 2 bp frameshift deletion mutation at codons L83N84 and corresponds to AnkGSoyta (group 2 in this study). Genome group IV representatives all contain the Ile to Leu exchange at residue 11 and a 1 bp frameshift insertion mutation at Gly29, leading to a truncated protein with 51 residues and correspond to AnkGF3 (group 3 in this study). Three subgroups (designated 3, 3b and 3c in Tab. S1) contain different additional mutations in the remaining 3′ section of ankG. Genome groups V and VI both encode a full-length protein with 2 either amino acid substitutions at residues 11 (Ile to Leu) and 72 (Gly to Glu) in genome group V, while genome group VI contains just the mutation at amino acid position 11. There are only three exceptions to this sequence/genome group correlation. The Guyana strain, assigned to genome group I, has a frameshift insertion mutation at codon Gly294, leading to a protein of 327 residues with 33 altered amino acids at its C-terminus. The Cb185 strain, classified as genome group IIa, has the wild type sequence otherwise present in the genome groups I, IIb and III. The strain Cb109, a member of genome group IIb, has a frameshift mutation at residue Asn287, resulting in a protein of 299 residues with 13 altered amino acids at its C-terminus.
Interestingly, only one C. burnetii isolate analyzed in this study, encodes AnkG and CaeA, namely C. burnetii Nine Mile. Nine isolates encode only the effector protein AnkG and four isolates encode only the effector protein CaeA. Thus, the majority of the strains analyzed encode none or only one of the two anti-apoptotic effector proteins AnkG and CaeA. There seems to be no correlation between the presence of a functional AnkG and/or CaeA effector protein and the geographic distribution of the isolate and its host species. However, this interpretation needs to be taken with caution, as the majority of the isolates were collected from infected ruminants in Germany. Importantly, AnkG and CaeA are not the only C. burnetii effector proteins involved in the regulation of host cell viability. So far, two others have also been characterized in more detail: CaeB was shown to inhibit intrinsic apoptosis23 and IcaA was described to prevent pyroptosis21. However, it can be expected that other members of the so far identified ~ 150 effector proteins might also inhibit host cell death43. In line with this assumption, a large-scale transposon mutagenesis coupled to a high-content multi-phenotypic screening in the laboratory of Matteo Bonazzi identified transposon mutants that exhibit a cytotoxic phenotype7. This suggests that the genes affected might also contribute to preventing host cell death. Under certain conditions C. burnetii also induces apoptosis44 or pyroptosis45, which might be due to the activity of pro-apoptotic effector proteins that still await identification and characterization. Therefore, the balance between bacterial anti- and pro-apoptotic effector proteins will determine the overall impact of C. burnetii on the host cell. In any case, the absence of AnkG and/or CaeA or the expression and translocation of the C-terminal part of AnkG might result in altered pathogenesis of C. burnetii. Interestingly, the three different groups of AnkG exhibited different cellular activities (Fig. 2c), although they all bound to the host cell proteins p32 (Fig. 2d) and importin-α1 (Fig. 2e), which was shown to be a prerequisite for AnkG-mediated apoptosis inhibition. Additionally, ectopically expressed AnkGSoyta and AnkGF3 are localized within the host cell nucleus, the site of protein activity (Fig. 2f). Furthermore, AnkGNM, AnkGSoyta and AnkGF3 contain the same first 28 amino acids, which were shown to be necessary and sufficient for anti-apoptotic activity (Fig. 4c, d). The reason for the different cellular activity of the three AnkG variants might be altered protein folding or the inability to interact with themselves or other proteins46,47. However, further research is required to identify the underlying reason(s) for the different activities.
To better understand bacterial pathogenesis, appropriate infection models are essential. The Galleria mellonella infection model has been recently used to study pathogenic Gram-positive and Gram-negative bacteria24,27,38,48,49,50,51,52. Importantly, several of these studies showed a good correlation between the G. mellonella and mammalian infection models. For example, the essential role of the type III and type IV secretion systems in virulence was also demonstrated in the G. mellonella infection model38,49,51. In order to examine the role of specific virulence factors in an in vivo model system, this factor might either be overexpressed or deleted. Here, we used C. burnetii NM overexpressing Flag-tagged AnkG33. Importantly, C. burnetii overexpressing Flag-tagged AnkG showed reduced killing of infected G. mellonella (Fig. 7a), although infection and replication were not reduced in comparison to wildtype bacteria (Fig. 7c, d). It can be only speculated how AnkG increased survival of the infected larvae; AnkG might adjust the host immune reaction, possibly by preventing destruction of hemocytes via its anti-apoptotic activity. Hemocytes are important phagocytic immune cells and depletion of circulating hemocytes upon infection correlates with G. mellonella mortality53. Similarly, the addition of AnkG to the repertoire of Legionella pneumophila effector proteins resulted in host tolerance to infection by preventing rapid pathogen-induced apoptosis in dendritic cells26.
Taken together, we have demonstrated that AnkG is an anti-apoptotic effector protein, which might mediate host tolerance to a C. burnetii infection in the G. mellonella model of infection. In addition C. burnetii lacking AnkG are attenuated in the G. mellonella infection model, indicating that AnkG might be an important virulence factor. Furthermore, we have further narrowed down the anti-apoptotic domain to amino acids 1 to 28 and showed that the effector function is not conserved in all C. burnetii isolates. Indeed, our analysis suggests that C. burnetii belonging to the genome group IIa and IV do not express a functional AnkG effector protein.
Methods
Reagents, cell and bacterial strains
Unless otherwise noted, chemicals were purchased from Sigma Aldrich as described in Schäfer et al.33. Complete Protease inhibitor cocktail mixture and Xtreme Gene 9 transfection reagent were purchased from Roche. Staurosporine was bought from Cell Signaling. Cell lines were cultured at 37 °C and 5% CO2 in media containing 10% heat-inactivated fetal bovine serum (Biochrom). CHO (Chinese hamster ovary) fibroblasts were grown in minimal essential medium alpha medium (Invitrogen), HEK293T (human embryonic kidney) and MEF cells were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen). E. coli strains DH5α and BL21(DE3) were cultivated in Luria–Bertani (LB) broth supplemented with kanamycin or ampicillin if indicated.
PCR, sequencing and sequence analysis
PCR, sequencing and sequence analysis were done similar to as described earlier in Bisle et al., 2016. Therefore, PCR primers (Table 2) for ankG were designed with Primer Select software implemented in Lasergene 9.0 (DNASTAR, Madison, WI). AnkG DNA was amplified according to the manufacturer´s protocol using 1 × OptiBuffer (Bioline, London, United Kingdom), 1.4 mM MgCl2, 200 μM deoxynucleoside triphosphates (Bioline), 25 pmol of each primer, 1.5 U Bio-X-Act Short DNA polymerase (Bioline), and 5 µl of DNA, in a final volume of 25 µl. The PCR was run with the following thermocycling profile: 5 min at 95 °C; 1 min at 56 °C; 35 cycles, each consisting of 1 min and 30 s at 95 °C, and 30 s at 56 °C, 45 s at 72 °C; and a final elongation step of 10 min at 72 °C. Cycle sequencing was performed using BigDye v1.1 chemistry according to the manufacturer’s instructions (Applied Biosystems) (see Sequencing primers in Table 2), purified with DyeEx 96 plates (Qiagen) and electrophoresed on a 3130XL Genetic Analyzer (Applied Biosystems). Sequence analysis and polymorphism evaluation was performed using Lasergene 9.0 (DNASTAR, Madison, WI) and MEGA554.
Isolation of genomic Coxiella burnetii DNA
Genomic DNAs of 37 paraformaldehyde-fixed or heat-killed C. burnetii isolates were prepared according to the manufacturer´s protocol with the illustra bacteria genomic Prep Mini Spin Kit (GE Health Care), with 3 × 10 min incubation at 55 °C in the beginning and a final elution with 30 µl H2O as described in Bisle et al., 2016.
Nuclear fragmentation assay
Nuclear fragmentation assays were performed as described previously23,31. In brief, CHO cells were plated on coverslips in 24-well dishes at a density of 2.5 × 104 cells per well. After overnight incubation, cells were transfected with the plasmids indicated. Eighteen hours post-transfection, the cells were incubated with staurosporine (2 mg/ml) for 4 h at 37 °C and 5% CO2. The cells were fixed with 4% paraformaldehyde (Alfa Aeser) in PBS (Biochrom) for 20 min at room temperature, permeabilized with ice-cold methanol for 30 s, quenched with 50 mM NH4Cl (Roth) in PBS for 15 min at room temperature. The cells were mounted using ProLong Diamond with DAPI (ThermoFisher) to visualize the nucleus.
Immunoblotting
As described in Schäfer et al.33, proteins were separated by SDS/PAGE and transferred to a PVDF membrane (Millipore). The membranes were probed with antibodies directed against GFP (Life Technologies) or HA (Covance Research Products). A chemiluminescence detection system (Thermo Scientific or Millipore) was used to visualize the proteins marked by HRP-conjugated secondary antibodies (Dianova).
Co-immunoprecipitation
A modified protocol from Schäfer et al.33, was used for co-immunoprecipitation. In short, HEK293T cells were transfected with the plasmids indicated. Cells were lysed in lysis buffer (10 mM Tris/HCl [pH 7.5], 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40) for 30 min on ice and centrifuged for 10 min, 20,000 × g at 4 °C. The supernatants were incubated with GFP-Trap®_A beads (ChromoTek) for 2 h at 4 °C. The beads were washed three times with wash buffer (10 mM Tris/HCl [pH 7.5], 150 mM NaCl, 0.5 mM EDTA). Precipitated proteins were analyzed by immunoblotting.
Pull-down
Pull-down experiments with E. coli BL21(DE3) transformed with a plasmid producing Flag-importin-α1 and lysates of transfected HEK293T cells were performed as described earlier33.
Indirect immunofluorescence
As in Schäfer et al.33, cells were seeded on coverslips, washed twice with PBS, fixed with 4% paraformaldehyde (Alfa Aeser) in PBS (Biochrom) for 20 min at room temperature, permeabilized with ice-cold methanol for 30 s, followed by quenching and blocking with 50 mM NH4Cl (Roth) in PBS/5% goat serum (Life Technologies) for 30 min at room temperature. The coverslips were incubated with primary antibodies directed against Flag M2 (Sigma Aldrich) and C. burnetii diluted in PBS/5% goat serum for 1 h at room temperature, washed three times with PBS and further incubated with secondary Alexa Fluor labelled antibodies Alexa 488 and Alexa 594 (Dianova) diluted in PBS/5% goat serum for 1 h. After three washes with PBS, the cells were mounted using ProLong Diamond with DAPI to visualize cell nuclei and bacterial DNA. Confocal fluorescence microscopy was performed using a Carl Zeiss LSM 700 Laser Scan Confocal Microscope.
Determination of transcript variations of AnkG
RNA from different AnkG strains was isolated as described in “Quantification of AnkG Level”. For DNase digestion and cDNA synthesis, the RNA was diluted to a 10 ng/µl final concentration. The following strains were used: NMI as member of the group one, 19/34 as member of the second group, and Z3574-1/92 as member of the third group (Table 1). To exclude DNA contamination, a no reverse transcriptase control was performed using 2 µl RNA as template in a PCR reaction with primers 760/761 (dotA).
PCR for different ankG sections was performed using the DreamTaq DNA polymerase kit (ThermoScientific). Per sample, 25 µl of PCR reagents were used containing 18.8 µl of DNase-free H2O, 2.5 µl of DreamTaq Green Buffer, 0.5 µl of dNTPs, 0.5 µl of each Primer (10 µM), and 0.2 µl of polymerase. The amplification was run with the following parameters: 2 min at 95 °C; 30 cycles, each consisting of 30 s, 95 °C, 30 s at 58 °C, and 1 min 72 °C; final elongation of 10 min at 72 °C. Primers were used for the 5´, N-terminus encoding part of the mRNA (a817/a818) and for the full length product (a817 and a822) (Table 3).
Amplified ankG fragments were analyzed using a 2% agarose (Bio&Sell) gel containing 0.004% midori green (Nippon genetics). The gels were run for 30 min with 120 V and were analyzed using UV light in a gel documentation chamber.
Axenic cultivation of C. burnetii and infection of MEFs
The following experimental procedures were described before33. In brief, C. burnetii Nine Mile phase II (NMII), transformed with the respective plasmid, was propagated for 6 days in 75 cm2 tissue culture flasks at 37 °C and 5% CO2, 2.5% O2 in 30 ml acidified citrate cysteine medium (ACCM-2), which supports host cell-free (axenic) growth of C. burnetii55. After 5 days, Isopropyl-β-D-1-thiogalactopyranoside (IPTG) was added to the media where appropriate. The bacteria were pelleted for 30 min at 4.500 × g, 4 °C, resuspended in PBS, pH 7.4, aliquoted and stored at -70 °C for further use. MEFs were seeded 1 day before infection on 10 mm coverslips in 24-well dishes at a density of 2 × 104 cells/well. MEFs were infected with C. burnetii at an MOI of 50 in cell culture media containing IPTG. The cells were centrifuged at 250 × g, for 10 min at 20 °C without braking and afterwards incubated at 37 °C and 5% CO2 for 48 h.
Infection of Galleria mellonella
C. burnetii, C. burnetii ΔdotA, C. burnetii pFlag-AnkG and C. burnetii pFlag-AnkF were grown in 15 ml of axenic ACCM-2 with or without 3 µg/ml chloramphenicol in 25 cm2 cell culture filter flasks at 37 °C, 95% CO2 and 2.5% O2 for 5 days. One day before infection, 2 mM IPTG was added to the C. burnetii pFlag-AnkG and C. burnetii pFlag-AnkF cultures to induce expression of 3xFlag-AnkG and 3xFlag-AnkF. At the day of infection, C. burnetii cultures were pelleted and resuspended in PBS at 5 × 104/µl. Galleria mellonella larvae, purchased from TruLarv (UK), were randomized by size into groups of 10 larvae on wood chips. For infection, 20 µl of PBS or C. burnetii-containing solutions were injected into the uppermost right leg of the larvae. The larvae were incubated at RT for 7 days with survival monitoring every 24 h. The larvae were regarded as dead when they were not able to move or appeared black.
Quantification bacterial load/larvae
At day 1 and day 5, two infected Galleria mellonella larvae were frozen at -80 °C. On the same day all larvae were disrupted using a BeadRuptor and lysed with Proteinase K overnight. The next day genomic DNA of C. burnetii was isolated with the Qiagen DNeasy Blood and Tissue Kit and an RT-PCR was performed to determine the bacterial load. For the fold replication analysis, the genome equivalents of day 5 larvae were compared to the genome equivalents of day 1 larvae. The experiment was performed three times.
Immunofluorescence of infected hemocytes
Hemocytes from 3 Galleria mellonella larvae infected with either C. burnetii, C. burnetii ΔdotA, C. burnetii pFlag-AnkG or C. burnetii pFlag-AnkF were collected at 1 day and 5 days post infection38. Cells were seeded and centrifuged on poly-l-lysine-coated coverslips, washed twice with PBS, fixed with 4% paraformaldehyde (Alfa Aeser) in PBS (Biochrom) for 20 min at room temperature, permeabilized with 0.1% Triton- X 100, followed by quenching and blocking with 50 mM NH4Cl (Roth) in PBS/5% goat serum (Life Technologies) for 30 min at room temperature. The coverslips were incubated with primary antibodies directed against C. burnetii and actin (phalloidin labeled with Alexa Fluor 674) diluted in PBS/5% goat serum for 20 min at room temperature, washed three times with PBS and further incubated with secondary Alexa Fluor labelled antibodies Alexa 488 diluted in PBS/5% goat serum for 20 min. After three washes with PBS, the cells were mounted using ProLong Diamond with DAPI to visualize cell nuclei and bacterial DNA. Confocal fluorescence microscopy was performed using a Carl Zeiss LSM 700 Laser Scan Confocal Microscope. In Z-Stack images with 1 µm distance 100 randomized cells were counted and determined whether they were infected or not. The experiment was performed three times.
Targeted DotA deletion in C. burnetii NMII
Targeted gene deletion of the NMII gene dotA was performed as described previously by homologous recombination with a suicide plasmid56. Briefly, 1 × 1010 electrocompetent C. burnetii NMII were transformed with the plasmid harboring 2 kb 5′ and 3′ flanking region of genomic dotA CDS flanking a PCom1-CAT cassette (BioRad Gene Pulser XCell, 18,000 V/cm2, 500 Ω, 25 µF in 0.1 cm gap cuvettes). Subsequently, transformed NMII were cultivated axenically in 6 ml 1xACCM-2 with 3 µg/ml chloramphenicol and/or 350 µg/ml kanamycin at 37 °C and 2.5% O2 atmosphere in T25 flasks for 6 days. Afterwards, cultures were passaged every 7 d for 3 weeks in 6 ml medium supplemented with chloramphenicol (CM) and kanamycin in T25 flasks. Identification of plasmid co-integrants was performed by colony PCR with primers as proposed56. Co-integrants were then subcultured in 3 ml medium supplemented with CM and 1% sucrose in 6-well plates for 4 d, followed by two passages for 6 d in 6 ml medium supplemented with CM in T25 flasks until late stationary phase. Identification of dotA knock-out (ΔdotA) strains was performed by colony PCR with specific primers56. Clonal isolation of ΔdotA strains was performed through plating on semi-solid agar plates.
Translocation assay
AnkGNM, AnkGSoyta and AnkGF3 were cloned into the pJB-CAT-CyaA plasmid. C. burnetii grown in ACCM-D medium were electroporated with 10 µg pJB-CAT-Cya-AnkGSoyta, pJB-CAT-Cya-AnkGNM or pJB-CAT-Cya-AnkGF3. Transformants were selected by culturing the bacteria in ACCM-D medium containing 3 µg/ml chloramphenicol for 5 days, plated onto ACCM-D/0.25% agarose plates supplemented with 3 µg/ml chloramphenicol. Single clones were picked after 7–10 days as previously described56. CHO cells were infected with the different C. burnetii mutants in a 24 well plate at an MOI of 200 for 3 days at 37 °C and 5% CO2. The translocation assay was performed by measuring the concentration of cAMP in cell lysates using the cAMP enzyme immunoassay (GE Healthcare) as previously described19. The threshold was set as 2.5-fold more cAMP than the controls (uninfected cells and cells infected with C. burnetii expressing CyaA alone)25.
Plasmid construction
Plasmids used in this study are listed in Table 3. The AnkGSoyta and AnkGF3 genes were amplified from C. burnetii Soyta or F3 genomic DNA by standard PCR using the primers listed in Table 4. The resulting PCR product was restricted with AscI, followed by ligation with likewise-restricted pEGFP-C3-AscI.
For creation of the constructs pJB-CAT-Cya-AnkGNM, pJB-CAT-Cya-AnkGSoyta and pJB-CAT-Cya-AnkGF3, the genes were amplified from pEGFP-AnkGNM, pEGFP-AnkGSoyta and pEGFP-AnkGF3 using the primers listed in Table 4, restricted with SalI, and ligated with likewise-restricted pJB-CAT-Cya.
For creation of the constructs pCMV-HA-AnkGSoyta and pCMV-HA-AnkGF3, the genes were amplified from pEGFP-AnkGSoyta and pEGFP-AnkGF3 using the primers listed in Table 4, restricted with BglII and NotI, and ligated with likewise-restricted pCMV-HA.
For cloning of the constructs pEGFP-AnkGNM 1–51, pEGFP-AnkGNM 1–92, pEGFP-AnkGF3 1–28, pEGFP-AnkGSoyta 1–84 and pEGFP-AnkGSoyta 1–86, the genes were amplified from pEGFP-AnkGNM, pEGFP-AnkGSoyta, pEGFP-AnkGF3, using the primers listed in Table 4, restricted as indicated, and ligated with likewise-restricted pEGFP-C1, C2, C3 or pEGFP-C3-AscI.
For creation of the constructs pcDNA–4TO–Flag–AnkGSoyta, and pcDNA–4TO–Flag-AnkGF3, the genes were amplified from pCMV–HA–AnkGSoyta and pCMV–HA–AnkGF3 using the primers listed in Table 4, restricted with BamHI and NotI, and ligated with likewise-restricted pcDNA–4TO–Flag.
For cloning of the constructs pFlag–AnkGSoyta and pFlag–AnkGF3, the genes were amplified from pcDNA–4TO–Flag–AnkGSoyta or pcDNA–4TO–Flag–AnkGF3 using the primers listed in Table 4, restricted with KpnI, and ligated with likewise-restricted pKM244mod.
The construct pEGFP-AnkG1-28 was produced using the primers in Table 4. As template pEGFP-AnkGNM was used and AnkG1-28 coupled to GFP was amplified and ligated in a pEGFP backbone lacking GFP. The restriction enzymes NdeI and BamHI were used for directed cloning.
For construction of pJC-CAT::ankG-5′3′-lysCA, the 5′ and 3′ regions of ankG were amplified by PCR from NMI genomic DNA using the specific primer sets (5′- CGGTACCCGGGGATCCGATCGATTACTGCAGAGAAGC and 3′- CACCCATATGCGACGCGAGCGTCGAGAGAATATCCTTATTTTTGATGTC) and (5′- CGTCGCATATGGGTGCGCATGTACGTCAAAAGATGAGGGGTGCTAATG and 3′- GAACCTGTTTGTCGACCAATAACGCTAAGAATAATAATATAG), respectively. The 5′ and 3′ PCR products were cloned into BamHI/SalI-digested pJC-CAT by In-Fusion, resulting in formation of an internal NdeI site between the 5′ and 3′ fragments and creation of pJC-CAT::ankG-5′3′. The 1169P-lysCA cassette was amplified from pJC-CAT::1169P-lysCA57 by PCR with specific primers (5′-GCTCGCGTCGCATATGGAGCTCGGTACCCGGGGATCC and 3′-CATGCGCACCCATATGGATTAATTAGAGAACCTGTTTGTCGAC) and cloned by In-Fusion into NdeI-digested pJC-CAT::ankG-5′3′ to create pJC-CAT::ankG-5′3′-lysCA.
Generation of a C. burnetii ΔankG mutant
C. burnetii Nine Mile phase II were electroporated with 10 µg pJC-CAT::ankG-5´3´-lysCA as previously described58. Co-integrants were selected by culturing the bacteria in ACCM-D media lacking lysine, but containing 2% sucrose for 4 days as previously described57. Surviving transformants were expanded in ACCM-D media lacking lysine for 7 days. After spreading the diluted culture on 0.25% ACCM-D agarose without lysine clonal ΔankG mutants were picked after 7 days of culture. The picked clones were expanded in ACCM-D media without lysine.
Quantification of the ankG expression level
RNA was isolated using the TriFast Reagent (VWR) according to the manufacturer’s instructions and treated with DNase (RNase-Free DNase Set, Qiagen). Therefore, 1,000 ng RNA were digested using 2 µl RDD Buffer and 1 µl DNase (ad 20 µl). cDNA synthesis was performed with SuperScriptII reverse transcriptase (Thermo Scientific) as recommend by the manufacturer using specific primers for ankG and dotA. Subsequent quantification by qRT-PCR was performed using the SybrGreen qPCR Mix (Thermo Scientific) with primers for ankG (65/66) and dotA (737/738) for normalization (Table 4).
Statistical analysis
An unpaired Student´s t-test was used for statistical analysis.
References
Maurin, M. & Raoult, D. Q fever. Clin.. Microbiol. Rev. 12, 518–553 (1999).
Ayres, J. G. et al. Post-infection fatigue syndrome following Q fever. QJM Mon. J. Assoc. Phys. 91, 105–123 (1998).
Marmion, B. P., Shannon, M., Maddocks, I., Storm, P. & Penttila, I. Protracted debility and fatigue after acute Q fever. Lancet 347, 977–978 (1996).
Kersh, G. J. Antimicrobial therapies for Q fever. Expert Rev. Anti-infect. Ther. 11, 1207–1214. https://doi.org/10.1586/14787210.2013.840534 (2013).
Stein, A. et al. Q fever pneumonia: virulence of Coxiella burnetii pathovars in a murine model of aerosol infection. Infect. Immun. 73, 2469–2477 (2005).
Capo, C. et al. Subversion of monocyte functions by coxiella burnetii: impairment of the cross-talk between alphavbeta3 integrin and CR3. J. Immunol. 163, 6078–6085 (1999).
Martinez, E., Cantet, F., Fava, L., Norville, I. & Bonazzi, M. Identification of OmpA, a Coxiella burnetii protein involved in host cell invasion, by multi-phenotypic high-content screening. PLoS Pathog. 10, e1004013 (2014).
Beron, W., Gutierrez, M. G., Rabinovitch, M. & Colombo, M. I. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect. Immun. 70, 5816–5821 (2002).
Howe, D. & Mallavia, L. P. Coxiella burnetii exhibits morphological change and delays phagolysosomal fusion after internalization by J774A.1 cells. Infect. Immun. 68, 3815–3821 (2000).
Schulze-Luehrmann, J. et al. LAMP proteins account for the maturation delay during the establishment of the Coxiella burnetii-containing vacuole. Cell Microbiol. 18, 181–194 (2016).
Flannagan, R. S., Jaumouille, V. & Grinstein, S. The cell biology of phagocytosis. Annu. Rev.. Pathol. 7, 61–98 (2012).
Hackstadt, T. & Williams, J. C. Biochemical stratagem for obligate parasitism of eukaryotic cells by Coxiella burnetii. Proc. Natl. Acad. Sci. U. S. A. 78, 3240–3244 (1981).
Lamkanfi, M. & Dixit, V. M. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 8, 44–54 (2010).
Gallucci, S., Lolkema, M. & Matzinger, P. Natural adjuvants: endogenous activators of dendritic cells. Nat. Med. 5, 1249–1255 (1999).
Lührmann, A. & Roy, C. R. Coxiella burnetii inhibits activation of host cell apoptosis through a mechanism that involves preventing cytochrome c release from mitochondria. Infect. Immun. 75, 5282–5289 (2007).
Voth, D. E., Howe, D. & Heinzen, R. A. Coxiella burnetii inhibits apoptosis in human THP-1 cells and monkey primary alveolar macrophages. Infect. Immun. 75, 4263–4271 (2007).
van Schaik, E. J., Chen, C., Mertens, K., Weber, M. M. & Samuel, J. E. Molecular pathogenesis of the obligate intracellular bacterium Coxiella burnetii. Nat. Rev. Microbiol. 11, 561–573 (2013).
Beare, P. A. et al. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. MBio 2, e00175-e1111 (2011).
Carey, K. L., Newton, H. J., Lührmann, A. & Roy, C. R. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog. 7, e1002056 (2011).
Alvarez-Martinez, C. E. & Christie, P. J. Biological diversity of prokaryotic type IV secretion systems. Microbiol. Mol. Biol. Rev. 73, 775–808 (2009).
Cunha, L. D. et al. Inhibition of inflammasome activation by Coxiella burnetii type IV secretion system effector IcaA. Nat. Commun. 6, 10205 (2015).
Graham, J. G., Winchell, C. G., Sharma, U. M. & Voth, D. E. Identification of ElpA, a Coxiella burnetii pathotype-specific Dot/Icm type IV secretion system substrate. Infect. Immun. 83, 1190–1198 (2015).
Klingenbeck, L., Eckart, R. A., Berens, C. & Lührmann, A. The Coxiella burnetii type IV secretion system substrate CaeB inhibits intrinsic apoptosis at the mitochondrial level. Cell Microbiol. 15, 675–687 (2013).
Kohler, L. J. et al. Effector protein Cig2 decreases host tolerance of infection by directing constitutive fusion of autophagosomes with the Coxiella-containing vacuole. MBio 7, e01327-e1416 (2016).
Larson, C. L., Beare, P. A., Howe, D. & Heinzen, R. A. Coxiella burnetii effector protein subverts clathrin-mediated vesicular trafficking for pathogen vacuole biogenesis. Proc. Natl. Acad. Sci. U. S. A. 110, E4770-4779 (2013).
Lührmann, A., Nogueira, C. V., Carey, K. L. & Roy, C. R. Inhibition of pathogen-induced apoptosis by a Coxiella burnetii type IV effector protein. Proc. Natl. Acad. Sci. U. S. A. 107, 18997–19001 (2010).
Martinez, E. et al. Coxiella burnetii effector CvpB modulates phosphoinositide metabolism for optimal vacuole development. Proc. Natl. Acad. Sci. U. S. A. 113, E3260-3269 (2016).
Weber, M. M. et al. Modulation of the host transcriptome by Coxiella burnetii nuclear effector Cbu1314. Microbes Infect. 18, 336–345 (2016).
Weber, M. M. et al. The Type IV secretion system effector protein CirA stimulates the GTPase activity of RhoA and is required for virulence in a mouse model of Coxiella burnetii infection. Infect. Immun. 84, 2524–2533 (2016).
Berens, C., Bisle, S., Klingenbeck, L. & Lührmann, A. Applying an inducible expression system to study interference of bacterial virulence factors with intracellular signaling. J. Vis. Exp. JoVE https://doi.org/10.3791/52903 (2015).
Bisle, S. et al. The inhibition of the apoptosis pathway by the Coxiella burnetii Effector Protein CaeA requires the EK repetition motif, but is independent of survivin. Virulence 7, 400–412 (2016).
Eckart, R. A. et al. Antiapoptotic activity of Coxiella burnetii effector protein AnkG is controlled by p32-dependent trafficking. Infect Immun 82, 2763–2771 (2014).
Schäfer, W. et al. Nuclear trafficking of the anti-apoptotic Coxiella burnetii effector protein AnkG requires binding to p32 and Importin-alpha1. Cell Microbiol. https://doi.org/10.1111/cmi.12634 (2017).
Voth, D. E. et al. The Coxiella burnetii ankyrin repeat domain-containing protein family is heterogeneous, with C-terminal truncations that influence Dot/Icm-mediated secretion. J. Bacteriol. 191, 4232–4242 (2009).
Kersh, G. J. et al. Genotyping and axenic growth of Coxiella burnetii isolates found in the United States environment. Vector Borne Zoonot. Dis. 16, 588–594 (2016).
Sandoz, K. M., Beare, P. A., Cockrell, D. C. & Heinzen, R. A. Complementation of arginine auxotrophy for genetic transformation of Coxiella burnetii by use of a defined axenic medium. Appl. Environ. Microbiol. 82, 3042–3051 (2016).
Voth, D. E. et al. The Coxiella burnetii cryptic plasmid is enriched in genes encoding type IV secretion system substrates. J. Bacteriol. 193, 1493–1503 (2011).
Norville, I. H. et al. Galleria mellonella as an alternative model of Coxiella burnetii infection. Microbiology 160, 1175–1181 (2014).
Selim, A., Yang, E., Rousset, E., Thiery, R. & Sidi-Boumedine, K. Characterization of Coxiella burnetii strains from ruminants in a Galleria mellonella host-based model. New Microbes New Infect. 24, 8–13 (2018).
Kuba, M. et al. Coxiella burnetii utilizes both glutamate and glucose during infection with glucose uptake mediated by multiple transporters. Biochem. J. 476, 2851–2867 (2019).
Beare, P. A. et al. Comparative genomics reveal extensive transposon-mediated genomic plasticity and diversity among potential effector proteins within the genus Coxiella. Infect. Immun. 77, 642–656 (2009).
Hemsley, C. M. et al. Extensive genome analysis of Coxiella burnetii reveals limited evolution within genomic groups. BMC Genomics 20, 441 (2019).
Lührmann, A., Newton, H. J. & Bonazzi, M. Beginning to understand the role of the type IV secretion system effector proteins in Coxiella burnetii pathogenesis. Curr. Top. Microbiol. Immunol. 413, 243–268 (2017).
Zhang, Y., Zhang, G., Hendrix, L. R., Tesh, V. L. & Samuel, J. E. Coxiella burnetii induces apoptosis during early stage infection via a caspase-independent pathway in human monocytic THP-1 cells. PLoS ONE 7, e30841 (2012).
Schoenlaub, L., Cherla, R., Zhang, Y. & Zhang, G. Coxiella burnetii avirulent nine mile phase II induces caspase-1-dependent pyroptosis in murine peritoneal B1a B cells. Infect. Immun. 84, 3638–3654 (2016).
Mei, G., Di Venere, A., Rosato, N. & Finazzi-Agro, A. The importance of being dimeric. FEBS J. 272, 16–27 (2005).
Gurlebeck, D., Szurek, B. & Bonas, U. Dimerization of the bacterial effector protein AvrBs3 in the plant cell cytoplasm prior to nuclear import. Plant J. 42, 175–187 (2005).
Bender, J. K., Wille, T., Blank, K., Lange, A. & Gerlach, R. G. LPS structure and PhoQ activity are important for Salmonella typhimurium virulence in the Galleria mellonella infection model [corrected]. PLoS ONE 8, e73287. https://doi.org/10.1371/journal.pone.0073287 (2013).
Harding, C. R. et al. Legionella pneumophila pathogenesis in the Galleria mellonella infection model. Infect. Immun. 80, 2780–2790 (2012).
Joyce, S. A. & Gahan, C. G. Molecular pathogenesis of Listeria monocytogenes in the alternative model host Galleria mellonella. Microbiology 156, 3456–3468 (2010).
Miyata, S., Casey, M., Frank, D. W., Ausubel, F. M. & Drenkard, E. Use of the Galleria mellonella caterpillar as a model host to study the role of the type III secretion system in Pseudomonas aeruginosa pathogenesis. Infect. Immun. 71, 2404–2413 (2003).
Ott, L. et al. Evaluation of invertebrate infection models for pathogenic corynebacteria. FEMS Immunol. Med. Microbiol. 65, 413–421 (2012).
Mukherjee, K. et al. Galleria mellonella as a model system for studying Listeria pathogenesis. Appl. Environ. Microbiol. 76, 310–317 (2010).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Omsland, A. et al. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc. Natl. Acad. Sci. U. S. A. 106, 4430–4434 (2009).
Beare, P. A., Sandoz, K. M., Omsland, A., Rockey, D. D. & Heinzen, R. A. Advances in genetic manipulation of obligate intracellular bacterial pathogens. Front.. Microbiol. 2, 97 (2011).
Beare, P. A., Jeffrey, B. M., Long, C. M., Martens, C. M. & Heinzen, R. A. Genetic mechanisms of Coxiella burnetii lipopolysaccharide phase variation. PLoS Pathog. 14, e1006922 (2018).
Beare, P. A. & Heinzen, R. A. Gene inactivation in Coxiella burnetii. Methods Mol. Biol. 1197, 329–345 (2014).
Lischka, P., Sorg, G., Kann, M., Winkler, M. & Stamminger, T. A nonconventional nuclear localization signal within the UL84 protein of human cytomegalovirus mediates nuclear import via the importin alpha/beta pathway. J. Virol. 77, 3734–3748 (2003).
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) through the grant LU1357/5-1 to AL. We thank Prof. Robert Heinzen, Dr. Alyssa Ingmundson, Prof. James Samuel and Prof. Thomas Stamminger for providing plasmids, and the Lührmann lab for discussion.
Funding
Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A.L. conceived the study. A.L. and J.P.G. designed the study. W.S., T.S., A.C., V.B., J.S.L., N.W. and J.H. performed the experiments. W.S., T.S., A.C., C.B., J.P.G. and A.L. analyzed the data. P.A.B., J.P., C.H. and C.B. provided resources. A.L. supervised the study. W.S., A.C. and A.L. wrote the manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schäfer, W., Schmidt, T., Cordsmeier, A. et al. The anti-apoptotic Coxiella burnetii effector protein AnkG is a strain specific virulence factor. Sci Rep 10, 15396 (2020). https://doi.org/10.1038/s41598-020-72340-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-72340-9
This article is cited by
-
STING dependent BAX-IRF3 signaling results in apoptosis during late-stage Coxiella burnetii infection
Cell Death & Disease (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
