
Abstract
Over the past 30 years, human disturbance and habitat fragmentation have severely endangered the survival of common wild rice (Oryza rufipogon Griff.) in China. A better understanding of the genetic structure of O. rufipogon populations will therefore be useful for the development of conservation strategies. We examined the diversity and genetic structure of natural O. rufipogon populations at the national, provincial, and local levels using simple sequence repeat (SSR) markers. Twenty representative populations from sites across China showed high levels of genetic variability, and approximately 44% of the total genetic variation was among populations. At the local level, we studied fourteen populations in Guangxi Province and four populations in Jiangxi Province. Populations from similar ecosystems showed less genetic differentiation, and local environmental conditions rather than geographic distance appeared to have influenced gene flow during population genetic evolution. We identified a triangular area, including northern Hainan, southern Guangdong, and southwestern Guangxi, as the genetic diversity center of O. rufipogon in China, and we proposed that this area should be given priority during the development of ex situ and in situ conservation strategies. Populations from less common ecosystem types should also be given priority for in situ conservation.
Similar content being viewed by others
Introduction
Common wild rice (Oryza rufipogon Griff.) is the putative progenitor of Asian cultivated rice, one of the most important food crops in the world. It is also an important source of germplasm for rice improvement1,2,3. Ding Ying found wild rice (O. rufipogon) in Guangzhou in 1926, and the wild × cultivated cross Zhong Shan No. 1 was widely planted in South China for more than 50 years. In 1970, Yuan Longping and his assistant discovered wild rice with male sterility in Hainan and used it to breed high yielding “three-line” hybrid rice varieties4,5. The increased yield of this hybrid rice saved thousands of lives in China and around the world. In recent years, wild rice has been used to introduce genes that confer agriculturally beneficial traits into cultivated species, and it holds great potential for future rice breeding efforts.
Before the 1970s, O. rufipogon was found in 113 counties of eight provinces in southern China, including Guangdong, Guangxi, Hainan, Yunnan, Hunan, Jiangxi, Fujian, and Taiwan, although the populations in Taiwan disappeared in 19786,7. Since the 1980s, many wild rice habitats have been converted to agricultural or industrial use because of the rapid development of the rural economy and the population expansion in rural China. Consequently, areas of wild rice cultivation have dramatically decreased. Our recent work indicates that approximately 70% of the O. rufipogon populations have disappeared, and all large populations (growth area > 33 hm2) have either disappeared or decreased dramatically (unpublished data).
The threatened status of wild rice has attracted increasing attention in China, and there is a desire to collect samples for ex situ conservation and to develop in situ conservation programs. The Chinese government began to invest in the construction of in situ conservation sites in 2001, and 15 such sites were established by the end of 2014. The government also established eight other in situ conservation sites using a mainstreaming approach, a new conservation strategy in which farmers are encouraged to participate in conservation activities that span physical boundaries such as hills and rivers. Most in situ conservation sites were selected based on scientific expertise or local government recommendations, although it was still necessary to justify their value and rationale. Detailed information on the population genetic structure of O. rufipogon is therefore useful to guide the selection of future sites.
Information generated using molecular methods has direct and indirect consequences for the practical management and conservation of germplasm. Genetic diversity data can be useful for understanding the taxonomy and evolution of crop species, and this basic knowledge supports their conservation8. More directly, genetic diversity studies can help us to adjust our strategies for collection, evaluation, and breeding. Although some studies have recently documented the population genetic structure of O. rufipogon in China9,10,11,12,13, few have focused on the development of conservation strategies based on this population genetic structure. Our study used SSR markers to examine the genetic diversity and population genetic structure of natural populations at three different levels: national (China), provincial (Guangxi Province), and local (Dongxiang population in Jiangxi Province).
Results
Genetic diversity of O. rufipogon populations
Twenty-four SSR primer pairs from previous studies14 with polymorphisms and a uniform distribution among chromosomes were selected for use in the analysis of population genetic diversity and genetic structure (Supplementary Table S1). All loci were found to be in Hardy–Weinberg equilibrium. High levels of genetic variability at 24 loci were detected in 628 individuals from 20 populations sampled across China (Table 1; Fig. 1a). A total of 340 alleles were detected across the loci, ranging from 23 alleles at RM253 to seven alleles at RM244 and RM345 (Supplementary Table S2). The average number of alleles was 14.17. The overall means of AE (the effective number of alleles), HO (the observed heterozygosity), HE (the expected heterozygosity) and I (the Shannon–Weaver information index) across all loci were 6.97, 0.58, 0.83, and 2.08, respectively. The values varied widely among loci: AE ranged from 2.80 (RM244) to 15.41 (RM336); HO ranged from 0.15 (RM244) to 0.88 (RM336); HE ranged from 0.64 (RM244) to 0.93 (RM253); and I ranged from 1.13 (RM244) to 2.85 (RM336).

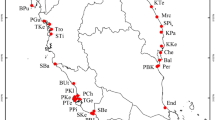
Locations of natural O. rufipogon populations used in this study (a) Map of 20 O. rufipogon populations in China generated by R version 3.5.2. The red triangular area represents the genetic diversity center identified in the present study, and the yellow ovals indicate the diversity center reported by Wang58. (b) The locations of 14 populations from Guangxi (URL: https://commons.wikimedia.org/wiki/File: China_topography_full_res.jpg). (c) The locations of populations from Dongxiang.
The genetic structure of populations across China was analyzed. The mean value of FST was 0.44, and it varied from 0.25 to 0.59, indicating that there was substantial genetic variation among populations (Supplementary Table S2). Genetic population differentiation was also measured by AMOVA analysis (Table 2) and pairwise population differentiation (Supplementary Table S3). AMOVA analysis showed that 41.2% of the variation occurred among populations. The significant differentiation (the P value of FST < 0.001) among populations also reflected larger differences between all populations from the whole country (Table 2). FIS ranged from − 0.49 to 0.44 with an average of − 0.20, consistent with the complex mating system of this species. Many studies have suggested that the mating system of O. rufipogon is a mix of outcrossing and selfing15. This result indicated that most populations deviated from the Hardy–Weinberg expectation within populations and had an excess of heterozygotes. The genetic diversity parameters of natural O. rufipogon populations in China were also summarized by province (Table 3). Among the provinces, Guangxi showed the highest levels of genetic diversity with AE = 5.14, HO = 0.77 and I = 1.80. The next highest levels of genetic diversity were found in Guangdong, Hainan, Hunan, Jiangxi, and Fujian. The correlation between FST and geographic distance in Chinese O. rufipogon populations was nearly significant (p = 0.07874).
Genetic structure of O. rufipogon populations in China
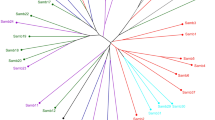
The genetic relationships among populations in China were analyzed with a population structure analysis in STRUCTURE, principal component analysis (PCA) and construction of a UPGMA tree (Fig. 2). In the structure analysis, the log-likelihood ln(P(D)) was largest when the number of populations, K, was equal to four (Fig. 2b), and four groups were therefore identified. Most populations from Hainan were placed in Group 1, together with two southern boundary populations N_GX2 and N_GD1. The N_HN3 population was placed by itself in Group 2. Two northern boundary populations from Guangdong and Guangxi were placed in Group 3 with N_HuN1 and N_JX1. Most populations from Guangdong and Guangxi were placed in Group 4, together with the northernmost population, N_HN5. Results of the PCA were consistent with those of STRUCTURE (Fig. 2c). The UPGMA dendrogram showed that the 20 populations formed three main clusters with a genetic similarity of approximately 0.27 (Fig. 2d). Three populations (N_HN1, N_HN3, and N_HN4) from Hainan were grouped into Cluster 1. N_HN2 from Hainan and N_GD1 and N_GD5 from Guangdong and N_GX2 from Guangxi were grouped into Cluster 2. Group 3 from the structure analysis corresponded to Cluster 4a, Group 4 from the structure analysis corresponded to Cluster 3 and Cluster 4b. Surprisingly, the two Hainan populations (N_HN2 and N_HN5) were placed in different clusters rather than forming a separate cluster. N_HN2 was placed in Cluster 2 with populations from Guangdong and Guangxi, and N_HN5 was separated away from the others.

Population structure analysis of 20 natural O. rufipogon populations in China. (a) Clustering of 20 populations inferred with STRUCTURE (K = 2–4). (b) Delta K values for STRUCTURE analysis. (c) Results of principal component analysis. (d) UPGMA dendrograms based on Nei’s distance.
Monmonier’s maximum difference algorithm was used to perform a genetic barrier prediction analysis with all populations included (Fig. 3). The first predicted barrier separated N_HN1 and N_HN2 from all other populations. The second barrier separated population N_FJ1, which was located in the easternmost sampled area. The third predicted barrier separated N_HN4, and the fourth barrier separated N_HN3 and N_HN5. Cluster 3b (Group 3) was separated by the fifth barrier, and N_GD1 by the sixth barrier.

Successive genetic barriers predicted with BARRIER software. The genetic barriers are shown as bold red lines with arrows. Lines a–f indicate genetic barriers, and alphabetical order indicates the sequence of boundary formation.
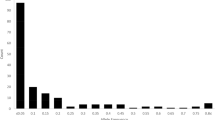
The populations with higher genetic diversity formed a triangular area (Fig. 1a) that reached from 19° N to 23° N and included northern Hainan, southern Guangdong, and southwestern Guangxi. It included the N_GX1, N_GX3, and N_GX2 populations from Guangxi, the N_GD1, N_GD2, N_GD5, and N_GD3 populations from Guangdong, and the N_HN5 population from Hainan. As shown in Table 1, the averages of the diversity parameters in this triangular area were significantly higher than those of other populations and higher than the overall population averages (Fig. 4). The higher diversity and greater number of private alleles indicate that this triangular area may be the genetic diversity center of O. rufipogon in China.

Comparison of genetic diversity parameters among 20 natural populations and between groups within and outside the genetic diversity center. (a) Mean for populations in the diversity center. (b) Mean for populations from across the whole country.
Population structure and differentiation of O. rufipogon in local regions
To understand the history of wild rice populations, we conducted an in-depth study of the largest (Guangxi) (Table 4) and smallest (Jiangxi) populations of wild rice in China. The values of the within-population genetic diversity (HS), the among-population genetic diversity (DST), and the coefficient of genetic differentiation (GST) were 0.38, 0.36 and 0.49, respectively, suggesting that most genetic variability occurred among populations, although GST showed somewhat higher differentiation than DST. These results agreed with the tests of pairwise genetic identity among populations (Supplementary Table S4).
We also used Monmonier’s maximum difference algorithm to perform a genetic barrier prediction analysis for 14 populations from Guangxi, revealing five predicted barriers to gene flow (Supplementary Fig. S1). The You River-Yu River-Xun River formed a primary barrier that isolated the populations into two parts. Five populations (R_GX1, R_GX2, R_GX3, R_GX4, and R_GX5) were from the Nanliu River (Fig. 1b) but were a long distance apart. They were isolated from the other populations by the Darong and Liuwan Mountains. R_GX6, located in Fangchenggang, was isolated by the Gutong Mountain. R_GX9 belonged to Baise City and was distributed in small areas along the You River. R_GX13 was located in the northernmost area of rice distribution and was isolated by Shanzhao Ling and the Liu River.
Dongxiang County is recognized as the northernmost habitat of O. rufipogon, and Anjiashan and Shuitaoshu are the only two sites in Dongxiang where O. rufipogon is found. Four populations were surveyed to gain a more complete understanding of the genetic structure of O. rufipogon populations in Dongxiang. At the Anjiashan site, the primary in situ conservation site for O. rufipogon in China, a relatively large population is divided by a concrete wall that was constructed in the 1980s. DXP1, DXP2 and DXP3 are separate populations that are located close together and have been isolated by the concrete wall from the outside. DXP2 is located in the southeast, DXP3 in the northwest, and DXP1 in the middle. DXP4 is from Shuitaoshu and is further isolated by a hill (Fig. 1c).
We first investigated the genetic structures of the three natural populations from Anjiashan (DXP1, DXP2, and DXP3). The mean HE estimates for DXP1, DXP2, and DXP3 were 0.47, 0.39 and 0.41, respectively, indicating that DXP1 had the highest genetic variation. However, the values of HT, DST, and GST were 0.47, 0.050, and 0.098, suggesting that there was little differentiation among the three populations after more than 20 years of isolation by the concrete wall.
Next, DXP4 was included in the genetic structure analysis, together with the other three populations. The mean DST was 0.06, indicating that there was little genetic variation among the populations. The genetic differentiation over loci assessed by GST (0.12) was slightly higher than DST, but it nonetheless indicated that there was minimal differentiation among the Dongxiang populations. The genetic similarity of all individuals from the four populations was 0.74, highlighting their close genetic relationship. Based on these analyses, the four Dongxiang populations can be considered a single population when collecting samples for ex situ conservation and the establishment of in situ conservation sites.
Discussion
We examined O. rufipogon population differentiation at the national, provincial, and local levels using SSR markers. FST calculations showed that almost half of the total variation occurred among populations. Previously, Zhou12 used SSR markers to investigate twelve Chinese wild rice populations from four provinces and found high genetic differentiation among them (RST = 0.52). Zheng16 analyzed the sequences of seven chloroplastic and nuclear loci and found that pairwise FST values between O. rufipogon populations at the nuclear loci ranged from 0.3175 to 0.5748. The AMOVA and pairwise FST results in the present study provide further evidence for relatively high genetic differentiation and corroborate previous results. Zhou12 concluded that population isolation caused by habitat fragmentation increased genetic differentiation by increasing the frequency of inbreeding and clonal growth. Previous studies have reported indica-like and japonica-like differentiation in the O. rufipogon population12,16,17. Wang18 also suggested that spatial or physical isolation and local adaptation may contribute to population differentiation within this species. The FIS of O. granulata was 0.402, suggesting that most populations deviated from Hardy–Weinberg expectation within populations and were deficient in heterozygotes. The FST of O. granulata was 0.859, indicating that 85.9% of the total genetic variation existed among populations19. For O. officinalis, FIS was 0.899 and FST was 0.88220. Compared with O. officinalis (selfing) and O. granulata (selfing), O. rufipogon has higher heterozygosity and greater genetic distances between populations. The value of FIS in our result was varied from -0.49 to 0.44, indicating that the diversity of mating system among O. rufipogon populations was very high. The mean value of FST was 0.44, and it varied from 0.25 to 0.59, indicating that there was substantial genetic variation among populations. The diversity patterns of species in the genera Leavenworthia21, Lycopersicon22 and Miscanthus23 with different mating systems are similar. The accumulation of mutations from generation to generation as a result of asexual reproduction may also contribute to high heterozygosity in O. rufipogon. In outbreeding species, a decrease in recombination rates is observed in certain regions of the genome, especially around centromeres24. On the contrary, in species with a high level of inbreeding, the rarity of double heterozygotes results in lowered effective recombination rates in the whole genome. Therefore, it is expected that both hitch-hiking and background selection will strongly affect genetic variability in inbreeding species24.
Here, we identified a triangular area, including northern Hainan, southern Guangdong, and southwestern Guangxi, as the genetic diversity center of O. rufipogon in China. Previously, a genetic diversity center for O. rufipogon in south China, including Guangdong and Guangxi provinces, was proposed based on random amplified polymorphic DNA analysis13, allozyme analysis9, and SSR data12. However, our samples were collected according to a more systematic sampling strategy25 in which sampled individuals were at least 12 m apart, and approximately 30 individuals were sampled from each population to encompass at least 95% of its genetic diversity. Our genetic diversity center included one population from Hainan that was not included in the previous diversity center. Gao11 found that, like Guangdong and Guangxi, Hainan also maintained higher levels of microsatellite diversity. Wang18 also found that Hainan ranked first in China with respect to its gene diversity index and gene richness. It is reasonable that the genetic diversity center includes Hainan because it has appropriate annual temperatures (16–23 °C) and precipitation (approximately 1,400 mm), as well as higher levels of outbreeding and diversity of ecological habitats26. In addition, populations in our proposed diversity center had more private alleles than did populations from other areas. Although the diversity of phenotypes in populations within and outside the proposed diversity center should be documented27,28 and compared with molecular data, our SSR data strongly suggest that the proposed triangular area is the diversity center of O. rufipogon in China. Southeastern China has generally enjoyed relative tectonic stability since the late Tertiary29, with perhaps the single exception of its two large islands. The high percentage of endemics in this region30 suggests that central and south China has played a significant role both as a center of survival but also as a center of plant differentiation and evolution during the Quaternary31. This may explain why the center of O. rufipogon genetic diversity is located in Southeastern China.
O. rufipogon is the most important genetic resource for rice breeding and the most endangered wild rice species in China; its collection and conservation are therefore increasingly important. At the national level, a region in southern China whose populations have higher genetic variation and more private alleles is likely to be the genetic diversity center of O. rufipogon. More valuable genes may exist in populations from this area, and its gene pool may be more useful for future variety improvement and biotechnology applications. Therefore, attention should be focused on O. rufipogon from this area for both the construction of in situ conservation sites and the collection of ex situ samples. However, populations outside the genetic center are also important for conservation: almost half of the genetic diversity and 32 out of 76 private alleles existed in these populations. Indeed, populations with relatively low genetic diversity may contain unique alleles that are absent from the diversity center (Table 5). More than 40% of the variation between populations also supported the conclusion that populations from outside the diversity center should receive attention. Based on our analysis of populations across the country, populations in the genetic diversity center should be given first priority when developing national strategies for O. rufipogon conservation. Nonetheless, populations in regions with special ecological conditions, such as unique soils, climates, or valley locations, should also be considered.
Based on the genetic structure analysis of wild rice in Guangxi and Jiangxi, local environmental conditions appear to have influenced gene flow to a greater extent than geographic distance during population genetic evolution. The geography of R_GX9 belonged region is unique, consisting of valleys surrounded by mountains, and the spread of wild rice has therefore been curtailed. The annual minimum temperature of the northernmost area of rice distribution where R_GX13 was located is often below 0 ℃ in this region. The unique local climate and geography have shaped the distinct characteristics of wild rice from the northern mountains. Moreover, The finding that populations from the lower and middle regions of a river contained more genetic variation than those from the upstream regions suggests that conservation efforts should be focused on the downstream populations. Similar genetic diversity results have been reported for plants growing in several important watersheds in China, including Myricaria laxiflora from the Changjiang River in the Three Gorges Region32 and Sophora moorcroftiana along the Yarlung Zhangbo River33.
Smaller population sizes that result from habitat fragmentation may lead to a loss of genetic variation through genetic drift, thereby increasing population differentiation34. However, we found that population divergence was more significantly correlated with environmental conditions than with geographic location and isolation by distance. Populations from similar ecosystems showed less genetic differentiation, and local environmental conditions rather than geographic distance appeared to have influenced gene flow during population genetic evolution. These results are consistent with the recently developed maximum genetic diversity (MGD) theory of molecular evolution35,36, which predicts that similar environments will select for similar genetic variants, regardless of geographic distance37. Environmental factors such as historical habitat fragmentation and local adaptation can cause divergence38, and adaptation to local conditions rather than simple geographic isolation appears to have driven O. rufipogon population differentiation. Our results suggest that ex situ sampling of multiple populations from similar ecosystems should not be a priority because such populations tend to be genetically similar even when they are separated by large distances. We should therefore reduce the number of samples for ex situ conservation collection to avoid duplication, no matter how far apart they are.
Methods
Population sampling
To analyze population genetic structure at the national level, 628 O. rufipogon accessions from 20 populations were collected at locations from 18° N to 28° N latitude. The locations of all 20 populations are shown in Fig. 1a. The sampling region covered all O. rufipogon’s natural distribution areas7 and spread across six provinces in southern China, including Hainan, Guangdong, Guangxi, Fujian, Hunan, and Jiangxi (Table 1). At each latitude, one to four representative populations were selected for analysis. Samples from the N_JX1 population in Jiangxi were collected from both Anjiashan and Shuitaoshu, as these are the only two locations in Jiangxi where O. rufipogon is found. Likewise, the N_FJ1 population is the only existing O. rufipogon population in Fujian. The N_JX1 and N_HuN1 populations are located at the northern boundary of O. rufipogon’s range in China. We expanded our sampling in Guangxi because this province contains the largest population of wild rice, and 380 accessions from 14 populations were collected from this province (Table 4). These samples included individuals from the most northern (R_GX14), southern (R_GX3, R_GX4, R_GX5), and western populations (R_GX7) in the province (Fig. 1b). Within each population, individuals were randomly collected at a distance of at least 5 m from one another to avoid collecting samples from a single genet.
DNA isolation and polymerase chain reaction
Genomic DNA was extracted using the CTAB method according to the protocol of Edwards39. The quality and quantity of DNA were assessed on 0.8% agarose gels. DNA concentrations were determined using an ultraviolet spectrophotometer, and the solutions were then diluted to 20 ng/μL with a Tris–EDTA buffer. PCR amplifications were performed with a 5700 thermocycler (PE Applied Biosystems, USA). The PCR reaction in a total volume of 20 μl consisted of 100 mmol/L Tris–HCl, 1 U Taq polymerase, 2.5 mmol/L MgCl2, 2.5 mmol/L dNTPs, 4 μmol/L forward and reverse SSR primers, and 100 ng DNA. The PCR program was 5 min at 94 °C, followed by 35 cycles of 1 min at 94 °C, 50 s at 56–61 °C, and 1 min at 72 °C, with 10 min at 72 °C for the final extension. PCR products were mixed with 6 μl of loading buffer, denatured at 95 °C for 5 min, and separated in 6% polyacrylamide denaturing gels (38 × 30 × 1 cm3). The banding patterns were visualized according to the manufacturer’s instructions (Promega, USA).
Statistical analysis
The original SSR data were preprocessed using DataFormater 2.740, which transformed SSR data to readable input files for STRUCTURE, PowerMarker, Tassel, GENEPOP and POPGENE. Micro-Checker 2.2.3 was used to check for scoring errors and null alleles41. Based on solid foundation on study of O. rufipogon diversity, we selected the markers without null alleles for analysis12,14,42,43,44,45.We used POPGENE 1.3146 and GENEPOP 3.447 to calculate the genetic diversity parameters: the mean number of alleles per locus (A), the effective number of alleles (AE), the Shannon–Weaver information index (I), the observed heterozygosity (HO), and the expected heterozygosity (HE)48,49. Deviation from Hardy–Weinberg equilibrium and population differentiation were assessed at each locus across all populations using F statistics, including the fixation index within populations (FIS), the fixation index across all populations (FIT) and the gene differentiation index (FST)50. STRUCTURE was used to infer genetic clusters (K) with the model-based clustering method51. We assessed K values from 2 to 9 by performing ten independent runs for each K value, and the model was run with a 10,000 burn-in period and 100,000 Monte Carlo Markov chain repetitions. CLUMPP version 1.152 was used to obtain the optimal clusters for each K. The relationships between populations were assessed by Nei’s53 standard genetic distance using the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) in PowerMarker54. Principal component analysis (PCA) was performed using Tassel 3.0 (https://www.maizegenetics.net/tassel) to summarize the major patterns of variation. Analysis of molecular variance (AMOVA) was performed using Arlequin 3.1155, and Mantel 2.056 was used to assess whether the data fit the hypothesis of isolation by distance, which predicts a significant relationship between geographic distance and genetic distance. A genetic barrier analysis was performed to suggest historical barriers to gene flow among or between collection sites using BARRIER57 (version 2.2, Syracuse University, USA) with Monmonier’s maximum difference algorithm, which takes the geographic coordinates and genetic distance (GD) of each population as inputs.
The presence of private alleles in each population and in groups within and outside the genetic diversity center were assessed, and the richness of private SSR alleles was defined as the average number of private alleles per genotype for each population.
References
Grant, V. & Oka, H. I. Origin of cultivated rice. Taxon 37, 935 (1988).
Khush, G. S. Origin, dispersal, cultivation and variation of rice. Plant Mol. Biol. 35, 25–34 (1997).
Xiao, J. et al. Genes from wild rice improve yield. Nature 384, 223–224 (1996).
Yuan L. P., Virmani S. S. & Mao C. X. Hybrid rice: Achievements and further outlook. in Progress in Irrigated Rice Research. 219–223 (International Rice Research Institute, Manila, 1989).
Yuan L. P. Advantages of and constraints to the use of hybrid rice varieties. in International Workshop on Apomixis in Rice (Wilson K. J. ed.). (Hunan Hybrid Rice Research Center, Changsha, 1993).
Kiang, Y. T., Antonovics, J. & Wu, L. The extinction of wild rice (Oryza perennis formosana) in Taiwan. J. Asian Ecol. 1, 1–9 (1979).
Pang H. H. & Chen C. B. in Wild Rice Resources in China. 204–214 (Science and Technology Press, Guangxi, 2001) (in Chinese).
Ramanatha Rao, V. & Hodgkin, T. Genetic diversity and conservation and utilization of plant genetic resources. Plant Cell. Tissue Organ Cult. 68, 1–19 (2002).
Gao, L. Z. & Hong, S. G. D. Allozyme variation and population genetic structure of common wild rice Oryza rufipogon Griff. in China. Theor. Appl. Genet. 101, 494–502 (2000).
Song, Z., Li, B., Chen, J. & Lu, B. R. Genetic diversity and conservation of common wild rice (Oryza rufipogon) in China. Plant Species Biol. 20, 83–92 (2005).
Gao, L. Population structure and conservation genetics of wild rice Oryza rufipogon (Poaceae): A region-wide perspective from microsatellite variation. Mol. Ecol. 13, 1009–1024 (2004).
Zhou, H., Xie, Z. & Ge, S. Microsatellite analysis of genetic diversity and population genetic structure of a wild rice (Oryza rufipogon Griff.) in China. Theor. Appl. Genet. 107, 332–339 (2003).
Xie Z.W. Population genetics and conservation strategies of Oryza rufipogon Griff. in China. PhD Dissertation (Institute of Botany, Chinese Academy of Sciences, Beijing, 1999).
Wang, C. et al. Genetic diversity and classification of Oryza sativa with emphasis on Chinese rice germplasm. Heredity 112, 489–496 (2014).
Morishima, H. & Barbier, P. Mating system and genetic structure of natural populations in wild rice Oryza rufipogon. Plant Species Biol. 5, 31–39 (1990).
Zheng, X. & Ge, S. Ecological divergence in the presence of gene flow in two closely related Oryza species (Oryza rufipogon and O. nivara). Mol. Ecol. 19, 2439–2454 (2010).
Wei, X. et al. Origin of Oryza sativa in China inferred by nucleotide polymorphisms of organelle DNA. PLoS ONE 7, e49546 (2012).
Wang, M. et al. Geographical genetic diversity and divergence of common wild rice (O. rufipogon Griff.) in China. Chin. Sci. Bull. 53, 3559–3566 (2008).
Gao, L.Z., Song, G. & Hong, D.Y. Low levels of genetic diversity within populations and high differentiation among populations of a wild rice, Oryza granulata Nees et Arn. ex Watt., from China. Int. J. Plant Sci. 161, 691–697(2000).
Gao, L.Z., Song, G. & Hong, D.Y. High levels of genetic differentiation of Oryza officinalis Wall. ex Watt. from China. J. Hered. 92, 511–516 (2001).
Liu, F., Zhang, L. & Charlesworth, D. Genetic diversity in Leavenworthia populations with different inbreeding levels. Proc. Biol. Sci. 265, 293–301. https://doi.org/10.1098/rspb.1998.0295 (1998).
Baudry, E., Kerdelhué, C., Innan, H. & Stephan, W. Species and recombination effects on DNA variability in the tomato genus. Genetics 158, 1725–1735 (2001).
Chiang, Y. C., Chou, C. H., Huang, S. & Chiang, T. Y. Possible consequences of fungal contamination on the RAPD fingerprinting in Miscanthus (Poaceae). Aust. J. Bot. 51, 197–201. https://doi.org/10.1071/BT02021 (2003).
Baudry, E. et al. Species and recombination effects on DNA variability in the tomato genus. Genetics 158, 1725–1735 (2001).
Zhou, Y. et al. Uncovering the dispersion history, adaptive evolution and selection of wheat in China. Plant Biotechnol. J. 16, 280–291 (2018).
Wang, X. et al. Genetic diversity of Oryza rufipogon Griff. in Hainan Province with SSR markers. J. Plant Genet. Resour. 8, 184–188 (2007).
Dong, Y. S., Zhuang, B. C., Zhao, L. M., Sun, H. & He, M. Y. The genetic diversity of annual wild soybeans grown in China. Theor. Appl. Genet. 103, 98–103 (2001).
Zeng, Y. et al. Evaluation of genetic diversity of rice landraces (Oryza sativa L.) in Yunnan, China. Breed. Sci. 57, 91–99 (2007).
Hsu, J. Late cretaceous and cenozoic vegetation in China, emphasizing their connections with North America. Ann. Mo. Bot. Gard. 70, 490–508 (1983).
Tang, C. Q. et al. Identifying long-term stable refugia for relict plant species in East Asia. Nat. Commun. 9, 4488 (2018).
López-Pujol, J., Zhang, F. M., Sun, H. Q., Ying, T. S. & Ge, S. Centres of plant endemism in China: Places for survival or for speciation?. J. Biogeogr. 38, 1267–1280 (2011).
Liu, Y., Wang, Y. & Huang, H. High interpopulation genetic differentiation and unidirectional linear migration patterns in Myricaria laxiflora (Tamaricaceae), an endemic riparian plant in the Three Gorges Valley of the Yangtze River. Am. J. Bot. 93, 206–215 (2006).
Liu, Z. M., Zhao, A. M., Kang, X. Y., Zhou, S. L. & López-Pujol, J. Genetic diversity, population structure, and conservation of Sophora moorcroftiana (Fabaceae), a shrub endemic to the Tibetan Plateau. Plant Biol. (Stuttg) 8, 81–92 (2006).
Ouborg, N. J., Vergeer, P. & Mix, C. The rough edges of the conservation genetics paradigm for plants. J. Ecol. 94, 1233–1248 (2006).
Hu, T. et al. The genetic equidistance result: misreading by the molecular clock and neutral theory and reinterpretation nearly half of a century later. Sci. China Life Sci. 56, 254–261 (2013).
Huang, S. New thoughts on an old riddle: What determines genetic diversity within and between species?. Genomics 108, 3–10 (2016).
Teske, P. R. et al. Mitochondrial DNA is unsuitable to test for isolation by distance. Sci. Rep. 8, 8448 (2018).
Zhao, Y. et al. Are habitat fragmentation, local adaptation and isolation-by-distance driving population divergence in wild rice Oryza rufipogon?. Mol. Ecol. 22, 5531–5547 (2013).
Edwards, K., Johnstone, C. & Thompson, C. A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res. 19, 1349 (1991).
Fan, W. et al. DataFormater, a software for SSR data formatting to develop population genetics analysis. Mol. Plant Breed. 14, 1029–1034 (2016).
Van Oosterhout, C., Hutchinson, W. F., Wills, D. P. M. & Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4, 535–538 (2004).
Wang, M. X. et al. Geographic diversity and differentiation of wild rice (O. rufipogon Griff.) in China. Chinese Sci. Bull. 053, 2768–2775 (2008).
Yang, Q. W. et al. The genetic differentiation of Dongxiang wild rice (Oryza rufipogon Griff.) and its implications for in-situ conservation. Sci. Agric. Sin. 40, 1085–1093 (2007).
Zheng, X.M. & Ge, S. Ecological divergence in the presence of gene flow in two closely related Oryza species (Oryza rufipogon and O. nivara). Mol. Ecol. 19, 2439–2454 (2010).
Zheng, X.M. et al. Genome-wide analyses reveal the role of noncoding variation in complex traits during rice domestication. Sci. Adv. 5, eaax3619 (2019).
Yeh, F.C., RC, Y. & Boyle, T. POPGENE version 1.31. Microsoft windows-based freeware for population genetic analysis. Univ. Alberta Cent. Int. For. Res. 11–23 (1998).
Glaubitz, J. CONVERT: A user-friendly program to reformat diploid genotypic data for commonly used population genetic software packages. Mol. Ecol. Notes 4, 309–310 (2004).
Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89, 583–590 (1978).
Hartl, D. L. & Clark, A. G. Principles of Population Genetics 3rd edn. (Sinauer, Sunderland, 1997).
Nei M. Molecular Evolutionary Genetics 159–164. (Columbia University Press, New York, 1987).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 164, 1567–1587 (2003).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806 (2007).
Nei, M. Genetic distances between populations. Am. Nat. 106, 283–292 (1972).
Liu, K. & Muse, S. V PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 21, 2128–2129 (2005).
Excoffier, L., Laval, G. & Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 1, 47–50 (2007).
Liedloff, A. Mantel Nonparametric Test Calculator (School of Natural Resource Sciences, Queensland University of Technology, Queensland, 1999).
Manni, F., Guérard, E. & Heyer, E. Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by “Monmonier’s algorithm”. Hum. Biol. 76, 173–190 (2004).
Wang, M. X. et al. Genetic structure of Oryza rufipogon Griff. in China. Heredity 101, 527–535 (2008).
Acknowledgements
We acknowledge the support received from the National Natural Science Foundation of China (Grant no. 31670211, 31970237), Shenyang Youth Science and Technology Project (no. RC190223) and the Agricultural Science and Technology Innovation Program of CAAS. In addition, we thank Prof. Chengbin Chen (Guangxi Academy of Agricultural Science), Prof. Xiaoming Wang (Hainan Academy of Agricultural Science), Prof. Luyuan Dai (Yunnan Academy of Agricultural Science), Prof. Dazhou Chen (Jiangxi Academy of Agricultural Science), Prof. Dajian Pan (Guangdong Academy of Agricultural Science), Prof. Chuan Jiang (Fujian Academy of Agricultural Science) and Prof. Xiaoxiang Li (Hunan Academy of Agricultural Science) for their assistance with sample collection. We would also like to acknowledge all of the people who made contributions to this work.
Author information
Authors and Affiliations
Contributions
Q.Y. and X.Z. were responsible for the study design. J.W., J.S., S.L., J.H. were involved in conducting the experiments. X.S., W.Q., Y.C. and L.Z. were involved in resources collection. J.W. and J.S. wrote the manuscript with editing help from all co‐authors. Q.Y. and X.Z. supervised. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Shi, J., Liu, S. et al. Conservation recommendations for Oryza rufipogon Griff. in China based on genetic diversity analysis. Sci Rep 10, 14375 (2020). https://doi.org/10.1038/s41598-020-70989-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-70989-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
