
Abstract
This study investigated the associations between image features extracted from tumor 18F-fluorodeoxyglucose (FDG) uptake and genetic alterations in patients with lung cancer. A total of 137 patients (age, 62.7 ± 10.2 years) who underwent FDG positron emission tomography/computed tomography (PET/CT) and targeted deep sequencing analysis for a tumor lesion, comprising 61 adenocarcinoma (ADC), 31 squamous cell carcinoma (SQCC), and 45 small cell lung cancer (SCLC) patients, were enrolled in this study. From the tumor lesions, 86 image features were extracted, and 381 genes were assessed. PET features were associated with genetic mutations: 41 genes with 24 features in ADC; 35 genes with 22 features in SQCC; and 43 genes with 25 features in SCLC (FDR < 0.05). Clusters based on PET features showed an association with alterations in oncogenic signaling pathways: Cell cycle and WNT signaling pathways in ADC (p = 0.023, p = 0.035, respectively); Cell cycle, p53, and WNT in SQCC (p = 0.045, 0.009, and 0.029, respectively); and TGFβ in SCLC (p = 0.030). In addition, SUVpeak and SUVmax were associated with a mutation of the TGFβ signaling pathway in ADC (FDR = 0.001, < 0.001). In this study, PET image features had significant associations with alterations in genes and oncogenic signaling pathways in patients with lung cancer.
Similar content being viewed by others

Introduction
Radiogenomics, merging medical imaging data and genomic information, has great potential in the era of personalized medicine1,2,3,4. Genomic information could enable physicians to select appropriate management strategies according to the genetic alterations in an individual patient with cancer5. Nevertheless, the application of genomic medicine in the field of oncology has shortcomings because tumors have genetic and phenotypic diversities even within a single mass5,6,7. Such intra-tumor heterogeneity eventually drives treatment failure and disease progression7,8,9. To overcome it, multiple and sequential biopsies should be conducted to identify all the genetic alterations within the whole tumor throughout the course of disease progression within a patient, which is not always feasible in routine clinical practice. Therefore, the search for noninvasive techniques that reflect genetic alterations in multiple sites and at multiple time points is important because such techniques could improve patient care. Various features from medical images of a tumor could be surrogate markers that predict the alteration of particular genetic pathways. A known association between image features and genetic alterations has a potential as a useful additional information to improve decision making of biopsies, which could create new, accessible management strategies for patients with cancer4,6,10.
Associations between image features extracted from 18F-fluorodeoxyglucose (FDG) positron emission tomography/computed tomography (PET/CT) and genetic alterations have not been fully investigated. Image features derived from tumor FDG uptake, which reflects a tumor’s metabolic status, might be associated with the tumor’s genetic characteristics, and such an association could complement the radiogenomic approach made possible by anatomical imaging modalities. We previously investigated the associations among genetic characteristics, heterogeneity, mutation burden, and FDG PET/CT features in patients with lung cancer11. However, that study did not consider whether FDG PET indices are associated with particular genetic alterations. Despite the potential of radiogenomics-based FDG PET images, only a few studies have focused on the relationships between FDG uptake and genetic alterations in patients with lung cancer12,13,14,15, and the study subjects and PET image features of the few studies that have been done were not sufficient to fully elucidate the association between FDG PET imaging and genomic information12,13,14,15. Therefore, the value of features extracted from tumor FDG uptake in predicting genetic alterations in tumors has not been established. Whether or not PET-derived features can reflect alterations in specific oncogenic pathways is not well known.
Therefore, we investigated the associations between features extracted from tumor FDG uptake and genetic alterations in patients with lung cancer.
Results
Characteristics of subjects
The characteristics of the study patients are summarized in Table 1. They had a mean age (± standard deviation) of 62.7 ± 10.2 years (range, 29–82 years), and ADC was the most common cancer type (44.5%).
The 137 patients underwent 1st line surgery treatments only (29 patients, 21.2%), chemotherapy only (54 patients, 39.4%), chemoradiotherapy (16 patients, 11.7%), surgery combined with adjuvant chemotherapy (24 patients, 17.5%), surgery combined with chemoradiotherapy (10 patients, 7.3%), and surgery with neoadjuvant chemoradiotherapy (4, 2.9%).
Patients were clinically followed-up for a median of 24.5 months (range, 0.3–133.0 months). Of the 137 patients, 76 patients (55.5%) died during the follow-up period, and 105 patients (76.6%) had disease progression, with a median PFS of 8.7 months.
Among the 137 tumor lesions, 106 lesions were from lung tissue or bronchus (77.4%, 106/137), and 26 lesions were from lymph nodes (18.9%, 26/137). The remaining 5 tumor lesions were from pleura or trachea (4%, 5/137). In the 106 lung tissue or bronchus lesions, 64 were obtained by surgical resection (60.4%, 64/106), 29 were percutaneous needle biopsy (27.3%, 29/106), and the other 13 were obtained by bronchoscopy biopsy (12.3%, 13/106). All lymph node lesions were obtained by fine needle aspiration biopsy. On average, tumor lesions of enrolled patients had a SUVmax of 11.8 ± 5.7 and an MTV of 65.7 ± 89.2 cm3 (These are measured values before the correction by harmonization method).
Genetic mutations and PET image features
We found that genetic mutations have an association with PET image features. Patients with mutations of particular genes showed different values in certain image features than patients without a mutation in those genes.
Due to limited space, the full dataset of the associations between PET image features and genetic mutations is presented only in Supplementary data 3 (FDR values). Briefly, in patients with ADC, 41 of the 381 target genes had an association with PET image features. The association varied from genes associated with 1 image feature to genes associated with 7 features. In terms of the PET image features, 24 features were associated with genetic mutation, and the number of genes associated with a particular feature ranged from 1 to 7. In SQCC, 35 genes had an association with image features (range from 1 to 10), and 22 image features had an association with genes (range from 1 to 9). In SCLC, 43 genes had an association with image features (range from 1 to 7), and 25 image features had an association with genes (range from 1 to 9).
The major associations (those genes which were mutated in more than 3 of the enrolled patients) are summarized in Table 2. Only 1 of the 41 associated genes in ADC, 4 of the 35 genes in SQCC, and 2 of the 43 genes in SCLC were mutated in more than 3 enrolled patients. In ADC, the PTCH2 gene were associated with 1 PET image feature; in SQCC, the ERCC2, IRS2, NOTCH1, and XPO1 genes were associated with 6 features; and in SCLC, the TSHR and ROS1 genes were associated with 2 features. Additionally, we calculated correlation of the PET image features in Supplementary data 4 because there are associations between the features.
Clusters of image features and oncogenic signaling pathways
The clusters based on PET image features showed an association with alterations in the oncogenic signaling pathways. We classified patients into different groups on the basis of patterns of PET image features by using a consensus clustering analysis, and then we assessed the association between those groups and oncogenic signaling pathways. The results are presented as heatmaps in Figs. 1, 2, and 3, which supports that PET radiomic signature has a potential to predict specific oncogenic pathway alteration. Although the current form of the result has a limitation in practical aspect, they can be based to build a novel image-based model for predicting genetic alteration. In ADC, patients were divided into 9 different clusters, which were mainly represented by 11 PET image features, and the incidence of mutations in the cell cycle and WNT signaling pathways differed significantly by cluster (p = 0.023, p = 0.035, respectively, Fig. 1). The patients with SQCC were divided into 7 clusters represented by 12 features. Those clusters had associations with the cell cycle, p53, and WNT pathway (p = 0.045, 0.009, and 0.029, respectively, Fig. 2). The patients with SCLC divided into 3 clusters represented by 7 features, which were associated with the TGFβ signaling pathways (p = 0.030, Fig. 3).

Visualization of the association between pathway alterations and clusters based on FDG PET features in ADC. (A) Patient clustering according to PET image features. The pathways on the right shows different incidence of alterations according to the clusters. (B) Heatmaps showing z-score of representative PET image features that were chosen based on statistical difference between clusters. MUT, mutation; NA, non applicable; T, texture feature; GLCM, gray level co-occurrence matrix; SUV, standardized uptake value; TFCCM, texture feature coding co-occurrence matrix.

Visualization of the association between pathway alterations and clusters based on FDG PET features in SQCC. (A) Patient clustering according to PET image features. The pathways on the right shows different incidence of alterations according to the clusters. (B) Heatmaps showing z-score of representative PET image features that were chosen based on statistical difference between clusters. CNV, copy number variation, MUT, mutation; NA, non applicable; T, texture feature; TFCCM, texture feature coding co-occurrence matrix; GLCM, gray level co-occurrence matrix; SUV, standardized uptake value; SD, standard deviation; GLSZM, gray level size zone matrix; NGLD, neighboring gray level dependence.

Visualization of the association between pathway alterations and clusters based on FDG PET features in SCLC. (A) Patient clustering according to PET image features. The pathways on the right shows different incidence of alterations according to the clusters. (B) Heatmaps showing z-score of representative PET image features that were chosen based on statistical difference between clusters. MUT, mutation; NA, non applicable; T, texture feature; T, texture feature; GLCM, gray level co-occurrence matrix; SUV, standardized uptake value; SUL; SUV normalized to lean body mass.
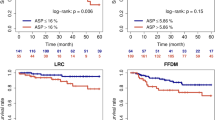
In addition, we assessed the survival value of the clusters. We selected the typical clusters based on their proximity to one another and the pattern of pathway alteration, and reclassified the clusters. The estimated survival over time according to the reclassified clusters is presented in Fig. 4. A Kaplan–Meier analysis showed that the patients in clusters 1 and 9 had better OS than the patients in the other clusters (p = 0.032) in ADC. Patients with SQCC and SCLC didn’t show any significant difference in survival according to the reclassified clusters. In addition, to evaluate survival value of the clusters in ADC, in relation to other factors such as TNM staging, therapeutic regimen, and other PET features, we performed a multivariate survival analysis using Cox proportional hazards regression model. However, the clusters didn't show any significance in multivariate analysis (supplementary table 5).

Prognostic value of the clusters in lung cancer. (A) OS differed significantly according to the reclassified clusters in ADC. The cluster A consists of the 1 and 9. The cluster B consists of the others (refer to Fig. 1). (B) In SQCC, there was no significant difference in survival according to the reclassified clusters. The cluster A consists of the 1, 4, 6, and 7. The cluster B consists of the 2, 3, and 5 (refer to Fig. 2). In SCLC, the reclassified cluster A of SCLC is the initial cluster 1 and the cluster B consists of the 2 and 3 (refer to Fig. 3), and there was also no significant difference in OS and PFS according to the reclassified clusters (Graph not shown, P = 0.194, P = 0.668, respectively). ADC, adenocarcinoma; SQCC, squamous cell carcinoma; SCLC, small cell lung cancer; OS, overall survival, PFS; progression free survival.
Metabolic intensity and oncogenic signaling pathways
Metabolic intensity is associated with a mutation of the TGFβ signaling pathway in ADC. SUVpeak was selected to indicate the metabolic intensity of tumor lesions and was found to be higher in patients with mutations in the TGFβ signaling pathway than in those without them. When SUVmax was used as an indicator of metabolic intensity, the results were similar to those with SUVpeak in the TGFβ signaling pathway (fold change = 0.130, FDR < 0.001, data not shown). SUVpeak also showed raw p-value less than 0.05 in correlation with PI3-Kinase mutation. However, they failed to show significance in the FDR method.
In SQCC, although SUVpeak showed raw p-values less than 0.05 in correlation with mutation of the Hippo signaling pathway, they lost significance when they were corrected by the FDR method. In SCLC, metabolic intensity was not associated with alterations in oncogenic signaling pathways. The correlations between metabolic intensity, presented as SUVpeak, and alterations in oncogenic signaling pathways are summarized in Table 3.
Discussion
We have here investigated the relationships between metabolic image features and genomic alterations, including oncogenic signaling pathways, from the dataset of our institution. Although the limitations of a heterogeneous dataset and incomplete methodology have hampered our ability to generalize our findings, some results are consistent with those of previous studies indicating that the FDG PET-based radiogenomic approach has the potential to play a significant role in cancer research and clinical practice.
In recent years, the search for relationships between imaging phenotypes and genomics, commonly called radiogenomics or image genomics, has emerged as a new direction in cancer research. Knowledge from this new approach has could lead to improved decision making in the management of cancer patients4,10,11. FDG PET-derived image features can reflect the metabolic status of a tumor in ways that anatomical imaging modalities cannot16,17. Genomic alterations underlie a pan-cancer metabolic shift, which is one of the hallmarks of cancer18,19. Many key neoplastic events are controlled by a host of mutational events in multiple cancer-associated genes that converge to alter tumor cell metabolism19,20,21. Therefore, there might be some relationships between the characteristics of tumor FDG uptake and the alteration of cancer-associated genes and pathways. However, despite the research interest in radiogenomics, the value of using an image analysis based on tumor FDG uptake to predict genetic alterations has not been established. Particularly, very few studies have focused on FDG PET–based radiogenomics in lung cancer12,13,14. Nair VS et al. explored differential genome-wide expression across 14 different SUV histogram features in 25 patients with non-small cell lung cancer (NSCLC) to identify individual genes and gene expression signatures associated with prognostically relevant features12. They reported that 8 single genes (BIRC2, FAP, FURIN, LOC648470, LY6E, MCM6, RNF149, and OBFC1) were associated with 7 different SUV histogram features, and eight co-expressed gene clusters (cell adhesion, protein catabolism, nucleic acid processing, metalloproteinase, TP53, RB1, protein processing, embryogenesis, apoptosis, extracellular matrix, and hypoxia ) were also associated with 7 different features. Gevaert O et al. also applied a radiogenomic approach to a cohort of 26 patients with NSCLC13. They extracted 180 image features from CT (179 features) and PET/CT (SUVmax), and identified 243 significant pairwise correlations between image features and co-expressed gene clusters in NSCLC. Among them, SUVmax was associated with 4 co-expressed gene clusters. Crespo-Jara A et al. developed a universal genomic signature predicting the FDG uptake of 84 patients with diverse metastatic tumors, including 7 with lung cancer14. They found that SUVmean correlated with biological processes beyond glycolysis, ribosome biogenesis, DNA replication, cytoskeleton reorganization, and autophagy. Although those researchers have elucidated meaningful results from the data available at the time in a sophisticated way, the relevance of PET image features to predicting gene alteration has not been proved. The patient populations included in the previous studies were too small to have sufficient statistical power. In addition, only a few of the many PET image features were considered12,13,14.
We explored differential genes and oncogenic signaling alterations across 27 PET features (SUV histogram features and textures features) in a cohort of 137 patients with lung cancer (61 ADC, 31 SQCC, and 45 SCLC patients) to identify individual genes and oncogenic signaling pathways associated with PET image features. One strength of the present study is that it considers a much larger study population than previous studies. Another is that it includes a much larger number of PET image features in its analysis. On the other hand, the number of genes analyzed in this study is relatively small compared with previous studies. We used data from a cohort of patients who underwent CancerSCAN, which targeted 83 or 381 genes, which is too few to identify hidden molecular mechanisms or pathways associated with tumor metabolism. In addition, mutation in a single gene may not be enough to guarantee an alteration in the specific pathway it belongs to, which may have led to misinterpretation in the present study. However, the selected target genes are associated with targeted cancer therapies or response to therapy in the literature and public databases22,23, Our data are, therefore, sufficient to identify clinically meaningful associations between genetic alterations and image features.
In this study, 41 single genes were associated with 24 PET image features in ADC. Among them, the only one gene, PTCH2, was arbitrarily classified according to the frequency of alteration among the study subjects. It has known to act as a tumor suppressor24, and showed significant association with GLCM normalized contrast in this study. In addition, image clusters comprising 11 features were associated with the alteration of the cell cycle and WNT signaling pathway. The association of PET features with single genes reported in a previous study was not reproduced in this study12. However, it is difficult to make a direct comparison with the result of the previous study because various factors that can affect the result, such as the PET image features used, the applied target genes, tumor segmentation method, scanner, image protocol, and analyzing strategy are all different. In SQCC, four major genes were associated with 6 PET image features: ERCC225, IRS226, NOTCH127, and XPO128, which functions as a DNA repair gene25, mediates effects of insulin and other cytokines26, plays a role in cell growth, division, differentiation, and apoptosis27, and mediates the nuclear export of proteins and RNAs28, respectively. Image clusters were associated with the p53, WNT, and cell cycle pathways. In SCLC, two major genes were associated with 2 PET image features: TSHR29 and ROS130, which mediates thyroid cell metabolism29 and functions as a growth or differentiation factor receptor30, respectively. Image clusters were associated with the TGFβ signaling pathways. To the best of our knowledge, this is the first study to assess associations between genetic alterations and PET image features, including texture features, in SQCC and SCLC.
However, it is quite questionable whether the association between the image feature/clusters and gene mutation will be reproduced in another cohort of patients. Many factors create obstacles to generalizing the findings of this study, but one of the biggest problems is that the image features are susceptible to various conditions and thus are not a robust indicator that produces consistent outcomes under different study conditions. PET image features are significantly affected by tumor size31, tumor segmentation method11,32, scanner, and image protocol33. In addition, measurement of the PET texture features is 5 times more sensitive to volume changes for small volumes below the proposed minimum than for those above it34. The textural features also depend on the conditions of image acquisition and the reconstruction method. Although there was a difference in degree, all such features exhibited variations according to different acquisition modes and reconstruction parameters17,35,36. Standardization and refinement of methodologies for producing objective and independent image features is required to ensure results of radiomic studies37. Consequently, unless features are obtained with a similar tumor size and the same segmentation method, acquisition modes, and reconstruction parameters, studies such as this one are difficult to compare with other studies. However, a collaboration works toward for standardization such as the image biomarker standardisation initiative (IBSI) has been proposed. Efforts toward standardizing all of these technical issues for the radiomic approach has been made6, which will facilitate the comparison of results between these kinds of studies and will be the basis for drawing reliable conclusions. In this study, to minimize the effect of PET scanners and segmentation methods, we adopted ComBat for harmonization38 recommended by IBSI and choose the robust features for segmentation methods11.
Even though the measurement of PET features is generally sensitive to various conditions, some indicators are relatively robust and can be used with some degree of certainty. SUVpeak and SUVmax, which indicate the metabolic intensity of the target tumor lesion, provide an observer-independent measurement and are less affected by conditions than other features39,40. These intensity features are related to certain genes or oncogenic signaling pathways and are likely to have potential as a universal surrogate marker that is reproducible even under different conditions. In this study, SUVpeak was associated with the TGFβ signaling pathway in ADC. This pathway is an important mediator of tumor invasion, and targeted inhibition of this pathway could be a new approach to lung cancer treatment41,42,43. This result is consistent with that of a previous study. Yamamoto S et al. conducted a differential expression analysis in a public NSCLC dataset that contained FDG PET and messenger RNA expression profile data (n = 26). They found that the genes which have been strongly implicated in epithelial-mesenchymal transition (EMT), including TGF-β (P-value = 0.007) were overexpressed in their high-normalized SUVmax group15. The results from both studies demonstrate a strong association between increased FDG uptake and gene alterations related to EMT in ADC. Given that SUVmax is indicator of relatively low variability that are not significantly affected by other conditions, the conclusion in that TGF-β signaling alteration are more likely to occur in ADC patient with high metabolism is highly reliable. Of course, it should be further confirmed in larger size populations.
This study has several limitations. First, because of an insufficient number of enrolled study subjects, internal validation was not performed, which diminished the quality of the research. Second, many of extracted features using CGITA were not identical to those provided by IBSI, which limits the generalizability and reproducibility of this study. In addition, the heterogeneity of the study subjects, including the scanner, reconstruction method, version of CancerSCAN, and different tissue type could restrict the generalizability of our study results. Validation studies with a large number of homogeneous subjects and standardized radiomic PET features are needed to confirm the findings of this study.
In conclusion, we found significant associations between PET image features and gene mutations in this study. Interestingly, the metabolic intensities of the tumor, presented as SUVpeak and SUVmax, and clusters of PET image features were associated with alterations in specific oncogenic signaling pathways, suggesting that it is not impossible to predict the presence of an alteration by using FDG PET in patients with lung cancer.
However, it is difficult to generalize our results due to the limitations given above and the methodological challenges of metabolic radiogenomics, including the reproducibility and reliability of the features. Further large-scale studies are needed to verify the findings of this study and evaluate whether PET image have predictive value for genetic alteration.
Methods
Subjects
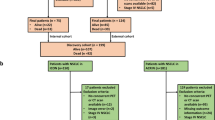
In this study, we enrolled novel study subjects from the candidates for our previous study11. The process of subject selection is summarized in Fig. 5. Briefly, the study candidates were 417 patients with histologically confirmed lung cancer who were enrolled in a database at the Samsung Genome Institute and who underwent 18F-FDG PET/CT. Gene profiles of their tumor tissue had supposed to be made using the CancerSCAN next generation sequencing (NGS)-based targeted-sequencing platform designed at our institution44. All patients had agreed that their data could be used in other studies. Within that candidate pool, we excluded 28 patients whose tumor tissues were obtained for genomic analysis after neoadjuvant therapy or more than 30 days prior to their PET/CT.

Flowchart of patient inclusion, with reasons for exclusion and the total study population. *, These patients were included in the analysis for the correlation between the intensity of tumor FDG uptake and alterations in oncogenic signaling pathways.
Of the remaining 389 patients, we excluded 95 patients whose CancerSCAN results failed quality control, 20 patients with cell line sequencing data, 55 patients who cancelled the CancerSCAN, and 11 patients with cancers other than adenocarcinoma (ADC), squamous cell carcinoma (SQCC), or small cell lung cancer (SCLC). All PET scans in these patients was performed before treatment. In addition, we excluded 75 patients with small tumor volume because small tumor volume affects the measurement of texture features in PET images31. The minimum metabolic tumor volume eligible for texture feature analysis is approximately 10 cm311,45. Thus, patients with less than 10 cm3 of tumor volume were excluded from the analysis. Therefore, a total of 137 patients were finally included and divided into 3 groups by their cancer type (ADC, SQCC, and SCLC).
All the clinical data for the enrolled patients were collected by a review of electronic medical records. Overall survival (OS) was defined as the time from the first date of 1st line treatment until death from any cause, with censoring at the date of the final follow-up in surviving patients. Progression-free survival (PFS) was defined as the time from the first date of 1st line treatment to the date of disease progression, with censoring at the date of the final follow-up if the patient had not progressed. This study was approved by Institutional Review Board of Samsung Medical Center, and the requirement for written informed consent was waived. In addition, all methods were performed in accordance with relevant guidelines and regulations.
PET/CT imaging
All patients were instructed to fast for at least 6 h, and blood glucose was < 200 mg/dl at the time of the FDG injection. PET/CT scans without intravenous or oral contrast were performed on a GE Healthcare (Milwaukee, WI, USA) Discovery LS (n = 41) or Discovery STe (n = 96) scanner. At 60 min after injecting 225–417 MBq of FDG, emission scans were acquired from the skull base to mid-thigh at 4 min per frame in 2D mode (Discovery LS) or 2.5 min per frame in 3D mode (Discovery STe). Whole-body spiral CT was performed with an 8-slice helical CT (140 keV, 40 to 120 mAs adjusted to body weight; section width = 5 mm) for the Discovery LS scanner and a 16-slice helical CT (140 keV, 30 to 170 mAs with AutomA mode; section width = 3.75 mm) for the STe scanner. Attenuation-corrected PET images (voxel size = 4.3 × 4.3 × 3.9 mm for Discovery LS, 3.9 × 3.9 × 3.3 mm for Discovery STe) were reconstructed using CT data and 2D (28 subsets, 2 iterations; Discovery LS) or 3D ordered-subset expectation maximization algorithms (20 subsets, 2 iterations; STe).
PET image analysis
To investigate the association between a PET image feature and a gene mutation or alteration in an oncogenic signaling pathway, we extracted PET image features from the tumors for which a tissue biopsy was performed using CancerSCAN.
Image feature extraction was based on a previous study and used the gradient-based segmentation method (‘PET Edge’) in MIM version 6.4 software (MIM Software Inc., Cleveland, OH, USA)11,46. The target tumor was identified by an experienced nuclear medicine physician (S.H.M) who was unaware of all clinical information except the target tumor site. As the physician drags the cursor out from the center of the target tumor to a point near the edge of the lesion, six axes interactively extend out, and the length of an axis is restricted when a large gradient is detected along that axis. Then, the software automatically outlines a three-dimensional volume of interest on the tumor. After creating gradient-based segmentation of the target tumor lesion, we extracted PET image features using the Chang-Gung Image Texture Analysis toolbox (CGITA, https://code.google.com/p/cigita), an open-source software package implemented in MATLAB (version 2012a; MathWorks Inc., Natick, MA, USA)47. A total of 86 PET features available in CGITA were measured on each segment11,47: 55 texture features and 31 non-texture features (standardized uptake value (SUV) and intensity histogram, 25; texture spectrum, 2; geometry, 4; see Supplementary data 1). To minimize the effect of different PET scanners with different image reconstruction method (2D vs. 3D), we adopted harmonization method which is recommended by IBSI (https://github.com/Jfortin1/Com-BatHarmonization)38, and obtained corrected values of extracted features. Among those corrected 86 image features, 27 features which have shown to be less affected by segmentation methods11 (Supplementary data 1) were selected for further analysis. The metabolic tumor volume (MTV), obtained through the gradient-based segmentation method, was used as a volume indicator to determine whether or not the measured target tumor would be included in the subject enrollment process (Fig. 5).
CancerSCAN and classification of oncogenic signaling pathway
CancerSCAN is an NGS-based targeted-sequencing platform designed at our institution. The reliability of this assay was proved by a robust analytic validation in previous studies, where the details of experimental procedures were described22,23,44.
CancerSCAN version 1 targeted 83 genes, and version 2 targeted 381 genes. The selected target genes for this customized platform were curated at the request of researchers and clinicians and associated in the literature and public databases with targeted cancer therapies or response to therapy. SNVs, small indels, CNVs, and gene fusions were detected using both existing and new algorithms. The genes contained in the two versions are listed in Supplementary data 2. The variant calls were classified into four categories to reflect mode and functional effect of mutations and then condensed at gene level. The four categories include (1) MUT: miss-sense mutation, (2) LoF: loss of function variant including frame-shift insertion/deletion and stop-gain mutation, (3) CNV: copy number variation, and (4) FUSION: known driver gene fusion event.
The genes in CancerSCAN were classified into ten canonical pathways according to the mechanism and pattern of alterations: cell cycle, Hippo, Myc, Notch, NF-E2 p45-related factor 2 (Nrf2), phosphoinositide-3-kinase–protein kinase (PI-3-Kinase)/Akt, receptor tyrosine kinases (RTK)-RAS, transforming growth factor-β (TGFβ) signaling, p53, and β-catenin/Wnt signaling, as proposed by F Sanchez-Vega et al.48. The genes corresponding to those pathways are listed in Supplementary data 2. In this study, patients with alterations in any of the genes corresponding to a particular pathway were considered to have alterations in that pathway.
Statistical analysis
Statistical analysis was performed using SAS version 9.4 (SAS Institute, Cary, NC, USA) and R 3.5.3 (Vienna, Austria; https://www.R-project.org/).
The associations between PET image features and genetic mutations and pathway alterations were assessed. In comparing texture features between patients with gene alterations and those without them, a total of 137 patients with tumor volume ≥ 10 cm3 were enrolled. In the comparison of FDG uptake intensity between patients with a pathway alteration and those without one, a total of 208 patients who met all the criteria of this study except for tumor volume were included because PET image features that represent FDG uptake intensity, such as SUVpeak or SUVmax, are not much affected by tumor volume11,31,45.
The PET image features were verified to determine whether they have a normal distribution. If the values were not normally distributed, they were log-transformed. T-testing was used when the values of the image features were normally distributed; otherwise, the Wilcoxon rank sum test was used. To assess how much a quantity changed, we calculated log fold changes of mean PET image feature between the patients having the alteration and the others. Before calculating the log fold change, if either numerator or denominator equals zero, rescaling was applied as follows:
where X is a PET image feature, and N is the number of the subjects. To correct random events that falsely appear significant, false discovery rate (FDR) was calculated using Benjamini Hochberg procedure, a statistical approach for multiple comparisons.
We clustered the study subjects based on all PET image features using consensus clustering analysis in which we used hierarchical clustering algorithm and Pearson correlation as distance metric. Based on the proportion of ambiguous clustering (PAC) with the interval of consensus index ranging from 0.1 to 0.9, we chose the optimal number of clusters (K). To identify the key image features that are distinctive between clusters, statistical tests were used; Firstly, either ANOVA, Welch’s ANOVA or Kruskal Wallis test depending on the normality of the distribution and homogeneity of variance. Secondly, pair-wise t-tests were performed and image features that were significant in all the tests were chosen in ADC and SQCC. In SCLC, there were no image features significant in all the 6 tests, thus we chose image features significant in five tests instead.
We also assessed the survival value of the clusters based on PET image features. The obtained clusters were reclassified according to their proximity to one another, which was based on dendrogram. The clusters split off together from the higher branches of the dendrogram were reclassified. They were defined in Figure Legends (Fig. 4). Progression free survival (PFS) and overall survival (OS) according to the reclassified clusters were estimated using the Kaplan–Meier survival analysis method and compared using the log-rank test.
All tests were two-sided, and p-values less than 0.05 were considered statistically significant.
References
Schillaci, O. & Urbano, N. Personalized medicine: a new option for nuclear medicine and molecular imaging in the third millennium. Eur. J. Nucl. Med. Mol. Imaging44, 563–566. https://doi.org/10.1007/s00259-017-3616-5 (2017).
Bai, H. X. et al. Imaging genomics in cancer research: limitations and promises. Br. J. Radiol.89, 20151030. https://doi.org/10.1259/bjr.20151030 (2016).
Jaffe, C. C. Imaging and genomics: is there a synergy?. Radiology264, 329–331. https://doi.org/10.1148/radiol.12120871 (2012).
Mazurowski, M. A. Radiogenomics: what it is and why it is important. J. Am. Coll. Radiol.12, 862–866. https://doi.org/10.1016/j.jacr.2015.04.019 (2015).
Vijay, P., McIntyre, A. B., Mason, C. E., Greenfield, J. P. & Li, S. Clinical genomics: challenges and opportunities. Crit. Rev. Eukaryot. Gene Expr.26, 97–113. https://doi.org/10.1615/CritRevEukaryotGeneExpr.2016015724 (2016).
Sala, E. et al. Unravelling tumour heterogeneity using next-generation imaging: radiomics, radiogenomics, and habitat imaging. Clin. Radiol.72, 3–10. https://doi.org/10.1016/j.crad.2016.09.013 (2017).
Gerlinger, M. et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med.366, 883–892. https://doi.org/10.1056/NEJMoa1113205 (2012).
Prasetyanti, P. R. & Medema, J. P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer16, 41. https://doi.org/10.1186/s12943-017-0600-4 (2017).
Burrell, R. A., McGranahan, N., Bartek, J. & Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature501, 338–345. https://doi.org/10.1038/nature12625 (2013).
Andreassen, C. N., Schack, L. M., Laursen, L. V. & Alsner, J. Radiogenomics—current status, challenges and future directions. Cancer Lett.382, 127–136. https://doi.org/10.1016/j.canlet.2016.01.035 (2016).
Moon, S. H. et al. Correlations between metabolic texture features, genetic heterogeneity, and mutation burden in patients with lung cancer. Eur. J. Nucl. Med. Mol. Imaging46, 446–454. https://doi.org/10.1007/s00259-018-4138-5 (2019).
Nair, V. S. et al. Prognostic PET 18F-FDG uptake imaging features are associated with major oncogenomic alterations in patients with resected non-small cell lung cancer. Cancer Res.72, 3725–3734. https://doi.org/10.1158/0008-5472.CAN-11-3943 (2012).
Gevaert, O. et al. Non-small cell lung cancer: identifying prognostic imaging biomarkers by leveraging public gene expression microarray data–methods and preliminary results. Radiology264, 387–396. https://doi.org/10.1148/radiol.12111607 (2012).
Crespo-Jara, A. et al. A novel genomic signature predicting FDG uptake in diverse metastatic tumors. EJNMMI Res.8, 4. https://doi.org/10.1186/s13550-017-0355-3 (2018).
Yamamoto, S. et al. Radiogenomic analysis demonstrates associations between (18)F-fluoro-2-deoxyglucose PET, prognosis, and epithelial-mesenchymal transition in non-small cell lung cancer. Radiology280, 261–270. https://doi.org/10.1148/radiol.2016160259 (2016).
Som, P. et al. A fluorinated glucose analog, 2-fluoro-2-deoxy-D-glucose (F-18): nontoxic tracer for rapid tumor detection. J. Nucl. Med.21, 670–675 (1980).
Hatt, M. et al. Characterization of PET/CT images using texture analysis: the past, the present... any future?. Eur. J. Nucl. Med. Mol. Imaging44, 151–165. https://doi.org/10.1007/s00259-016-3427-0 (2017).
Haider, S. et al. Genomic alterations underlie a pan-cancer metabolic shift associated with tumour hypoxia. Genome Biol.17, 140. https://doi.org/10.1186/s13059-016-0999-8 (2016).
Yizhak, K., Chaneton, B., Gottlieb, E. & Ruppin, E. Modeling cancer metabolism on a genome scale. Mol. Syst. Biol.11, 817. https://doi.org/10.15252/msb.20145307 (2015).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol.31, 213–219. https://doi.org/10.1038/nbt.2514nbt.2514[pii] (2013).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature505, 495–501. https://doi.org/10.1038/nature12912 (2014).
Lee, J. et al. Rare mechanism of acquired resistance to Osimertinib in Korean patients with EGFR-mutated non-small cell lung cancer. Cancer Res. Treat.51, 408–412. https://doi.org/10.4143/crt.2018.138 (2019).
Lee, H. et al. Performance evaluation method for read mapping tool in clinical panel sequencing. Genes Genomics40, 189–197. https://doi.org/10.1007/s13258-017-0621-9621[pii] (2018).
Smyth, I. et al. Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene inbasal cell carcinoma and medulloblastoma on chromosome 1p32. Hum. Mol. Genet.8, 291–297. https://doi.org/10.1093/hmg/8.2.291 (1999).
Rump, A. et al. Identification and functional testing of ERCC2 mutations in a multi-national cohort of patients with familial breast- and ovarian cancer. PLoS Genet.12, e1006248. https://doi.org/10.1371/journal.pgen.1006248 (2016).
Lee, M. S. et al. IRS2 amplification as a predictive biomarker in response to ceritinib in small cell lung cancer. Mol. Ther. Oncolytics16, 188–196. https://doi.org/10.1016/j.omto.2019.12.009 (2020).
Aster, J. C., Pear, W. S. & Blacklow, S. C. The varied roles of notch in cancer. Annu. Rev. Pathol.12, 245–275. https://doi.org/10.1146/annurev-pathol-052016-100127 (2017).
Kohler, A. & Hurt, E. Exporting RNA from the nucleus to the cytoplasm. Nat. Rev. Mol. Cell. Biol.8, 761–773. https://doi.org/10.1038/nrm2255 (2007).
Huang, W. L. et al. Thyrostimulin-TSHR signaling promotes the proliferation of NIH:OVCAR-3 ovarian cancer cells via trans-regulation of the EGFR pathway. Sci. Rep.6, 27471. https://doi.org/10.1038/srep27471 (2016).
Lin, J. J. & Shaw, A. T. Recent advances in targeting ROS1 in lung cancer. J. Thorac. Oncol.12, 1611–1625. https://doi.org/10.1016/j.jtho.2017.08.002 (2017).
Hatt, M. et al. Impact of tumor size and tracer uptake heterogeneity in (18)F-FDG PET and CT non-small cell lung cancer tumor delineation. J. Nucl. Med.52, 1690–1697. https://doi.org/10.2967/jnumed.111.092767 (2011).
Hatt, M. et al. Classification and evaluation strategies of auto-segmentation approaches for PET: report of AAPM task group No 211. Med. Phys.44, e1–e42. https://doi.org/10.1002/mp.12124 (2017).
Boellaard, R. Standards for PET image acquisition and quantitative data analysis. J. Nucl. Med.50(Suppl 1), 11S-20S. https://doi.org/10.2967/jnumed.108.057182 (2009).
Brooks, F. J. & Grigsby, P. W. The effect of small tumor volumes on studies of intratumoral heterogeneity of tracer uptake. J. Nucl. Med.55, 37–42. https://doi.org/10.2967/jnumed.112.116715 (2014).
Galavis, P. E., Hollensen, C., Jallow, N., Paliwal, B. & Jeraj, R. Variability of textural features in FDG PET images due to different acquisition modes and reconstruction parameters. Acta Oncol.49, 1012–1016. https://doi.org/10.3109/0284186X.2010.498437 (2010).
Desseroit, M. C. et al. Reliability of PET/CT shape and heterogeneity features in functional and morphologic components of non-small cell lung cancer tumors: a repeatability analysis in a prospective multicenter cohort. J. Nucl. Med.58, 406–411. https://doi.org/10.2967/jnumed.116.180919 (2017).
Welch, M. L. et al. Vulnerabilities of radiomic signature development: the need for safeguards. Radiother. Oncol.130, 2–9. https://doi.org/10.1016/j.radonc.2018.10.027 (2019).
Orlhac, F. et al. A postreconstruction harmonization method for multicenter radiomic studies in PET. J. Nucl. Med.59, 1321–1328. https://doi.org/10.2967/jnumed.117.199935 (2018).
Moon, S. H., Hyun, S. H. & Choi, J. Y. Prognostic significance of volume-based PET parameters in cancer patients. Korean J. Radiol.14, 1–12. https://doi.org/10.3348/kjr.2013.14.1.1 (2013).
Soret, M., Bacharach, S. L. & Buvat, I. Partial-volume effect in PET tumor imaging. J. Nucl. Med.48, 932–945. https://doi.org/10.2967/jnumed.106.035774 (2007).
Neuzillet, C. et al. Targeting the TGFbeta pathway for cancer therapy. Pharmacol. Ther.147, 22–31. https://doi.org/10.1016/j.pharmthera.2014.11.001S0163-7258(14)00201-0[pii] (2015).
Toonkel, R. L., Borczuk, A. C. & Powell, C. A. Tgf-beta signaling pathway in lung adenocarcinoma invasion. J. Thorac. Oncol.5, 153–157. https://doi.org/10.1097/JTO.0b013e3181c8cc0cS1556-0864(15)32270-X[pii] (2010).
Shi, J. et al. Targeted blockade of TGF-beta and IL-6/JAK2/STAT3 pathways inhibits lung cancer growth promoted by bone marrow-derived myofibroblasts. Sci. Rep.7, 8660. https://doi.org/10.1038/s41598-017-09020-810.1038/s41598-017-09020-8[pii] (2017).
Shin, H. T. et al. Prevalence and detection of low-allele-fraction variants in clinical cancer samples. Nat. Commun.8, 1377. https://doi.org/10.1038/s41467-017-01470-y10.1038/s41467-017-01470-y[pii] (2017).
Hatt, M. et al. 18F-FDG PET uptake characterization through texture analysis: investigating the complementary nature of heterogeneity and functional tumor volume in a multi-cancer site patient cohort. J. Nucl. Med.56, 38–44. https://doi.org/10.2967/jnumed.114.144055 (2015).
Hyun, S. H. et al. Intratumoral heterogeneity of (18)F-FDG uptake predicts survival in patients with pancreatic ductal adenocarcinoma. Eur. J. Nucl. Med. Mol. Imaging43, 1461–1468. https://doi.org/10.1007/s00259-016-3316-610.1007/s00259-016-3316-6[pii] (2016).
Fang, Y. H. et al. Development and evaluation of an open-source software package “CGITA” for quantifying tumor heterogeneity with molecular images. Biomed. Res. Int.2014, 248505. https://doi.org/10.1155/2014/248505 (2014).
Sanchez-Vega, F. et al. Oncogenic signaling pathways in the cancer genome atlas. Cell173, 321-337e310 (2018).
Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) Grant funded by the Korean government (MSIP) (No. NRF-2016R1C1B2013411).
Author information
Authors and Affiliations
Contributions
G.K., J.K., H.C., S.L., and S.M.: Study concept and experimental design; G.K., J.K., H.C., W.P., J.A., M.A., K.P., S.L., and S.M.: Data collection; G.K., J.K., S.L., and S.M.: Analysis and interpretation of data; G. K., J.K., J.A., M.A., K.P., Y.P., J.C., K.L., S.L., and S.M: Writing of the manuscript, preparation of figures and statistical analysis; S.M.: Acquisition of funding; All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, G., Kim, J., Cha, H. et al. Metabolic radiogenomics in lung cancer: associations between FDG PET image features and oncogenic signaling pathway alterations. Sci Rep 10, 13231 (2020). https://doi.org/10.1038/s41598-020-70168-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-70168-x
This article is cited by
-
Comparative analysis of batch correction methods for FDG PET/CT using metabolic radiogenomic data of lung cancer patients
Scientific Reports (2023)
-
Glucose metabolic heterogeneity correlates with pathological features and improves survival stratification of resectable lung adenocarcinoma
Annals of Nuclear Medicine (2023)
-
A review of radiomics and genomics applications in cancers: the way towards precision medicine
Radiation Oncology (2022)
-
Tumor glycolytic heterogeneity improves detection of regional nodal metastasis in patients with lung adenocarcinoma
Annals of Nuclear Medicine (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
