
Abstract
Faecal lipopolysaccharides (LPS) have attracted attention as potent elements to explain a correlation between the gut microbiota and cardiovascular disease (CVD) progression. However, the underlying mechanism of how specific gut bacteria contribute to faecal LPS levels remains unclear. We retrospectively analysed the data of 92 patients and found that the abundance of the genus Bacteroides was significantly and negatively correlated with faecal LPS levels. The controls showed a higher abundance of Bacteroides than that in the patients with CVD. The endotoxin units of the Bacteroides LPS, as determined by the limulus amoebocyte lysate (LAL) tests, were drastically lower than those of the Escherichia coli LPS; similarly, the Bacteroides LPS induced relatively low levels of pro-inflammatory cytokine production and did not induce sepsis in mice. Fermenting patient faecal samples in a single-batch fermentation system with Bacteroides probiotics led to a significant increase in the Bacteroides abundance, suggesting that the human gut microbiota could be manipulated toward decreasing the faecal LPS levels. In the clinical perspective, Bacteroides decrease faecal LPS levels because of their reduced LAL activity; therefore, increasing Bacteroides abundance might serve as a novel therapeutic approach to prevent CVD via reducing faecal LPS levels and suppressing immune responses.
Similar content being viewed by others
Introduction
Increasing evidence suggests that a strong correlation exists between the gut microbiota and development of many diseases, such as coronary artery disease (CAD), heart failure (HF), obesity and related metabolic diseases, and rheumatoid arthritis1,2,3,4,5,6. Next-generation sequencing techniques and multi-omics approaches have revealed the function of the gut microbiota and improved understanding of the mechanisms underlying disease progression7,8,9,10. For instance, the short-chain fatty acid metabolites produced by the gut microbiota are known to play a central immune-metabolic role11, where trimethylamine and trimethylamine N-oxide are associated with cardiovascular disease12. In addition, the gut microbial lipopolysaccharide (LPS) has recently attracted attention, because chronic immune cell activation is, in part, caused by the LPS-mediated stimulation of toll-like receptor 4 (TLR4) in the gu13,14. Until now, researchers have focused on plasma LPS, and have reported that plasma LPS plays an important role in the process of activating pro-inflammatory cytokines during heart failure, and may be used to predict the development of atherosclerosis and other major advanced cardiac events15,16,17. In this context, faecal LPS levels are now considered potent elements that may be used to explain a strong correlation between the gut microbiota and disease progression and as a novel dysbiosis marker or risk factor for inflammatory diseases18,19,20,21,22. However, how specific gut bacteria contribute to faecal LPS levels remains unclear.
Interestingly, each bacterial species has distinct LPS structures13,14,23,24,25. In particular, structural variations in the lipid A moiety, characterised by diversity in the acylation pattern, are associated with different immunogenicity. Specifically, the penta- and tetra-acylated lipid A moieties of the Bacteroides species elicits reduced TLR4 responses compared to the hexa-acylated lipid A moiety of Escherichia coli13,14. A recent study indicates that a variety of innate immune responses induced by structural differences in lipid A moiety are associated with the incidence of autoimmune diseases13. Although the limulus amoebocyte lysate (LAL) tests are the gold standard for measuring LPS levels, it is important to remember that LPS demonstrates its own unique LAL activity due to the structural differences in lipid A moiety,therefore, faecal LPS levels measured by the LAL tests do not correctly reflect the absolute mass of LPS26. In other words, the variation of LAL activity among bacterial lipid A types might play a key role in the interpretation of faecal LPS levels. However, the impact of structural differences between the lipid A moieties of different gut bacteria on faecal LPS levels has not yet been measured using the LAL tests.
Here, we aimed to elucidate which gut bacteria contribute the most to faecal LPS levels measured by LAL tests and the reason for this impact. We also aimed to investigate whether probiotics manipulate the human gut microbiota toward decreasing faecal LPS levels using a single-batch anaerobic culturing system, Kobe University Human Intestinal Microbiota Model (KUHIMM). The KUHIMM is capable of hosting more than 500 bacterial species and can effectively and accurately mimic the healthy human gut microbiota and a patients’ impaired colonic microbiota; this system simulates the human gut microbiota metagenomically, and can overcome ethical and safety considerations of human intervention clinical trials27,28,29. We fermented patient faecal samples in the KUHIMM with probiotics to facilitate evaluation of the effect of probiotics on the gut microbiota before clinical studies.
Methods
Retrospective analysis: study design and participants
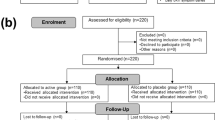
In our retrospective analysis, we accessed 93 clinical data sets from two independent studies (UMIN000015703 and UMIN000022414) (Fig. 1)1,30. As one patient was excluded because of the lack of detectable faecal LPS levels, we finally analysed the data of 92 patients. All study participants were recruited at Kobe University Hospital between October 2014 and April 2017. Faecal samples were collected while participants were hospitalised and consumed a hospital diet for more than 1 day. Patients with HF were admitted for de novo acute decompensated HF or acute worsening of chronic HF. HF was defined based on the modified Framingham criteria31. The CAD patients, who underwent coronary artery bypass graft (CABG) surgery or percutaneous coronary intervention, demonstrated > 75% stenosis on diagnostic coronary angiography, with preserved left ventricular ejection fraction and no history of HF. Patients with acute coronary syndrome were excluded. Control patients, who were hospitalised at the same hospital, had hypertension, diabetes mellitus, or dyslipidemia as per the guidelines32, but no history of CAD and HF. Patients who had undergone antibiotic treatment within a month before admission and during hospitalization were excluded. All participants provided written informed consent, and the study was conducted according to the principles of the Declaration of Helsinki. This study was approved by the Medical Ethics Committee at the Kobe University (No. B190088) and was registered with the University Hospital Medical Information Network (UMIN) Clinical Trials Registry (UMIN000036752).

Overview of experimental design. This investigation consisted of three separate studies: study A, study B, and study C. The goal of study A was to clarify which and how specific gut bacteria contribute to faecal LPS levels using the human data. The goal of study B was to examine the LPS bioactivity of the gut bacteria determined in study A in vitro and in vivo. The goal of study C was to elucidate that probiotics could change the gut flora of patients to symbiotic states toward decreasing their faecal LPS levels prior to the clinical trials. CAD coronary artery disease; E. coli, Escherichia coli; HF heart failure; LAL Limulus amoebocyte lysate; LPS lipopolysaccharide.
Faecal LPS levels
Faecal supernatant was obtained according to a previously described protocol, with some modifications18. Briefly, the faecal samples were suspended in sterile PBS to a concentration of 1 g per 10 mL and vortexed mildly to avoid disruption of bacterial cells. After centrifugation for 15 min at 3,000 rpm, the supernatant was collected and sterilised by filtration through a 0.45-μm filter, followed by re-filtration through a 0.22-μm filter and inactivation for 15 min at 90 °C. The faecal LPS levels were determined using the LAL assay (#K50-643 J; Lonza Inc., Basel, Switzerland), according to the manufacturer’s instructions. The LPS measurements were performed in pyrogen-free glass tubes, Eppendorf tubes, and 96-well plates.
DNA extraction, 16S rRNA gene amplification, and pyrosequencing
The collected faecal samples were stored at − 80 °C. DNA extraction from human faecal samples was performed by the Nihon Gene Research Laboratories Inc., according to a previously established procedure33.
Parts of the 16S rRNA genes (the V3–V4 region, corresponding to positions 342 to 806 in the E. coli numbering system) were PCR-amplified using our non-degenerate universal primer set of 342F and 806R. A detailed description of the primer set and PCR conditions is available elsewhere34. After adding the sequencing adapters, the amplicons were sequenced by Takara Bio Inc. using an Illumina MiSeq platform (Illumina Inc., San Diego, CA) according to the manufacturer’s protocol. To make the bacterial composition matrix, we used USEARCH version 10.0.240. Our previous protocol was also used to select high-quality 16S rRNA gene amplicon sequences generated using Trimmomatic35 version 0.33 with the parameters ‘LEADING:17 TRAILING:17 AVGQUAL:25 MINLEN:100’. The remaining reads were processed using the -fastq_mergepairs command of USEARCH, with default parameters. Next, we removed the sequences lacking the primer region using Tagcleaner version 0.16, with parameters “-tag5 CTACGGGGGGCAGCAG -mm5 3 -tag3 AGATACCCCGGTAGTCC -mm3 3 -nomatch 3”. After removing the primer, the sequences with unknown nucleotides (N) were removed using an in-house python script. To remove the PhiX reads, we used the -filter_phix command of USEARCH. Thereafter, we removed the short sequences (< 300 nucleotides) using the USEARCH command—sort_by_length, with the parameter ‘-minseqlength 300’. Finally, we generated operational taxonomic unit (OTU) tables using the UPARSE algorithm (-fastx_unique and otu_cluster commands with the parameter ‘-minsize 1’). The representative sequences of each OTU were annotated to bacterial genus using Ribosomal Database Project (RDP) Classifier version 2.12, with a bootstrap value ≥ 0.5. Moreover, we annotated each representative sequence of each OTU to the reference database Silva Living Tree Project version 12336 using BLASTN version 2.2.25, with the identity threshold ≥ 97% and coverage ≥ 80%.
Animals
Wild-type C57BL/6J mice were purchased from CLEA Japan (Tokyo, Japan), and Tlr4−/− mice on a C57BL/6J background were purchased from Charles River Japan (Yokohama, Japan). The mice were housed in a specific pathogen-free animal facility at the Kobe University Institute. They were fed a standard chow diet (CE-2, CLEA, Tokyo, Japan) and water ad libitum under a strict 12-h light cycle. The E. coli LPS, purified by phenol extraction, was purchased from Sigma-Aldrich (St. Louis, MO, #L2630), and Bacteroides cells were prepared for LPS purification as outlined in Sect. "LPS extraction from Bacteroides vulgatus and B. dorei". The 8-week-old wild-type and Tlr4−/− mice were treated with LPS intraperitoneally to induce septic shock37. Survival was monitored daily after LPS was injected. This study was approved by the Animal Ethics Committee at the Kobe University (P190706), and was performed according to the Guidelines for Animal Experiments in effect at Kobe University School of Medicine.
LPS extraction from Bacteroides vulgatus and B. dorei
LPS was purified by hot phenol-water method as previously described with minor modification38. Bacteria were cultivated in Gifu anaerobic medium (Nissui pharmaceutical, Tokyo, Japan) at 37 °C for 24 h under anaerobic conditions. Subsequently, 100 mL of cultured bacteria were washed twice with 15 mL of PBS containing 0.15 mM CaCl2 and 0.5 mM MgCl2. The washed cells were resuspended in 8 mL of PBS and disrupted by sonication on ice for 10 min (Insonator model 201 M; KUBOTA, Tokyo). Thereafter, 80 μL of proteinase K (10 mg/mL; Wako Pure Chemical, Osaka, Japan) was added to the cell suspension and incubated for 1 h at 65 °C. This was followed by treatment with DNase I (20 μg/mL; FUJIFILM Wako Pure Chemical, Osaka, Japan) and RNase A (40 μg/mL; Macherey–nagel, Düren, Germany) in the presence of 0.81 mM MgSO4 and 4 μL/mL chloroform at 37 °C overnight with shaking. Equal volume of phenol was added to the cell suspension and shook for 15 min at 65 °C. The resulting mixture was centrifuged (8,500×g, 4 °C, 15 min), and the water layer was collected. Sodium acetate was added to the water layer to a final concentration of 0.5 M. Ethanol was then added 10 times the volume of the water layer and placed at − 20 °C overnight. The resulting mixture was centrifuged (2000×g, 4 °C, 10 min), and the pellet was resuspended in water. The residual phenol was removed by dialysis against water at 4 °C using Spectra/Por 6 (MWCO 1,000; Spectrum Laboratories Inc., Ranch Dominquez, CA, USA). After purification, the materials were separated by SDS-PAGE using 4% stacking gel and 15% separating gel and stained with silver stain (Silver Stain Kit II, Wako Pure Chemical) and Coomassie brilliant blue to confirm purity. The signals were detected using ApeosPort-V C4475 (Fujixerox, Tokyo, Japan) with molecular size marker (EzStandard AE-1440, ATTO, Tokyo, Japan). No image processing software was used. The purified LPS was lyophilised and stored at − 20 °C.
In vitro cytokine stimulation assays
RAW 264.7 macrophages were cultured in RPMI-1640 medium supplemented with 10% foetal bovine serum. The cells (1 × 104) were plated in 96-well flat-bottom plates (Corning Costar, Corning, NY) and incubated in the presence of E. coli LPS or Bacteroides LPS for 12, 24, 48, and 72 h at 37 °C with 5% CO2. The cytokine levels were analysed using the Cytometric Bead Array Kit (BD Biosciences, San Diego, CA, USA), according to the manufacturer’s instructions.
Faecal culture in the KUHIMM
Faecal samples were collected from seven patients with CAD during their hospital stay at Kobe University Hospital between August 2018 and May 2019. Each faecal sample was collected with an anaerobic culture swab (#212550; BD BBL, NJ, USA) and fermented for 24 h27. Patients who had undergone antibiotic treatment within a month before admission and during hospitalization were excluded. The sample size was calculated using the R software (power = 0.9, significance level = 0.05, mean difference = 8, SD = 5; N = 7 per group). All study participants provided written informed consent, and the study was conducted according to the principles of the Declaration of Helsinki. This study was approved by the Medical Ethics Committee at the Kobe University (No.160191) and was registered with the UMIN Clinical Trials Registry (UMIN000024555).
The Kobe University Human Intestinal Microbiota Model (KUHIMM) is a small-scale multi-channel fermenter (Bio Jr.8; ABLE, Tokyo, Japan) composed of eight parallel and independent anaerobic culturing vessels (Fig. 1)27,28,29. Each vessel contained 100 mL Gifu anaerobic medium (Nissui Pharmaceutical Co, Tokyo, Japan) at pH 6.5. The cultures were kept at 37 °C with consistent stirring at 300 rpm. Filtered N2:CO2 (80:20) gas was supplied continuously at a constant flow rate of 15 mL/min to maintain anaerobiosis. The faecal samples were suspended in 2 mL of 0.1 M phosphate buffer (2:1 of NaH2PO4 and 0.1 M Na2HPO4) supplemented with 1% L-ascorbic acid. One hundred microliters of faecal suspension was inoculated in one vessel (culture group). Immediately after faecal inoculation, 1 × 108 cells each of B. vulgatus and B. dorei were added to the vessel (Bacteroides group). The faecal cultures were fermented for 48 h.
Gut microbial analysis of faecal culture
The gut microbial profiles of faecal culture samples were determined by 16S rRNA gene sequencing, as described in detail elsewhere28,29. Briefly, the V3-V4 region of the 16S rRNA gene was amplified, and index primers were added to the gene-specific sequences. After PCR, the products were purified and eluted in 25 μL of 10 mM Tris buffer. The 16S rRNA genes and an internal control (PhiX control v3; Illumina) were subjected to 600 cycles of paired-end sequencing using a MiSeq sequencer and reagent kit v3 (Illumina, Inc.). The PhiX sequences were removed, and the paired-end reads with Q scores ≥ 20 were joined using the MacQIIME software, version 1.9.1 (Werner Lab, Cortland, NY, USA). The UCLUST algorithm was used to cluster the filtered sequences into OTUs based on a similarity threshold of ≥ 97%. Chimeric sequences were identified and removed from the library using ChimeraSlayer. Representative sequences from each OTU were taxonomically classified using the GreenGenes taxonomic database and the RDP Classifier.
Statistical analysis
Statistical analyses were performed using Prism version 7.0 (GraphPad Software; San Diego, CA), R version 3.1.0, and JMP version 14 (SAS Institute, Cary, NC). The Shapiro–Wilk test was used to determine the normality of data. The results were expressed as mean ± standard error of the mean for the normally distributed data and as median ± interquartile range (25th–75th percentiles) for the non-normally distributed data. The significance of differences between the two groups was evaluated using the two-tailed Student’s t-test for the normally distributed data and Mann–Whitney U-test for the non-normally distributed data. Differences in continuous parameters among the three groups were calculated using one-way analysis of variance for the parametric data and Kruskal–Wallis test for the non-normally distributed data. The matched-pair samples were compared using the Wilcoxon signed-rank test. The Fisher’s exact test or Chi-square test was used to compare categorical variables. The Kaplan–Meier survival curves were constructed and analysed using a log-rank test. For all tests, a value of P < 0.05 was considered to indicate statistical significance. To discover the strength and direction of a link between two parameters, the Spearman’s rank correlation coefficient was calculated. A principal component analysis and dendrogram analysis was performed using the JMP software version 14 (SAS Institute). The random forest model was built using the random Forest package of the R software.
Results
Bacteroides contributes to faecal LPS levels
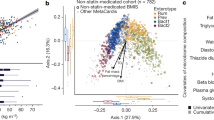
To examine which gut microbiota contribute to faecal LPS levels, we used our previous two clinical data sets1,30. As shown in Fig. 1, the data of 92 patients (39 control patients, 31 CAD patients, and 22 HF patients) were analysed. We first used a random forest classifier to rank 25 gut microbiota organisms at the genus level, according to the importance of their contribution to faecal LPS levels. We found that the genus Bacteroides ranked first among the top 25 genera of the gut microbiota (Fig. 2a).

Bacteroides decrease faecal LPS levels. (a) Twenty-five genera of the gut microbiota were ranked according to the importance of their contribution to faecal LPS levels, as determined by a random forest classifier. (b) Patients were divided into three groups according to the abundance of Bacteroides in the gut microbiota. The relative abundance of the top 25 genera are indicated. (c) A principal coordinate analysis was performed to compare the distribution of each genus in the gut microbiota. Only genera with larger weight on principal coordinate analysis are shown. (d) Relative abundance of Bacteroides (percentage of total gut bacteria) among three groups. (e) Spearman’s rank correlation coefficient was calculated between faecal LPS levels and relative abundance of Bacteroides. (f) Distribution of controls and patients with CAD or HF in each cluster. Data are shown as median ± interquartile range (25th–75th percentile) (d). CAD, coronary artery disease; EU, endotoxin units; HF, heart failure; LPS, lipopolysaccharide.
We then divided the study population into three groups according to the tertile of abundance of Bacteroides (Fig. 1). Baseline characteristics, including age, sex, body mass index, comorbidities, medications, and laboratory data are listed in Table 1. The age of patients in tertile 2 was significantly lower compared to the other groups (70.1 ± 2.1 (tertile 1) vs. 62.8 ± 2.1 (tertile 2) vs. 65.8 ± 1.7 (tertile 3), P = 0.04.). The plasma LPS levels were not different among the groups. No significant differences were observed in other characteristics among the three groups. The relative abundance of the top 25 genera detected in the gut microbiota in the three groups is shown in Fig. 2b. The principal coordinate analysis, at the genus level, showed that the three groups differed in the abundance of major gut bacteria, where tertile 1 showed a relative enrichment in the Prevotella and Escherichia/Shigella genera (Fig. 2c). The abundance of Bacteroides among the groups was significantly different (8.01% [range, 0.71–14.9%] vs. 22.55% [range, 15.90–30.48%] vs. 43.25% [range, 30.77–59.22%]; P < 0.0001.) (Fig. 2d). Of note, the faecal LPS levels in tertile 1 were significantly higher than those in tertile 3 (3.075 × 105 EU/g [range, 0.313–15.810 × 105 EU/g] vs. 1.523 × 105 EU/g [range, 0.183–8.944 × 105 EU/g]; P < 0.05.). Surprisingly, plotting faecal LPS levels with Bacteroides abundance revealed a significant negative correlation between these two parameters (r = − 0.292, P = 0.0047) (Fig. 2e). Although not significant, the control patients were more likely to be categorised into tertile 3, whereas the patients with CAD and HF were more likely to be categorised into tertile 1 or tertile 2 (Fig. 2f).
Bacteroides LPS showed drastically low endotoxicity
We next extracted the Bacteroides LPS to examine its biological activity and compare with E. coli LPS, which has a strong biological activity. We cultured B. dorei and B. vulgatus, the two most abundant Bacteroides species in human gut microbiota1, and purified LPS from these species. The biological activity of LPS was evaluated by three different methods: endotoxin units via LAL tests, cytokine secretion from RAW cells, and survival rate after intraperitoneal injection of mice.
The results of the silver staining and Coomassie brilliant blue staining indicated that the purified Bacteroides LPS was structurally different from the E. coli LPS (Fig. S1). The Bacteroides LPS endotoxin units, as measured by the LAL tests, were remarkably lower than those measured for the E. coli LPS (Fig. 3a). RAW cells stimulated with the Bacteroides LPS produced lower cytokines when stimulated with higher LPS concentration. Moreover, stimulation of RAW cells with the E. coli LPS induced a dose-dependent increase in the secretion of pro-inflammatory cytokines IL-6 and TNF-α, whereas the secretion of cytokines did not increase with the Bacteroides LPS concentration (Fig. 3b). Interestingly, both E. coli LPS and Bacteroides LPS showed inflammatory potency in a time-dependent manner (Fig. 3c). Furthermore, dose-dependent sepsis-related death was observed in mice injected with E. coli LPS (10 mg/kg group, 43%; 20 mg/kg, 100%) by day 7 after injection (Fig. 3d). In contrast, none of the mice injected with the Bacteroides LPS or the Tlr4−/− mice injected with E. coli LPS died (Fig. 3d,e). Plasma LPS level in mice injected with E. coli LPS, as measured by the LAL tests, was significantly higher than that in mice injected with Bacteroides LPS (221.2 ± 73.5 EU/mL vs. 2.2 ± 0.5 EU/mL, P < 0.001) (Fig. 3f)..

Bacteroides LPS shows lower LAL activity and immunogenicity. (a) LAL activity of the indicated LPS preparation (1 ng) was measured. The results were the sum of three independent experiments. (b) RAW 264.7 cells were stimulated with the indicated LPS preparation for 12 h. The cytokine levels in supernatants were quantified. (c) RAW 264.7 cells were stimulated with the indicated LPS (10 ng/mL) for 24, 48, and 72 h. The cytokine levels in supernatants were quantified. (d) Eight-week-old wild-type mice were treated with LPS intraperitoneally to induce septic shock. Bacteroides LPS consisted of half each of B. dorei LPS and B. vulgatus LPS. Survival was monitored daily. The log-rank test was used to determine statistically significance. N = 5 to 8 per group. (e) Eight-week-old toll-like receptor 4-deficient (Tlr4−/−) mice were treated with E. coli LPS intraperitoneally. N = 5. (f) Plasma LPS levels 12 h after LPS injection intraperitoneally. Bacteroides LPS consisted of half each of B. dorei LPS and B. vulgatus LPS. N = 5 per group. EU; endotoxin units, LAL; Limulus amoebocyte lysate. LPS, lipopolysaccharide. *P < 0.05, ***P < 0.001.
Bacteroides probiotics increased the abundance of Bacteroides in a gut model
Finally, we simulated gut microbiota alteration in the KUHIMM to investigate whether Bacteroides probiotic (B. dorei and B. vulgatus) administration could increase the Bacteroides abundance in the human gut. We fermented the patient faecal samples with or without Bacteroides probiotics in the KUHIMM and then evaluated the microbiota. The mean age and body mass index of seven patients were 72.7 ± 2.2 and 24.2 ± 0.6 (Table 2). All patients had CAD with coronary risk factors. The relative abundance of the top 15 genera detected in the gut microbiota of the two groups is shown in Fig. 4a, which reveals the gut microbial alteration induced by Bacteroides supplementation. To visualise the impact of Bacteroides probiotics, we performed dendrogram analysis and found that Bacteroides probiotics changed the global composition of the gut microbiota to a certain extent, but the effect did not induce a drastic change in the composition of any single flora (Fig. 4b).The principal coordinate analysis at the genus level suggested increased Bacteroides abundance after Bacteroides probiotics supplementation (Fig. 4c). The abundance of Bacteroides was significantly higher with Bacteroides probiotics supplementation (30.7% [range, 24.1–36.8%]) than with non-Bacteroides probiotics supplementation (25.5% [range, 14.2–37.4%], P < 0.05.), though one faecal sample (No. 7) showed decreased abundance of Bacteroides after probiotics supplementation (Fig. 4d).

Alteration of the gut microbiota in the KUHIMM. The V3–V4 regions of the bacterial 16S rRNA gene in faecal sample cultures from seven patients were sequenced.(a) Relative abundance of the top 15 gut bacterial genera in faecal sample cultures from seven patients without (black letters) or with (blue letters) Bacteroides probiotics. (b) Dendrogram analysis at the genus level without (white circle) or with (blue dots) Bacteroides probiotics. (c) Principal component analysis score plots at the genus level without (white circle) or with (blue dots) Bacteroides probiotics. (d) The abundance of the genus Bacteroides without (white circle) or with (blue dots) Bacteroides probiotics. The Wilcoxon signed-rank test was used to compare the matched-pair samples. KUHIMM, Kobe University Human Intestinal Microbiota Model.
Discussion
Many clinical and basic scientific reports have stated that faecal LPS, which is derived from the gut microbiota, has a clinical impact on patients with irritable bowel syndrome, autoimmune disease, and cardiovascular disease1,13,19,22. However, experimental studies have yet to delineate the impact of specific gut bacteria on faecal LPS levels. Furthermore, though LPS-lipid A structure differs between bacteria, the impact of LPS-lipid A structural differences on faecal LPS levels has not yet been measured using the LAL tests. In the present study, we indicated that the abundance of the genus Bacteroides is negatively correlated with the faecal LPS levels measured by the LAL tests. Taken that the LPS purified from the two species of Bacteroides showed low biological activity in the LAL tests, it is logical that the increased abundance of Bacteroides should exhibit lower faecal LPS levels.
Bacteroides is one of the predominant genera in the human gut and plays an important role in maintaining the stability of a healthy gut ecosystem39. Interestingly, the LPS component, lipid A, which activates TLR4, exhibits structural variation. The Bacteroides species do not express lpxM, which yields the hexa-acylated lipid A moiety found in E. coli; instead, the lipid A component of Bacteroides LPS are penta- or tetra-acylated13,14. The penta- and tetra-acylated lipid A moieties are known to elicit reduced TLR4 responses13,14, this is consistent with our findings and suggests that the penta- or tetra-acylated Bacteroides LPS provides a beneficial effect on the progression of inflammatory diseases. We also investigated the impact of LPS structural variation on the LAL activity and found that the Bacteroides LPS demonstrated significantly lower LAL activity compared to the E. coli LPS. Of note, the mice injected with Bacteroides LPS did not develop sepsis, indicating that the Bacteroides LPS did not exhibit endotoxicity in vivo. These results further suggest that the decreased abundance of Bacteroides in the gut microbiota might predispose people to more inflammatory diseases. Importantly, our results imply that it would be insufficient to analyse the gut metagenome data to evaluate LPS biosynthesis in the gut; LPS immunogenicity should be considered while interpreting the gut microbial LPS data.
We also showed that the administration of Bacteroides as probiotics in the KUHIMM manipulated the human gut microbiota toward increasing the abundance of Bacteroides in all except one sample (Fig. 4d). This results indicate that Bacteroides prebiotics did not always increase the abundance of Bacteroides as the human gut is home to trillions of microorganisms. Each gut microbial profile depends on age, sex, local food or lifestyle, drugs, and other many factors8. As the KUHIMM is designed to monitor changes in the bacterial composition and facilitates evaluations of the impact of probiotics in independent vessels, culture samples in the KUHIMM are unaffected by such factors. In that sense, we believe that the KUHIMM might be useful for carrying out preclinical evaluations of the effect of Bacteroides prebiotics on the gut microbiota. Our results at least suggest that the gut microbial alterations aimed at preventing and treating inflammatory diseases could be feasible in humans. These data could therefore lead to a novel strategy for the prevention and treatment of inflammatory diseases via manipulation of the gut microbiota.
From a clinical perspective, our results pave the way for further studies investigating faecal LPS levels and Bacteroides abundance. It was interesting that patients with CAD or HF were more likely categorised in tertile 1, which showed less Bacteroides abundance. A previous cross-sectional study also shows that the abundance of Bacteroides is lower in patients with atherosclerotic cardiovascular disease compared to healthy controls9. A cohort study is warranted to assess causality and to provide the strongest scientific evidence of the relationship between Bacteroides abundance and faecal LPS levels and disease progression; this will aid deeper understanding and acceptance of the importance of Bacteroides abundance and faecal LPS levels in human diseases. Further research is necessary to understand whether gut microbial composition directly reflects faecal LPS composition. Advances in mass spectrometry technology might expand our knowledge of LPS composition.
Fig. S1 shows the schematic illustration of the present study. In summary, we identified the Bacteroides genus as the primary constituent of the human gut microbiota and determined that the abundance of organisms of this genus was negatively correlated to LPS endotoxicity. We speculate that this reduced immunogenicity is linked to the lower levels of cytokine stimulation and, therefore, Bacteroides might present a valuable therapeutic option in certain inflammatory conditions. We also demonstrated that supplementation with Bacteroides as a probiotic in a model gut system increased the abundance of this genus.
Data availability
All data supporting the findings of our study are available from the corresponding author upon reasonable request. The sequencing data were deposited to the Japan BioProject database with links to the BioProject accession number PRJDB8454.
References
Yoshida, N. et al. Bacteroides vulgatus and Bacteroides dorei reduce gut microbial lipopolysaccharide production and inhibit atherosclerosis. Circulation 138, 2486–2498. https://doi.org/10.1161/CIRCULATIONAHA.118.033714 (2018).
Turnbaugh, P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031. https://doi.org/10.1038/nature05414 (2006).
Xavier, R. J. & Podolsky, D. K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 448, 427–434. https://doi.org/10.1038/nature06005 (2007).
Maeda, Y. et al. Dysbiosis contributes to arthritis development via activation of autoreactive T cells in the intestine. Arthritis Rheumatol. 68, 2646–2661. https://doi.org/10.1002/art.39783 (2016).
Pedersen, H. K. et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 535, 376–381. https://doi.org/10.1038/nature18646 (2016).
Schiattarella, G. G. et al. Gut microbe-generated metabolite trimethylamine-N-oxide as cardiovascular risk biomarker: a systematic review and dose–response meta-analysis. Eur. Heart J. 38, 2948–2956. https://doi.org/10.1093/eurheartj/ehx342 (2017).
The Human Microbiome Project. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. https://doi.org/10.1038/nature11234 (2012).
Qin, J. et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60. https://doi.org/10.1038/nature11450 (2012).
Jie, Z. et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 8, 845. https://doi.org/10.1038/s41467-017-00900-1 (2017).
Liu, R. et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 23, 859–868. https://doi.org/10.1038/nm.4358 (2017).
Furusawa, Y. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. https://doi.org/10.1038/nature12721 (2013).
Wang, Z. et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63. https://doi.org/10.1038/nature09922 (2011).
Vatanen, T. et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell 165, 842–853. https://doi.org/10.1016/j.cell.2016.04.007 (2016).
d’Hennezel, E., Abubucker, S., Murphy, L. O. & Cullen, T. W. Total lipopolysaccharide from the human gut microbiome silences toll-like receptor signaling. mSystems 2, e0004617. https://doi.org/10.1128/mSystems.00046-17 (2017).
Wiedermann, C. J. et al. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the Bruneck Study. J. Am. Coll. Cardiol. 34, 1975–1981. https://doi.org/10.1016/s0735-1097(99)00448-9 (1999).
Anker, S. D. & von Haehling, S. Inflammatory mediators in chronic heart failure: an overview. Heart 90, 464–470. https://doi.org/10.1136/hrt.2002.007005 (2004).
Pastori, D. et al. Gut-derived serum lipopolysaccharide is associated with enhanced risk of major adverse cardiovascular events in atrial fibrillation: effect of adherence to Mediterranean diet. J. Am. Heart Assoc. 6, 5784. https://doi.org/10.1161/JAHA.117.005784 (2017).
Kim, K. A., Jeong, J. J., Yoo, S. Y. & Kim, D. H. Gut microbiota lipopolysaccharide accelerates inflamm-aging in mice. BMC Microbiol. 16, 9–9. https://doi.org/10.1186/s12866-016-0625-7 (2016).
Zhou, S. Y. et al. FODMAP diet modulates visceral nociception by lipopolysaccharide-mediated intestinal inflammation and barrier dysfunction. J. Clin. Invest. 128, 267–280. https://doi.org/10.1172/JCI92390 (2018).
Jang, S. E. et al. Gastrointestinal inflammation by gut microbiota disturbance induces memory impairment in mice. Mucosal Immunol. 11, 369–379. https://doi.org/10.1038/mi.2017.49 (2018).
Neff, C. P. et al. Fecal microbiota composition drives immune activation in HIV-infected individuals. EBiomedicine 30, 192–202. https://doi.org/10.1016/j.ebiom.2018.03.024 (2018).
Fabbiano, S. et al. Functional gut microbiota remodeling contributes to the caloric restriction-induced metabolic improvements. Cell Metab. 28, 907-921.e7. https://doi.org/10.1016/j.cmet.2018.08.005 (2018).
Mancuso, G. et al. Bacteroides fragilis-derived lipopolysaccharide Produces Cell Activation and Lethal Toxicity via toll-like receptor 4. Infect. Immun. 73, 5620–5627. https://doi.org/10.1128/IAI.73.9.5620-5627.2005 (2005).
Munford, R. S. Sensing gram-negative bacterial lipopolysaccharides: a human disease determinant?. Infect. Immun. 76, 454–465. https://doi.org/10.1128/IAI.00939-07 (2008).
Gronbach, K. et al. Endotoxicity of lipopolysaccharide as a determinant of T-cell−mediated colitis induction in mice. Gastroenterology 146, 765–775. https://doi.org/10.1053/j.gastro.2013.11.033 (2014).
Poxton, I. R. & Edmond, D. M. Biological activity of Bacteroides lipopolysaccharide - reappraisal. Clin. Infect. Dis. 20(Suppl 2), S149–S153. https://doi.org/10.1093/clinids/20.supplement_2.s149 (1995).
Takagi, R. et al. A single-batch fermentation system to simulate human colonic microbiota for high-throughput evaluation of prebiotics. PLoS ONE 11, e0160533. https://doi.org/10.1371/journal.pone.0160533 (2016).
Sasaki, D. et al. Low amounts of dietary fibre increase in vitro production of short-chain fatty acids without changing human colonic microbiota structure. Sci. Rep. 8, 435. https://doi.org/10.1038/s41598-017-18877-8 (2018).
Yoshida, N. et al. Effect of resistant starch on the gut microbiota and its metabolites in patients with coronary artery disease. J. Atheroscler. Thromb. 26, 705–719. https://doi.org/10.5551/jat.47415 (2019).
Hayashi, T. et al. Gut microbiome and plasma microbiome-related metabolites in patients with decompensated and compensated heart failure. Circ. J. 83, 182–192. https://doi.org/10.1253/circj.CJ-18-0468 (2018).
Senni, M. et al. Congestive heart failure in the community: a study of all incident cases in Olmsted County, Minnesota, in 1991. Circulation 98, 2282–2289. https://doi.org/10.1161/01.cir.98.21.2282 (1998).
Mancia, G. et al. 2013 ESH/ESC Guidelines for the management of arterial hypertension: the Task Force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur. Heart J. 34, 2159–2219. https://doi.org/10.1093/eurheartj/eht151 (2013).
Furet, J. P. et al. Comparative assessment of human and farm animal faecal microbiota using real-time quantitative PCR. FEMS Microbiol. Ecol. 68, 351–362. https://doi.org/10.1111/j.1574-6941.2009.00671.x (2009).
Mori, H. et al. Design and experimental application of a novel non-degenerate universal primer set that amplifies prokaryotic 16S rRNA genes with a low possibility to amplify eukaryotic rRNA genes. DNA Res. 21, 217–227. https://doi.org/10.1093/dnares/dst052 (2014).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. https://doi.org/10.1093/bioinformatics/btu170 (2014).
Yarza, P. et al. The all-species living tree project: a 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst. Appl. Microbiol. 31, 241–250. https://doi.org/10.1016/j.syapm.2008.07.001 (2008).
Yamashita, T. et al. Resistance to endotoxin shock in transgenic mice overexpressing endothelial nitric oxide synthase. Circulation 101, 931–937. https://doi.org/10.1161/01.cir.101.8.931 (2000).
Rezania, S. et al. Extraction, purification and characterization of lipopolysaccharide from Escherichia coli and Salmonella typhi. Avicenna J. Med. Biotechnol. 3, 3–9 (2011).
Wexler, A. G. & Goodman, A. L. An insider’s perspective: Bacteroides as a window into the microbiome. Nat. Microbiol. 2, 17026. https://doi.org/10.1038/nmicrobiol.2017.26 (2017).
Acknowledgements
We thank all study participants and are grateful to the medical staff for their cooperation with collecting the faecal samples. We also thank Ayami Fujino, Yasunobu Takeshima, Yasuko Koura, Shoko Sakai, Hiromi Nishiki, Ryohei Shinohara, Hitomi Nakashima and Emiko Yoshida for their excellent technical support and laboratory management. We would like to thank Editage for English language editing. This work was supported by the Japan Society for the Promotion of Science KAKENHI (16K09516, 17K09497, 19H03653, 19K23944), PRIME from the Japan Agency for Medical Research and Development (18069370), Japan Innovative Bioproduction Kobe from the Ministry of Education, Culture, Sports and Technology, Hyogo Science and Technology Association (T.Y. and K.H.), Grant for Basic Research of the Japanese Circulation Society, and Grant for Clinical Research of the Japanese Circulation Society (N.Y.).
Author information
Authors and Affiliations
Contributions
Conceptualization: N.Y., T. Yamashita, S.K., J.O.; Methodology: N.Y., H.W., T. Tabata, K.S., D.S., Y.S., N.K., T.E., T.H., and T. Yamada; Formal Analysis: N.Y., H.W., T. Tabata, K.S., D.S., and T. Takahashi; Writing (Original Draft): N.Y. and S.K.; Writing (Review & Editing): N.Y., T. Yamashita, S.K., K.S., D.S., M.S., R.O., A.K., T. Yamada, J.O., and K.H; Resources: N.Y., S.K., A.K., and R.O.; Supervision: T. Yamashita, S.K., M.S., R.O., A.K., T. Yamada, J.O., and K.H; Funding Acquisition: N.Y., T. Yamashita, and K.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yoshida, N., Yamashita, T., Kishino, S. et al. A possible beneficial effect of Bacteroides on faecal lipopolysaccharide activity and cardiovascular diseases. Sci Rep 10, 13009 (2020). https://doi.org/10.1038/s41598-020-69983-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-69983-z
This article is cited by
-
Murine sterile fecal filtrate is a potent pharmacological agent that exerts age-independent immunomodulatory effects in RAW264.7 macrophages
BMC Complementary Medicine and Therapies (2023)
-
Microbiota in health and diseases
Signal Transduction and Targeted Therapy (2022)
-
Fecal microbiota transplantation from patients with rheumatoid arthritis causes depression-like behaviors in mice through abnormal T cells activation
Translational Psychiatry (2022)
-
Altered Gut Microbiota Profile in Lin28a Transgenic Mice Can Improve Glucose Tolerance
Bulletin of Experimental Biology and Medicine (2021)
-
The Kobe University Human Intestinal Microbiota Model for gut intervention studies
Applied Microbiology and Biotechnology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
