
Abstract
Mycobacterium tuberculosis and M. smegmatis form drug-tolerant biofilms through dedicated genetic programs. In support of a stepwise process regulating biofilm production in mycobacteria, it was shown elsewhere that lsr2 participates in intercellular aggregation, while groEL1 was required for biofilm maturation in M. smegmatis. Here, by means of RNA-Seq, we monitored the early steps of biofilm production in M. bovis BCG, to distinguish intercellular aggregation from attachment to a surface. Genes encoding for the transcriptional regulators dosR and BCG0114 (Rv0081) were significantly regulated and responded differently to intercellular aggregation and surface attachment. Moreover, a M. tuberculosis H37Rv deletion mutant in the Rv3134c-dosS-dosR regulon, formed less biofilm than wild type M. tuberculosis, a phenotype reverted upon reintroduction of this operon into the mutant. Combining RT-qPCR with microbiological assays (colony and surface pellicle morphologies, biofilm quantification, Ziehl–Neelsen staining, growth curve and replication of planktonic cells), we found that BCG0642c affected biofilm production and replication of planktonic BCG, whereas ethR affected only phenotypes linked to planktonic cells despite its downregulation at the intercellular aggregation step. Our results provide evidence for a stage-dependent expression of genes that contribute to biofilm production in slow-growing mycobacteria.
Similar content being viewed by others

Introduction
In nature, microbial species are often found within a matrix, forming multicellular communities that attach to surfaces or air–liquid interfaces, called biofilms1. Biofilms are relevant to human health, as a majority of bacterial pathogens employ these structures to modify the host response2 contributing to persistence3. In this regard, a link exists between in vitro biofilm production and in vivo persistence for BCG4 and M. tuberculosis5. Biofilm formation occurs via a series of well-defined steps. These include the attachment of single-cell planktonic microbes onto a substratum; aggregation and growth of the adherent cells into three-dimensionally organized structures; and encapsulation of the structures by a self-produced matrix of extracellular polymeric substance1.
Mycobacterium tuberculosis and M. smegmatis form drug-tolerant biofilms through dedicated genetic programs6,7. In support of a stepwise process regulating biofilm production in mycobacteria, it was recently shown that in M. smegmatis, lsr2 participates in intercellular aggregation, while groEL1 was required for biofilm maturation1. Additionally, it was found that multiple genes that are necessary for fitness of M. tuberculosis cells within biofilms, were also implicated in tolerance to a diverse set of stressors and antibiotics8, something not observed for planktonic cells, further supporting a role for M. tuberculosis biofilms in drug tolerance7.
To date, a number of genes have been shown to affect the capacity of mycobacteria to produce biofilms in vitro, with few reports describing their specific participation in the stepwise process of regulating biofilm production. Here, by means of whole transcriptome analysis, we monitored the early steps of biofilm production in M. bovis BCG, to distinguish intercellular aggregation from attachment to a surface. We identified a number of genes being differentially expressed at these stages, including genes encoding for the transcriptional regulators linked to oxygen availability, dosR and BCG0114 (Rv0081), which were expressed in a temporal order during mycobacterial biofilm formation. Our results also provide a potential explanation for a stage-dependent expression of additional genes previously reported to contribute to biofilm production in mycobacteria and suggest new targets that can be assessed for their particular contribution to this phenotype.
Results
Transcriptional profiling during intercellular aggregation and substrate attachment
Our model of biofilm production by BCG consists of four distinct stages based on visual inspection as cultures progressed in Sauton medium from planktonic cells to mature biofilms. There, BCG starts as free-swimming bacteria (24 h) that in the absence of detergent, forms microcolonies of aggregated cells that can be readily visible (7 days). Later, these aggregates attach to the plastic wells (10 days), to finally produce mature surface pellicles that cover all the air–liquid interphase as well as part of the plastic wells (14 days). We previously demonstrated that it is possible to visually detect these steps during BCG biofilm formation at these time points9. To reduce potential variation from experiment to experiment, we always started biofilm cultures with cells adjusted at OD600nm 0.03.
To investigate how BCG responds to intercellular aggregation, differential gene expression analysis was used to compare the transcriptome of 7-day-old cultures (visible intercellular aggregation) as compared with 24 h cultures (basal transcriptome of BCG planktonic cells). Next, we interrogated how BCG specifically responds to substrate attachment, by comparing the transcriptome of 10 days-old cultures (visible attachment to the plastic walls) as compared with 7 days cultures (visible intercellular aggregation). The BCG Pasteur 1173P2 genome, used as a reference, has 4,109 protein and RNA-encoding genes. In our assays, we were able to detect differential gene expression [considered as significant (when both Log2-fold change ≥ 1 or ≤ − 1 plus p < 0.05) or not] when comparing intercellular aggregation versus growth as planktonic cells. We found mostly gene downregulation during this transition [1503 upregulated (37.5% of the coding potential), 2,605 downregulated (65%), Supplementary Table 1]. For substrate attachment, as compared with intercellular aggregation, there was less of a biased response [1975 (49.3%) upregulated and 2,132 (53.2%) downregulated, Supplementary Table 1].
For illustrative purposes, the 30 most significantly up- or down-regulated genes are shown in Table 2. Among the most upregulated genes with a known function, we found BCG3184c (Rv3160c, probable TetR-family transcriptional regulator), sigE, fadE23 (fatty-acid-CoA ligase, involved in sulfolipid production)10, hupB (DNA binding protein), BCG3929 (Rv3866, espG), ppsC (involved in PDIM synthesis), BCG1191 (Rv1130, prpD, 2-methylcitrate dehydratase), and BCG1826 (Rv1794, part of the ESX-5 secretion system)11. Of note, 9 out of the 30 most downregulated genes in the transition from planktonic cells to intercellular aggregation were members of the DosR regulon (Dormancy Survival Regulon). The DosR-regulon is composed of 48 genes that respond to in vitro microaerophilic or hypoxic conditions as well as exposure to nitric oxide or replication within macrophages12,13. Genes BCG0115 (Rv0082), BCG0112 (Rv0079), BCG0114 (Rv0081), BCG0113 (Rv0080), BCG3157c (Rv3134c), TB31.7 (Rv2623), hspX (Rv2031c), BCG2049c (Rv2030c), and BCG3154 (Rv3131) were downregulated when BCG changed from planktonic cells to the intercellular aggregation step.
Interestingly, 14 out of the 30 most upregulated genes in the transition from intercellular aggregation to surface attachment were also part of the DosR regulon: BCG0112 (Rv0079), BCG2051 (acg), BCG2653c (Rv2626c), BCG3157c (Rv3134c), BCG1777 (Rv1738), BCG1772c (Rv1733c), TB31.7 (Rv2623), BCG0115 (Rv0082), BCG0614 (Rv0569), hspX (Rv2031c), BCG3154 (Rv3131), and BCG2049c (Rv2030c). Genes downregulated when BCG changed from intercellular aggregation to surface attachment were mostly encoding for hypothetical, conserved hypothetical, or membrane-associated proteins, with the exception of the one encoding for DNA-directed RNA polymerase subunit α, rpoA.
The total of differentially expressed genes showing a statistically significant difference (Log2-fold change ≥ 1 or ≤ − 1, p < 0.05) with respect to the reference, previous growth stage are shown in Supplementary Table 2 [upregulated during intercellular aggregation, 248 genes (6.2%)], Supplementary Table 3 [downregulated during intercellular aggregation, 764 genes (19.1%)], Supplementary Table 4 [upregulated during substrate attachment, 474 genes (11.8%)], and Supplementary Table 5 [downregulated during substrate attachment, 683 genes (17%)].
Pang et al. described some genes as relevant for biofilm production in M. tuberculosis, such as PE1, nirB, PPE5, mycP1, and pks1, among others14, although their temporal requirement during biofilm production was not elucidated. Later, lsr2 was implicated in intercellular aggregation1. We found that PE1, nirB, and lsr2 were moderately upregulated (FC = 0.6, 0.7, and 0.95 Log2, respectively, Supplementary Table 1) during the transition from planktonic to intercellular aggregation, while their expression moderately decreased (FC = − 0.88, − 0.64, and − 0.8 Log2, respectively, Supplementary Table 1) during substratum attachment. In both instances, the FC set up in our screening to find differentially expressed genes (Log2 ≥ 1) was not reached, and therefore these 3 genes were not considered as DE in our analyses, although we acknowledge that the p-value found for these genes was statistically significant and below the threshold of p ≤ 0.05. As for PE5 and pks1, neither of these genes complied with FC and p value criteria set up here to be considered as DE.
GroEL1 was reported to be required for biofilm production in M. smegmatis via its binding to KasA and regulation of mycolic acids synthesis and biofilm maturation6. On the other hand, in BCG GL2, deletion of groEL1 produced thinner surface pellicles, devoid of PDIM and with 2-carbon longer mycolic acids15, therefore implicating a more complex role for this chaperone in modulation of the cell surface for biofilm production in mycobacteria. In our work, transcription of groEL1 was found to be significantly repressed during the transition from planktonic to intercellular aggregation (Supplementary Table 1), while it was significantly induced after substratum attachment (Supplementary Table 1). In agreement with our recent report9, genes involved in mycolic acid biosynthesis (kasA, kasB, acpM, fas) were significantly induced after substratum attachment (Supplementary Table 1), therefore confirming their upregulation during biofilm formation in BCG.
Taken together, the most significant changes that BCG experiences during the early stages of biofilm production were the downregulation of genes of the DosR-regulon during intercellular aggregation, and their upregulation upon substrate attachment. Further, we found a plausible explanation for the temporary requirement of genes already reported to be required for biofilm production (PE1, nirB, lsr2, groEL1, kasA, kasB, fas, and acpM) in mycobacteria.
We then evaluated the expression of BCG0114, dosR, BCG0642c, ethR, and BCG3766c by RT-qPCR. dosR and BCG0114 were specifically downregulated at the intercellular aggregation step while both of them were upregulated at the substrate attachment stage (Supplementary Table 1), and as part of the DosR-regulon their role in regulating expression of other genes have been described12,13,16. BCG0642c, which encodes for a conserved hypothetical protein with a PhdYeFM antitoxin domain, was significantly upregulated only during intercellular aggregation and downregulated (p = 0.06) at the substrate attachment step (Supplementary Table 1). ethR was specifically downregulated during surface attachment, while BCG3766c, which encodes for a conserved hypothetical proline rich protein, was also was close to significant downregulation during surface attachment and almost reached the criteria to be considered as significantly affected during the intercellular aggregation step (FC 0.76, p = 0.022).
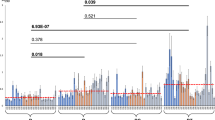
We sought to evaluate and validate the expression of these 5 selected genes by RT-qPCR at the same stages as we did for RNA-Seq analyses (Fig. 1a). Given that the mean Ct value for each gene of interest with respect to the reference gene, rrs, showed a statistically significant difference between the 24 h time point compared to the remaining ones (p < 0.0001 compared with 7 days, 10 days, and 14 days, one-way ANOVA followed by Tukey’s multiple comparison test), we were able only to compare differential gene expression as measured by RNA-Seq to that of the substrate attachment (10 days) versus intercellular aggregation (7 days) step as determined by RT-qPCR. Transcription of the reference gene, rrs, was found to be non-significantly different for biofilm samples (mean Ct values of 9.9, 9.7, and 10.7 at days 7, 10, and 14, respectively, with p values 0.9724 for the 10 vs. 7 days comparison, 0.2921 for the 14 vs. 7 days comparison, and 0.282 for the 10 vs. 14 days comparison, determined by the Brown–Forsythe and Welch ANOVA test followed by Dunnett’s post-hoc comparison). Similarly, rrs transcription was not statistically different for planktonic cultures, with mean Ct values of 8.9 (log phase) and 9.4 (stationary phase cultures, p = 0.1409 after a two-tailed, unpaired t test with Welch correction).

RT-qPCR analyses of a set of genes specifically regulated at the intercellular and substrate attachment steps during biofilm formation in BCG. Relative expression of genes at different steps during biofilm formation (a) and as planktonic cultures at early logarithmic and stationary phases (b). Error bars represent standard deviations of the mean from three biological replicates, each with technical duplicates, for six total data per gene. Brown–Forsythe and Welch ANOVA followed by Dunnett’s multiple comparison test was used to compare samples obtained from biofilms. Multiple t tests followed by Holm–Sidak comparison was used for samples obtained from planktonic cultures. Brackets encompass the comparisons for which statistically significant p values are shown on top of the bars depicting the means.
We found an agreement in expression for 4 out of 5 genes, with a slight variation in the magnitude of the change measured: BCG0114 (FC = 6.72 by RNA-seq; FC = 2.66 by RT-qPCR), dosR (FC = 3.62 by RNA-Seq; FC = 2.4 by RT-qPCR), ethR (FC = 0.35 by RNA-Seq; FC = 0.39 by RT-qPCR), and BCG3766c (FC = 0.48 by RNA-Seq; FC = 0.67 by RT-qPCR). Only expression of BCG0642c showed discrepancy between RNA-Seq (2.1 mean FC) and RT-qPCR (0.46 mean FC).
Further evaluating gene expression by RT-qPCR we noticed that upregulation of BCG0114 and dosR, or downregulation of ethR and BCG0642c, which both started at the substrate attachment step, were maintained during biofilm maturation (Fig. 1a). Whereas induction of BCG3766c was only found during substrate attachment with no further change during biofilm maturation (Fig. 1a).
To complete our gene expression analyses, we used RT-qPCR to monitor the expression of BCG0114, dosR, BCG0642c, ethR, and BCG3766c in planktonic cultures of BCG at early-log and stationary phase (Fig. 1b). Using early-log phase cultures as a reference, we observed that expression of all genes in stationary cultures followed the same pattern as they did during intercellular aggregation (BCG0114 and dosR upregulated; BCG0642c, ethR, and BCG3766c downregulated). Hence, RT-qPCR validated differential expression for 4 out of 5 genes selected from RNA-Seq assays. Moreover, it showed that expression of the 5 selected targets in stationary phase planktonic cultures resembled the pattern found during intercellular aggregation.
Phenotypic changes in multicellular BCG aggregates derived from increased expression of dosR, BCG0114, BCG0642c, ethR, and BCG3766c
Having confirmed that dosR, BCG0114, BCG0642c, ethR, and BCG3766c showed differential expression specifically at either the intercellular aggregation or substrate attachment steps, we next evaluated the effect of increasing their expression by inserting an additional single copy of each one of them into BCG Pasteur via pMV361, under the control of the strong promoter hsp6017. Expression of other genes from this promoter has already been shown by us to result in downstream changes at the transcriptomic18 and proteomic levels, altered infectivity in vitro19, and improved immunogenicity or vaccine efficacy in vivo20. Using this approach, we uncoupled gene transcription from the temporary differential expression observed in RNA-Seq and RT-qPCR assays.
Our initial assessment of the phenotypic changes in BCG was focused on those related to multicellular aggregates, such as colony morphology, surface pellicle appearance, Ziehl–Neelsen staining of biofilm samples, and biofilm formed at 10 (substrate attachment) and 14 days (biofilm maturation) as measured by crystal violet staining. Regarding colony morphology (Fig. 2a), we found morphological characteristics of the M. tuberculosis complex, such as: irregular form, waxy dry appearance, wrinkled and rough surface with irregular margins. We also found some differences among the strains, as follows: BCG strains harboring additional copies of BCG0114 (BCG::BCG0114), and dosR (BCG::dosR) showed a grayish color, unlike the other strains that presented a light yellowish color. Also, both strains produced smaller and flatter colonies compared with the others. The strain with an additional copy of BCG3766c (BCG::BCG3766c) was the one most similar to the wild type strain harboring the empty vector (BCG::pMV361), the only difference was its smaller size, with both of them showing an irregular elevation in the center of the colony. Finally, strains harboring an additional copy of ethR (BCG::ethR) or BCG0642c (BCG::BCG0642c) were very similar to each other, the main difference being their size (Fig. 2a).

Phenotypic changes in multicellular BCG aggregates derived from increased expression of dosR, BCG0114, BCG0642c, ethR, and BCG3766c. BCG strains harboring each one of the indicated genes, under the control of the strong hsp60 promoter, or the empty vector (pMV361), were evaluated for different multicellular phenotypes. Isolated, single colonies obtained after 3 weeks of incubation at 37 °C on 7H10 OADC agar plates (a). Surface pellicles formed in Sauton media with no detergent at 37 °C, 5% CO2, for 2-weeks in tissue culture flasks (b), 10 days (c) or 2 weeks in 48-well plates. Ziehl–Neelsen staining of the different BCG strains sampled from 2 weeks-old surface pellicles (e). Biofilm quantification of the different BCG at 10 days (f) or 2 weeks of culture in Sauton media, in 48-well plates. Biofilm quantification of M. tuberculosis H37Rv wild type, its isogenic deletion mutant devoid of the Rv3134c-dosS-dosR operon (H37Rv dosR KO) and its complemented derivative (H37Rv dosR KO::Comp) in 24-well plates using Sauton medium with no detergent. For colonies, a 2 mm scale bar is shown, with the mean diameter and standard deviation indicated (a). For surface pellicles, images at 10× are shown (b–d). For Ziehl–Neelsen staining, images at 100× are shown (e). All experiments were performed three different times, with duplicates (a), or four to six biological duplicates (b–e), and one representative image is shown in all instances. For biofilm quantification, the experiment was repeated independently three times and one representative set of results is shown; error bars represent standard deviations of the mean from four to six biological replicates, indicated as individual dots (f–h). One-Way ANOVA followed by Dunnett’s multiple comparison test was used to assess significance of changes between each recombinant BCG strain and wild type BCG harboring the empty vector (f–g), or among the three M. tuberculosis H37Rv strains (h). Brackets encompass the comparisons for which statistically significant p values are shown on top of the bars depicting the means.
We also observed changes in mature surface pellicle appearance formed in tissue culture flasks, as both BCG::BCG0114 and BCG::dosR produced smooth pellicles as opposed to the rugose ones produced by BCG::pMV361, BCG::ethR, BCG::BCG0642c, and BCG::BCG3766c, with slight variations among strains in apparent rugosity (Fig. 2b).
When we followed biofilm formation in multiwell plates, we noticed that at the substrate attachment step, no noticeable difference was found during biofilm formation (10 days, Fig. 2c), and minor variations in surface pellicles were observed when biofilms were mature (14 days, Fig. 2d), although a lower rugosity was consistently observed for both BCG::BCG0114 and BCG::dosR cultures (Fig. 2d). No major changes were observed in acid-fastness or intercellular adherence of the different BCG strains in mature biofilms, with the exception of some metachromatic-like granules present in BCG::ethR and BCG::BCG0642c (Fig. 2e).
Biofilm formed by any of the BCG strains at the substrate attachment step showed no quantitative difference compared with wild type BCG (Fig. 2f), and only BCG::BCG0642c produced more mature biofilm than wild type BCG (p < 0.0001) with BCG::BCG3766c almost reaching significance for a reduced production of this structure (p = 0.0521, One-Way ANOVA followed by Dunnett’s multiple comparison test) (Fig. 2g). We also evaluated the capacity to produce biofilms by M. tuberculosis strains with different dosR contents and found that a mutant lacking this operon (H37Rv dosR KO) was affected in its biofilm formation (p = 0.0426, One-Way ANOVA followed by Dunnett’s multiple comparison test) with capacity being restored to wild-type levels upon reinsertion of the gene into the chromosome (H37Rv dosR KO::Comp, Fig. 2h).
In summary, compared to wild type BCG, hsp60-driven expression of dosR and BCG0114, led to smaller colonies on agar, with changes in color and elevation, and also produced smoother surface pellicles with no quantitative effect on biofilm production. Moreover, deletion of dosR in M. tuberculosis H37Rv reduced biofilm production (Fig. 2). hsp60-driven expression of BCG0642c enhanced biofilm production with formation of metachromatic-like granules in acid-fast bacteria (Fig. 2). hsp60-driven expression of BCG3766c reduced colony size and surface pellicle rugosity, although less than dosR or BCG0114, and tended to decrease biofilm production (Fig. 2). ethR expression from hsp60 did not produce any detectable difference in the assays performed here (Fig. 2).
Phenotypic changes in planktonic BCG cells derived from increased expression of dosR, BCG0114, BCG0642c, ethR, and BCG3766c
After evaluating the effect of hsp60-driven expression of dosR, BCG0114, BCG0642c, ethR, and BCG3766c in BCG multicellular phenotypes, we next evaluated the effect of these genes in planktonic BCG cultures. We did not see any major difference in acid-fastness of early-log phase bacteria, except for the presence of metachromatic-like granules in BCG::BCG0642c (Fig. 3a) as it occurred for this strain in biofilm cultures (Fig. 2). An apparent tendency to form tight bundles was also noticed for BCG::ethR and BCG::BCG3766c, although this was not quantified.

Phenotypic changes in planktonic BCG cells derived from increased expression of dosR, BCG0114, BCG0642c, ethR, and BCG3766c. Ziehl–Neelsen staining of the different BCG strains sampled from mid-log phase planktonic cultures in 7H9 OADC 0.05% Tween 80 (a). Growth (as OD600nm readings) of each recombinant BCG was compared with that of parental BCG harboring the empty vector pMV361 (BCG WT::pMV361) (b). Two-Way ANOVA followed by Dunnett’s multiple comparison test was used to compare apparent growth, using four biological duplicates; p values for the recombinant strains compared with BCG::pMV361 being described in results. Bacterial replication (CFU/mL) (c) was compared by 2-Way ANOVA followed by Tukey’s multiple comparison test, using four biological duplicates. Each experiment was repeated independently two times, and one representative result is shown. Brackets encompass the comparisons for which statistically significant p values are shown on top of the bars depicting the means.
Regarding the growth curve of planktonic BCG cultures, we monitored apparent growth by reading OD600nm every 24 h. There, we found that the most pronounced differences observed as an apparent growth delay as observed in OD600nm readings occurred when BCG had hsp60-driven expression of either dosR (significant differences in days 2 and 3, and from day 5 to day 12) or BCG0114, which significantly differed at all time points except at the start of the culture (Fig. 3b). Differences in OD600nm coincided with changes in doubling time for the BCG::dosR strain as compared with BCG::pMV361 at days 5 and 10 (46.94 ± 5.96 h vs. 35.6 ± 4.18, p = 0.0176; and 67.79 ± 11.42 h vs. 223.17 ± 22.96 h, p = 0.0086, respectively).
Even though OD600nm for BCG::BCG0114 suggested a marked growth defect as compared with wild type BCG harboring the empty vector (BCG::pMV361), doubling time indicated that there was indeed a significant difference between these strains, but only at day 6 of culture (BCG::pMV361, 55.17 ± 8 h vs. BCG::BCG0114, 88.65 ± 6.66 h; p = 0.0131). On the other hand, apparent growth of BCG::ethR significantly differed from wild type BCG in days 0 (start of the culture) and day 4 (early log-phase) (Fig. 3b), although their doubling time was different only at day 5 of culture (BCG::pMV361, 35.6 ± 4.18 h vs. BCG::ethR, 43.37 ± 5.04 h; p = 0.038).
OD600nm readings of BCG0642c significantly differed in days 0, 2, 4, and from day 10 to day 12 (stationary phase) (Fig. 3b); however, doubling time differed at day 1 (BCG::pMV361, 20.72 ± 1.33 h vs. BCG::BCG0642c, 29.05 ± 2 h; p = 0.0181) and day 3 only (BCG::pMV361, 21 ± 0.76 h vs. BCG::BCG0642c, 14.3 ± 1.97 h; p = 0.0067). hsp60-driven expression of BCG3766c resulted in significant differences in OD600nm readings at day 2, and from day 6 to day 8 (mid-log phase), with doubling time being significantly different at days 2 and 3 of culture (day 2, BCG::pMV361, 20.72 ± 1.33 h vs. BCG::BCG3766c, 26.08 ± 1.29 h; p = 0.0065. Day 3, BCG::pMV361, 21 ± 0.76 h vs. BCG::BCG3766c, 18.19 ± 0.75 h; p = 0.0111).
Next, we compared bacterial replication as colony-forming units (CFU) per milliliter at 3 stages: day 0 (start of the culture), day 4 (early-log phase), and day 10 (stationary phase). We found that despite the apparent growth delay produced in BCG upon hsp60-driven expression of dosR or BCG0114, specifically in the mid-log to stationary phase transition (Fig. 3b), no significant difference in terms of bacterial numbers was found in any of the three stages evaluated here (Fig. 3c). On the other hand, in agreement with differences found in the growth curve at day 4, BCG::ethR had higher CFU/mL than wild type BCG (p < 0.0001 for early-log cultures, Fig. 3c). Also in agreement with changes observed in growth curve was the replication of early-log and stationary phase cultures of BCG::BCG0642c (p = 0.0022 for early-log, and p = 0.0125 for stationary phases cultures, Fig. 3c). No significant change was detected in replication of BCG::BCG3766c compared with BCG::pMV361 (Fig. 3c).
Discussion
Bacteria must accurately regulate growth and stress resilience. The formation of biofilms contributes to stress survival, since these dense multicellular aggregates, in which cells are embedded in an extracellular matrix of self-produced polymers, represent a self-constructed protective ‘niche’21 that yet remains metabolically active even after reaching maturity22.
In M. tuberculosis complex bacteria, biofilm production in vitro has been shown to harbor drug-tolerant bacteria7 and to be genetically linked to this phenotype8. Drug-tolerant mycobacteria may comprise a fraction of the population, as mature M. tuberculosis biofilms showed sensitivity to antibiotic treatment when assessed with microcalorimetry (IMC) and tunable diode laser absorption spectroscopy22. Drug tolerance has also been found to occur in ex vivo caseum23 and is thought to contribute to persistence after drug treatment in vivo24. In fact, the lungs of chronically infected mice harbored a subpopulation of nongrowing but metabolically active M. tuberculosis25.
Evidence reported over the last decade associate the capacity of M. tuberculosis-complex bacteria with virulence in ex vivo or in vivo models26. Pang et al.14 found several genes that were required for biofilm production using a “formation/no formation” readout, while Yang et al.1 utilized a genetic approach to propose a temporal order for development of mycobacterial biofilms using M. smegmatis as model.
In this work, we performed an unbiased, whole transcriptome analysis, aimed to find genes differentially expressed during intercellular aggregation and substrate attachment. This followed the rationale that upon affecting their expression levels, this may result in either major or subtle changes during biofilm formation by BCG. This contrasts with both Pang et al.14 strategy based on “formation/no formation” readout in microtiter plates, and the one used by Yang et al.1 that relied on a clever yet static approach, as these authors screened only one time point to look for M. smegmatis mutants with altered capacity to produce biofilms within a syringe-based model.
It is worth noting that we utilized a panel of 5 DE tools to identify gene expression changes. Selection of genes with potential relevance for the intercellular aggregation and substrate attachment steps during biofilm production by BCG were based on their up- or down-regulation by a twofold or greater change coupled with p < 0.05 after multivariate analysis. Using these criteria, we observed that the most significant changes that BCG experiences at the early stages of biofilm production are the downregulation of part of the DosR-regulon during intercellular aggregation, and their upregulation upon substrate attachment. Therefore, we selected 2 genes from this regulon: dosR itself, and BCG0114 (homologous to Rv0081), and characterized the effects of the strong expression of these genes from the hsp60 promoter both during multicellular and planktonic growth.
DosR has been shown to respond to oxygen limitation and to the presence of nitric oxide in vitro12,13. We found that only the absence of dosR in M. tuberculosis, as opposed to its increased expression in BCG, result in reduced biofilm production. We hypothesize that this may be explained by the fact of slow growing mycobacteria requiring DosR to metabolically adapt to low O2 levels as already reported27. This result is also in agreement with reduced dosR transcription and reduced biofilm production observed in a double mutant devoid of the exopolyphosphatases genes28 and also in a mtrB mutant29.
Changes in surface pellicle appearance during biofilm production upon dosR or BCG0114 expression from hsp60, constitute a subtle phenotype that may go unnoticed when assessing transposon insertion mutant for “formation/no formation” readout, as we found no quantitative effect on biofilm production. The relevance of this biofilm-specific change for other in vitro or in vivo phenotypes remains to be determined. We contend that altering either dosR or BCG0114 expression results in unique, biofilm-specific phenotypes, as during planktonic growth, their expression from hsp60 delayed logarithmic growth (Fig. 3b) but with no effect on bacterial replication at the beginning, early-log, and stationary phases (Fig. 3c). This difference might be explained, at least to some extent, by differences in oxygen availability in biofilms as has been suggested9. In support of this hypothesis, it is worth noting that the already complex regulatory network utilized by M. tuberculosis to respond to hypoxia16 has just been refined using a comprehensive genome-wide transcription factor binding map and network topology analysis. This unraveled M. tuberculosis response during the adaptation to varying oxygen levels (normoxia, depletion, early-, mid-, and late hypoxia, and resuscitation) and contributed to further support the role of Rv0081 (BCG0114), DosR, and Lsr2 in adaptation to oxygen availability30, regulators that were differentially expressed at distinct stages of biofilm production in BCG (Table 1).
BCG0642c, which encodes for a conserved hypothetical protein with a PhdYeFM antitoxin domain, was significantly upregulated only during the intercellular aggregation step and tended towards downregulation (p = 0.06) at the substrate attachment step (Supplementary Table 1). To date, only the structure and function as an antitoxin of VapB4 (Rv0596c, the orthologous of BCG0642c) has been described31 but with no other role identified thus far. However, toxin-antitoxin modules have been shown to play a major role in persister formation in many model systems32, therefore it seems reasonable to find at least one of these genes as differentially expressed and contributing to biofilm production in BCG (Fig. 2). We acknowledge that a unique, biofilm-specific role for BCG0642c cannot be claimed at this point, given that its expression from hsp60 also affected planktonic replication, positively during early-log phase, and negatively in stationary phase planktonic BCG cultures (Fig. 3).
ethR was specifically downregulated during surface attachment (Supplementary Table 1) but its expression from hsp60 resulted in no change during biofilm production (Fig. 2) yet it favored growth and replication during early-log phase (Fig. 3). In this regard, a M. bovis BCG ethA-ethR KO mutant showed increased adherence to mammalian cells33 but its capacity to produce biofilm was not evaluated in that work. Nevertheless, another report stated that ethR did not participate in biofilm production in M. tuberculosis34, which seems in agreement with our findings in BCG.
The last gene we evaluated was BCG3766c, which encodes for a conserved hypothetical proline rich protein (Supplementary Table 1). This gene was significantly downregulated during surface attachment and was also downregulated during the intercellular aggregation step as well (FC 0.76, p = 0.022). This may explain why its expression from hsp60 tended to reduce biofilm production (Fig. 2).
We also found differential expression for a number of other genes that we did not further evaluate in this work, including among the most significantly upregulated genes sigE, fadE23 (fatty-acid-CoA ligase, involved in sulfolipid production)10, hupB (DNA binding protein), BCG3929 (Rv3866, espG), ppsC (involved in PDIM synthesis), BCG1191 (Rv1130, prpD, 2-methylcitrate dehydratase), and BCG1826 (Rv1794, part of the ESX-5 secretion system)11. For BCG3929 (Rv3866, espG), a deletion of espG in M. marinum reduced sliding motility and biofilm formation35. BCG3929 upregulation during intercellular aggregation as compared to planktonic growth (Table 2) may explain the defect of the M. marinum mutant in biofilm formation.
Biofilm formation by BCG in the presence of the histone methyltransferase SUV39H1 was reduced, an effect proposed to occur via trimethylation of HupB36. This suggests a positive effect for this DNA-binding protein for biofilm production, and it is in agreement with hupB upregulation during intercellular aggregation (Table 2).
Rv3385c (orthologous to BCG3454c) was shown to be repressed in mature biofilms formed upon exposure to DTT as compared to late-log cultures of M. tuberculosis37. Redox conditions intervene in modulating M. tuberculosis pathogenesis, including activity of DosR38, which, to add further complexity to the mechanisms of gene regulation driven by this transcriptional regulator, was recently shown to be positively affected by c-di-GMP binding in M. smegmatis in response, precisely, to oxidative stress39.
Biofilm-specific proteins were recognized by antibodies present in sera from M. tuberculosis infected guinea pigs40. Of the antigenic proteins reported in that study, we found ceoB and BCG2013 (Rv1996) significantly repressed during the transition from planktonic to intercellular aggregation (Supplementary Table 3), while they were significantly induced after substratum attachment (Supplementary Table 4). Moreover, BCG2232 (Rv2216) and TB39.8 (BCG0050c, Rv0020c, FhaA) were affected specifically after substratum attachment (FC = 1, and − 0.74 Log2, respectively, Supplementary Tables 4 and 5). The fact that some biofilm-specific proteins that were recognized in vivo had their encoding genes differentially expressed during biofilm production in vitro by BCG further strengthen the notion of biofilms mimicking aspects found during TB pathogenesis26. Taken together, our results show that dosR and BCG0114 were expressed in a temporal order during mycobacterial biofilm formation to produce biofilm-specific changes, which most likely are triggered in response to varying oxygen levels within biofilms. Furthermore, we also provide a potential explanation for a stage dependent expression of additional genes previously reported to contribute to biofilm production in mycobacteria and suggest new targets that can be assessed for their particular contribution to this phenotype.
Methods
Bacterial strains, growth conditions and RNA extraction
M. bovis BCG Pasteur strain (ATCC 35734) or M. tuberculosis H37Rv and derivatives with deletion of the Rv3134c-dosR-dosS operon (referred to as H37Rv dosR KO in Fig. 2) and its complemented strain (referred to as H37Rv dosR::KO::Comp in Fig. 2)27 were used in this study. Planktonic cultures were performed in Middlebrook 7H9 liquid media (BD) with 10% OADC, 0.2% glycerol, 25 µg/mL of kanamycin, at 37 °C, 100 rpm. Serial dilutions of samples were followed by plating onto Middlebrook 7H10 agar plates supplemented with 10% OADC, 0.5% glycerol, and 25 µg/mL kanamycin served to determine colony-forming units per milliliter (CFU/mL). Biofilms (which include bacteria attached to the plastic wells and surface pellicles) for RNA extraction of BCG strains were cultured in Sauton media as already reported9. After 1, 7, 10 and 14 days of incubation, two culture flasks were used to harvest, with a scraper, the whole surface pellicle and biofilm attached to the wells (these samples are referred to as “biofilms”), and transferred into 50 mL tubes that were immediately frozen at − 70 °C. From frozen samples, we proceeded to perform RNA extraction and purification as already reported41, to ship these samples to Arizona State University for RNA-Seq analyses. The experiment was repeated three (7, 10, and 14 days cultures) or four times (24 h cultures, because of the low biomass present at this time point), to produce independent replicates.
Temporal expression profiling during biofilm production by BCG
RNA was used to prepare cDNA using Nugen’s Ovation RNA-Seq System via single primer isothermal amplification (Catalogue # 7102-A01) and automated on the (BRAVO NGS liquid handler from Agilent). cDNA was quantified on the Nanodrop (Thermo Fisher Scientific). Using Kapa Biosystem’s DNA Hyper Plus library preparation kit, (KK8514) cDNA was enzymatically sheared to approximately 150 bp fragments, end repaired and A-tailed. Adapters with unique indexes compatible with Illumina (IDT #00989130v2) were ligated on each sample individually, then, these were cleaned using Kapa pure beads (Kapa Biosciences, KK8002), followed by amplification with Kapa’s HIFI enzyme (KK2502). Using Agilent’s Tapestation, we analyzed fragment size of each library, and quantified them by qPCR (KAPA Library Quantification Kit, KK4835) on a Quantstudio 5 (Thermo Fisher Scientific). Next, we multiplex pooled and sequenced a 2 × 75 flow cell on the NextSeq500 platform (Illumina) at the ASU’s Genomics Core facility.
RNA-seq analysis
Raw FASTQ read data were processed using the R package DuffyNGS as described previously42. Briefly, raw reads were passed through a 3-stage alignment pipeline: (1) a prealignment step, to remove unwanted transcripts, such as rRNA; (2) a main genomic alignment step against the genome of interest; and (3) a splice junction alignment step, compared with an index of standard and alternative exon splice junctions. Reads were aligned to M. bovis BCG str. Pasteur (1173P2) with Bowtie243, using the command line option “very-sensitive.” BAM files from stages (2) and (3) were combined into read depth wiggle tracks that recorded both multiply mapped and uniquely mapped reads to each of the forward and reverse strands of the genome of reference at single-nucleotide resolution. Next, gene transcript abundance was measured by summing total reads found inside annotated gene boundaries, expressed as both RPKM and raw read counts. RNA-seq data (raw fastq files and read counts) have been deposited in the GEO repository under accession number GSE150030.
Differentially expressed genes
A panel of 5 DE tools was used to identify gene expression changes between 1-week old biofilm samples and 24-h samples (to determine genes affected or necessary for intercellular aggregation or cell-to-cell attachment) or 10-day old biofilm samples and 1-week old biofilm samples (to determine genes affected or necessary for substratum attachment to start building up the mature biofilm). The tools included (1) RoundRobin (in-house); (2) RankProduct44; (3) significance analysis of microarrays (SAM)45 ; (4) EdgeR46; and (5) DESeq247. Appropriate default parameters were used to call each DE tool and operated on the same set of transcription results. For RankProduct, RoundRobin, and SAM, we used RPKM abundance units and for DESeq2 and EdgeR, raw read count abundance units were used. Next, we combined gene DE rank positions across all 5 DE tools. This consists of averaging a gene’s rank position in all 5 results, using a generalized mean to the 1/2 power, to yield the gene’s final net rank position. Similarly, explicit measurements of differential expression (fold change -FC-) and significance (p value) determined by each DE tool were combined via appropriate averaging (arithmetic and geometric mean, respectively). Genes with averaged absolute Log2 FC bigger than one and multiple hypothesis adjusted p value below 0.05 were considered differentially expressed. When gene function was predicted for those Conserved Hypothetical Proteins (CHP) and Hypothetical Proteins (HP) found among the 30 most up- or down-regulated genes during biofilm formation, we took the whole predicted protein sequence from BCG Pasteur 1173P2 (https://www.genome.jp/kegg-bin/show_organism?org=mbb), and used to search for Conserved Domains using the CD-Search tool from NCBI (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi).
Reverse transcription coupled to quantitative PCR (RT-qPCR)
In order to independently determine the relative expression of genes selected from RNA-Seq experiments, total RNA samples obtained from additional cultures grown as biofilms (days 1, 7, 10, and 14) or planktonic cells (OD600nm 0.3 and ≈ 1.7) were quantified by UV spectroscopy and shipped to Centro Médico Siglo XXI for real time qPCR assays.This was conducted essentially as described9. Briefly, cDNA was synthesized using 500 ng of RNA, 0.2 µg/µl of random hexamer primers and 2 U/µl of Revertaid M-MulV-RT (Thermo Scientific). Specific gene primers are listed in Table 1. Control reactions were run in all experiments, with no transcript detected. Quantitative real-time PCR was performed in a LightCycler 480 instrument (Roche) and 16S rRNA (rrs) was used as a reference gene for normalization. The relative gene expression was calculated using the 2−ΔΔCt method48.
Construction of recombinant plasmids to drive expression of selected genes under the strong promoter hsp60
Primers used to amplify the open reading frame (ORF) of each one of the selected genes, recombinant plasmids generated, bacterial strains used in this work, and primers used for real time qPCR are indicated in Table 1. ORFs were amplified from genomic DNA obtained from BCG Pasteur by PCR using high fidelity Q5 DNA polymerase (New England Biolabs), digested with specific endonucleases and cloned under the hsp60 promoter in pMV36117. Identity and fidelity of the ORFs was confirmed by DNA sequencing. Amplify4 for MacOS was used to design and test primers. DNA Strider 3.0 for MacOS was used for virtual cloning and plasmid characterizations. Sequence fidelity of cloned ORFs was evaluated using BLAST alignments both locally with DNA Strider 3.0 for MacOS and by direct comparison with genome sequences of BCG Pasteur 1173P2 (https://www.genome.jp/kegg-bin/show_organism?org=mbb). Recombinant plasmids were transformed into BCG by electroporation and selected on Middlebrook 7H10 (BD) OADC (BD-BBL) with 0.5% glycerol (Sigma) agar plates containing 25 µg/mL of kanamycin (Sigma).
Growth curve and bacterial enumeration
Planktonic cultures were performed in Middlebrook 7H9 liquid media (BD) with 10% OADC, 0.2% glycerol, 25 µg/mL of kanamycin, at 37 ºC, 100 rpm, with a starting OD600nm of 0.03, to read OD600nm every 24 h. When each strain reached OD600nm of ≈ 0.3 (early-log), ≈ 0.6 (mid-log) and ≈ 1.7 (stationary phase), we took 1 mL samples. Serial dilutions of these samples were followed by plating 50 µL aliquots onto Middlebrook 7H10 agar plates supplemented with 10% OADC, 0.5% glycerol, and 25 µg/mL kanamycin served to determine colony-forming units per milliliter (CFU/mL).
Ziehl–Neelsen staining
We used 10 µL aliquots from mid-log phase samples (planktonic cultures) or a loop of fully mature biofilms (2 weeks-old) taken with a disposable inoculating loop (BD 220,215). After fixation, staining was performed with a Ziehl–Neelsen kit (Hycel, https://www.hycel.com.mx/productos/kit-colorantes/) according to the manufacturer´s instructions.
Quantification of biofilm by crystal violet staining
All mycobacterial strains were cultured in Sauton media, started at OD600nm 0.03, in 24-well (M. tuberculosis) or 48-well non-treated tissue culture plates (BCG strains), and were incubated at 37 °C, 5% CO2. Each strain was inoculated into 6 different wells, and experiments were repeated three times for statistical analysis. After 10 days (BCG strains) and 14 days (BCG strains and M. tuberculosis strains) of incubation, liquid media was removed and the whole surface pellicle and biofilm attached to the wells (these samples are referred to as “biofilms”) was maintained. Plates were baked at 30 °C for 24 h and 1 ml of 100% methanol was added to each well and incubated at room temperature for 15 min. Then, methanol was removed, and plates were dried at 37 °C for 15 min. Crystal violet (CV) was added to each well and incubated at 37 °C for 5 min. CV was removed and each well was washed four times with deionized water. Plates were dried at 37 °C for 15 min. Dye was extracted with 30% acetic acid for 15 min at 37 °C. Then the extract from each well was diluted 1:10 (M. tuberculosis), 1:20 (BCG, 10 days cultures), or 1:40 (BCG, 14 days cultures) in 30% acetic acid and read for OD550nm.
Statistics
Data distribution for qPCR, CFU, and biofilm quantification were analyzed using the Anderson–Darling and Shapiro–Wilk tests, and found to follow a normal distribution in all instances. Growth (as OD600nm readings) of rBCG strains was compared with that of parental BCG harboring the empty vector pMV361 (BCG WT::pMV361) using Two-Way ANOVA followed by Dunnett’s multiple comparison test. Growth (as doubling time) was compared by One-Way ANOVA (logarithmic phase cultures) or Brown–Forsythe and Welch ANOVA (stationary phase cultures) followed by Dunnett’s multiple comparison tests. Bacterial replication (CFU/mL) was compared by 2-Way ANOVA followed by Tukey’s multiple comparison test. For quantification of biofilms, statistical significance was determined using One Way ANOVA followed by Dunnett’s test for multiple comparison. For RT-qPCR analyses, Brown-Forsythe and Welch ANOVA followed by Dunnett’s multiple comparison test was used for comparing biofilm samples; multiple t tests followed by Holm–Sidak multiple comparison test was used for comparing planktonic samples. GraphPad Prism 8 for MacOS was used for performing statistical analyses. Assays were conducted in three independent times, and the number of replicates per experiment is indicated in each figure legend.
References
Yang, Y. et al. Defining a temporal order of genetic requirements for development of mycobacterial biofilms. Mol. Microbiol. 105, 794–809. https://doi.org/10.1111/mmi.13734 (2017).
Anderson, G. G. & O’Toole, G. A. Innate and induced resistance mechanisms of bacterial biofilms. Curr. Top. Microbiol. Immunol. 322, 85–105. https://doi.org/10.1007/978-3-540-75418-3_5 (2008).
Prabhakara, R., Harro, J. M., Leid, J. G., Harris, M. & Shirtliff, M. E. Murine immune response to a chronic Staphylococcus aureus biofilm infection. Infect. Immun. 79, 1789–1796. https://doi.org/10.1128/IAI.01386-10 (2011).
Flores-Valdez, M. A. et al. The cyclic Di-GMP phosphodiesterase gene Rv1357c/BCG1419c affects BCG pellicle production and in vivo maintenance. IUBMB Life 67, 129–138. https://doi.org/10.1002/iub.1353 (2015).
Lefebvre, C. et al. Discovery of a novel dehydratase of the fatty acid synthase type II critical for ketomycolic acid biosynthesis and virulence of Mycobacterium tuberculosis. Sci. Rep. 10, 2112. https://doi.org/10.1038/s41598-020-58967-8 (2020).
Ojha, A. et al. GroEL1: a dedicated chaperone involved in mycolic acid biosynthesis during biofilm formation in mycobacteria. Cell 123, 861–873. https://doi.org/10.1016/j.cell.2005.09.012 (2005).
Ojha, A. K. et al. Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol. Microbiol. 69, 164–174. https://doi.org/10.1111/j.1365-2958.2008.06274.x (2008).
Richards, J. P., Cai, W., Zill, N. A., Zhang, W. & Ojha, A. K. Adaptation of Mycobacterium tuberculosis to biofilm growth is genetically linked to drug tolerance. Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.01213-19 (2019).
Cruz-Villegas, M. A. et al.. Transcriptional and mycolic acids profiling in Mycobacterium bovis BCG in vitro show an effect for c-di-GMP and overlap between dormancy and biofilms. J. Microbiol. Biotechnol. 30(6), 811-821 (2020).
Lynett, J. & Stokes, R. W. Selection of transposon mutants of Mycobacterium tuberculosis with increased macrophage infectivity identifies fadD23 to be involved in sulfolipid production and association with macrophages. Microbiology 153, 3133–3140. https://doi.org/10.1099/mic.0.2007/007864-0 (2007).
Bottai, D. et al. Disruption of the ESX-5 system of Mycobacterium tuberculosis causes loss of PPE protein secretion, reduction of cell wall integrity and strong attenuation. Mol. Microbiol. 83, 1195–1209. https://doi.org/10.1111/j.1365-2958.2012.08001.x (2012).
Park, H. D. et al. Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol. Microbiol. 48, 833–843. https://doi.org/10.1046/j.1365-2958.2003.03474.x (2003).
Voskuil, M. I. et al. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 198, 705–713. https://doi.org/10.1084/jem.20030205 (2003).
Pang, J. M. et al. The polyketide Pks1 contributes to biofilm formation in Mycobacterium tuberculosis. J. Bacteriol. 194, 715–721. https://doi.org/10.1128/JB.06304-11 (2012).
Wang, X. M. et al. Biochemical and immunological characterization of a cpn60.1 knockout mutant of Mycobacterium bovis BCG. Microbiology 157, 1205–1219. https://doi.org/10.1099/mic.0.045120-0 (2011).
Galagan, J. E. et al. The Mycobacterium tuberculosis regulatory network and hypoxia. Nature 499, 178–183. https://doi.org/10.1038/nature12337 (2013).
Stover, C. K. et al. New use of BCG for recombinant vaccines. Nature 351, 456–460. https://doi.org/10.1038/351456a0 (1991).
Flores Valdez, M. A. & Schoolnik, G. K. DosR-regulon genes induction in Mycobacterium bovis BCG under aerobic conditions. Tuberculosis (Edinb) 90, 197–200. https://doi.org/10.1016/j.tube.2010.04.001 (2010).
Pedroza-Roldan, C. et al. The adenylyl cyclase Rv2212 modifies the proteome and infectivity of Mycobacterium bovis BCG. Folia Microbiol. (Praha) 60, 21–31. https://doi.org/10.1007/s12223-014-0335-1 (2015).
Pedroza-Roldan, C. et al. BCG constitutively expressing the adenylyl cyclase encoded by Rv2212 increases its immunogenicity and reduces replication of M. tuberculosis in lungs of BALB/c mice. Tuberculosis (Edinb) 113, 19–25. https://doi.org/10.1016/j.tube.2018.08.012 (2018).
Hengge, R. Linking bacterial growth, survival, and multicellularity: small signaling molecules as triggers and drivers. Curr. Opin. Microbiol. 55, 57–66. https://doi.org/10.1016/j.mib.2020.02.007 (2020).
Solokhina, A., Bruckner, D., Bonkat, G. & Braissant, O. Metabolic activity of mature biofilms of Mycobacterium tuberculosis and other non-tuberculous mycobacteria. Sci. Rep. 7, 9225. https://doi.org/10.1038/s41598-017-10019-4 (2017).
Sarathy, J. P. et al. Extreme drug tolerance of Mycobacterium tuberculosis in Caseum. Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.02266-17 (2018).
Lenaerts, A. J. et al. Location of persisting mycobacteria in a Guinea pig model of tuberculosis revealed by r207910. Antimicrob. Agents Chemother. 51, 3338–3345. https://doi.org/10.1128/AAC.00276-07 (2007).
Manina, G., Dhar, N. & McKinney, J. D. Stress and host immunity amplify Mycobacterium tuberculosis phenotypic heterogeneity and induce nongrowing metabolically active forms. Cell Host Microbe 17, 32–46. https://doi.org/10.1016/j.chom.2014.11.016 (2015).
Flores-Valdez, M. A. Vaccines directed against microorganisms or their products present during biofilm lifestyle: can we make a translation as a broad biological model to tuberculosis?. Front. Microbiol. 7, 14. https://doi.org/10.3389/fmicb.2016.00014 (2016).
Bartek, I. L. et al. The DosR regulon of M. tuberculosis and antibacterial tolerance. Tuberculosis (Edinb) 89, 310–316. https://doi.org/10.1016/j.tube.2009.06.001 (2009).
Tiwari, P. et al. Inorganic polyphosphate accumulation suppresses the dormancy response and virulence in Mycobacterium tuberculosis. J. Biol. Chem. 294, 10819–10832. https://doi.org/10.1074/jbc.RA119.008370 (2019).
Banerjee, S. K. et al. The sensor kinase MtrB of Mycobacterium tuberculosis regulates hypoxic survival and establishment of infection. J. Biol. Chem. 294, 19862–19876. https://doi.org/10.1074/jbc.RA119.009449 (2019).
Peterson, E. J. R. et al. Intricate genetic programs controlling dormancy in Mycobacterium tuberculosis. Cell Rep. 31, 107577. https://doi.org/10.1016/j.celrep.2020.107577 (2020).
Jin, G., Pavelka, M. S. Jr. & Butler, J. S. Structure-function analysis of VapB4 antitoxin identifies critical features of a minimal VapC4 toxin-binding module. J. Bacteriol. 197, 1197–1207. https://doi.org/10.1128/JB.02508-14 (2015).
Harms, A., Maisonneuve, E. & Gerdes, K. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science https://doi.org/10.1126/science.aaf4268 (2016).
Ang, M. L. et al. An ethA-ethR-deficient Mycobacterium bovis BCG mutant displays increased adherence to mammalian cells and greater persistence in vivo, which correlate with altered mycolic acid composition. Infect. Immun. 82, 1850–1859. https://doi.org/10.1128/IAI.01332-13 (2014).
Zhang, H. N. et al. Cyclic di-GMP regulates Mycobacterium tuberculosis resistance to ethionamide. Sci. Rep. 7, 5860. https://doi.org/10.1038/s41598-017-06289-7 (2017).
Lai, L. Y., Lin, T. L., Chen, Y. Y., Hsieh, P. F. & Wang, J. T. Role of the Mycobacterium marinum ESX-1 secretion system in sliding motility and biofilm formation. Front. Microbiol. 9, 1160. https://doi.org/10.3389/fmicb.2018.01160 (2018).
Yaseen, I., Choudhury, M., Sritharan, M. & Khosla, S. Histone methyltransferase SUV39H1 participates in host defense by methylating mycobacterial histone-like protein HupB. EMBO J. 37, 183–200. https://doi.org/10.15252/embj.201796918 (2018).
Trivedi, A., Mavi, P. S., Bhatt, D. & Kumar, A. Thiol reductive stress induces cellulose-anchored biofilm formation in Mycobacterium tuberculosis. Nat. Commun. 7, 11392. https://doi.org/10.1038/ncomms11392 (2016).
Pacl, H. T., Reddy, V. P., Saini, V., Chinta, K. C. & Steyn, A. J. C. Host-pathogen redox dynamics modulate Mycobacterium tuberculosis pathogenesis. Pathog. Dis. https://doi.org/10.1093/femspd/fty036 (2018).
Hu, Q. et al. Cyclic di-GMP co-activates the two-component transcriptional regulator DevR in Mycobacterium smegmatis in response to oxidative stress. J. Biol. Chem. 294, 12729–12742. https://doi.org/10.1074/jbc.RA119.008252 (2019).
Kerns, P. W., Ackhart, D. F., Basaraba, R. J., Leid, J. G. & Shirtliff, M. E. Mycobacterium tuberculosis pellicles express unique proteins recognized by the host humoral response. Pathog. Dis. 70, 347–358. https://doi.org/10.1111/2049-632X.12142 (2014).
Flores-Valdez, M. A. et al. The BCGDeltaBCG1419c vaccine candidate reduces lung pathology, IL-6, TNF-alpha, and IL-10 during chronic TB infection. Front. Microbiol. 9, 1281. https://doi.org/10.3389/fmicb.2018.01281 (2018).
Vignali, M. et al. NSR-seq transcriptional profiling enables identification of a gene signature of Plasmodium falciparum parasites infecting children. J. Clin. Invest. 121, 1119–1129. https://doi.org/10.1172/JCI43457 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Breitling, R., Armengaud, P., Amtmann, A. & Herzyk, P. Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 573, 83–92. https://doi.org/10.1016/j.febslet.2004.07.055 (2004).
Tusher, V. G., Tibshirani, R. & Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 98, 5116–5121. https://doi.org/10.1073/pnas.091062498 (2001).
Robinson, M. D. & Smyth, G. K. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9, 321–332. https://doi.org/10.1093/biostatistics/kxm030 (2008).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25, 402–408. https://doi.org/10.1006/meth.2001.1262 (2001).
Acknowledgements
M.A.F.V. is grateful to CIATEJ for internal funding to complete this project, and also to Jason Steel and Shanshan Yang (Arizona State University) for technical support regarding RNA-Seq and preliminary bioinformatics analyses, respectively. M.J.A.S. received a Ph.D. fellowship from CONACYT (Number 745841).
Author information
Authors and Affiliations
Contributions
Conceptualization M.A.F.V.; Methodology M.A.F.V., M.J.A.S., E.P., N.B., M.A.C., M.A., and M.V.; Investigation M.A.F.V., M.J.A.S., J.B., M.A., S.B., N.P.P., M.B.F., T.A.C.V., and M.G.E.J.; Formal analysis M.A.F.V., M.J.A.S., E.P., N.B., M.A.C., M.V., J.B., T.A.C.V., S.B., and M.V.; Writing M.A.F.V., E.P., S.B., and M.V.; Resources M.A.F.V., N.B., and M.V.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Flores-Valdez, M.A., Aceves-Sánchez, M.d., Peterson, E.J.R. et al. Transcriptional portrait of M. bovis BCG during biofilm production shows genes differentially expressed during intercellular aggregation and substrate attachment. Sci Rep 10, 12578 (2020). https://doi.org/10.1038/s41598-020-69152-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-69152-2
This article is cited by
-
Lsr2 acts as a cyclic di-GMP receptor that promotes keto-mycolic acid synthesis and biofilm formation in mycobacteria
Nature Communications (2024)
-
Diminishing of Helicobacter pylori adhesion to Cavia porcellus gastric epithelial cells by BCG vaccine mycobacteria
Scientific Reports (2023)
-
Evaluation of early innate and adaptive immune responses to the TB vaccine Mycobacterium bovis BCG and vaccine candidate BCGΔBCG1419c
Scientific Reports (2022)
-
Three-dimensional low shear culture of Mycobacterium bovis BCG induces biofilm formation and antimicrobial drug tolerance
npj Biofilms and Microbiomes (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
