
Abstract
We recently showed that the anti-helminthic compound mebendazole (MBZ) has immunomodulating activity in monocyte/macrophage models and induces ERK signalling. In the present study we investigated whether MBZ induced ERK activation is shared by other tubulin binding agents (TBAs) and if it is observable also in other human cell types. Curated gene signatures for a panel of TBAs in the LINCS Connectivity Map (CMap) database showed a unique strong negative correlation of MBZ with MEK/ERK inhibitors indicating ERK activation also in non-haematological cell lines. L1000 gene expression signatures for MBZ treated THP-1 monocytes also connected negatively to MEK inhibitors. MEK/ERK phosphoprotein activity testing of a number of TBAs showed that only MBZ increased the activity in both THP-1 monocytes and PMA differentiated macrophages. Distal effects on ERK phosphorylation of the substrate P90RSK and release of IL1B followed the same pattern. The effect of MBZ on MEK/ERK phosphorylation was inhibited by RAF/MEK/ERK inhibitors in THP-1 models, CD3/IL2 stimulated PBMCs and a MAPK reporter HEK-293 cell line. MBZ was also shown to increase ERK activity in CD4+ T-cells from lupus patients with known defective ERK signalling. Given these mechanistic features MBZ is suggested suitable for treatment of diseases characterized by defective ERK signalling, notably difficult to treat autoimmune diseases.
Similar content being viewed by others


Introduction
Mebendazole (MBZ) is an anti-parasitic drug that has gained considerable interest as a potential anticancer agent. MBZ is generally perceived as a tubulin polymerase inhibitor1,2 but several other mechanisms have been suggested to explain the anticancer activity observed in vitro and in vivo. Mechanisms suggested include angiogenesis inhibition3,4, apoptosis induction5,6 and inhibition of the Hedgehog signalling pathway7. MBZ has also been shown to potently bind to several protein kinases involved in oncogenic signalling8. Anticancer activity has also occasionally been observed in patients9,10.
Recently, we demonstrated that MBZ induces a pro-inflammatory tumour-suppressive M1 phenotype in THP-1 monocytes and macrophages which could potentially explain tumour cell killing11. It was also noted that MBZ induced activation of ERK potentially contributes to the immune effects observed.
ERK is part of a pathway which comprises proteins referred to as mitogen-activated protein kinases (MAPKs)12. The MAPKs can be categorised into three major protein families: extracellular signal-regulated kinases (ERK), stress-activated protein kinases/c-Jun N-terminal kinase (SAPK/JNK) and the p38 proteins12. Whereas SAPK/JNK is associated with stress response, apoptosis and inflammation, increased ERK is generally suggested to mediate cell survival, activation and differentiation13. Proximal proteins involved in the ERK pathway include Ras, Raf and MEK12.
In the present study we investigated whether MBZ induced ERK activation is shared by other tubulin binding agents (TBAs) and if the ERK activation is observable also in other human cell types.
Methods
Cell culture
Monocytoid THP-1 cells were obtained from ATCC (Manassas, VA, USA) and were cultured in RPMI-1640 medium, supplemented with 10% heat-inactivated foetal bovine serum (HIFBS), 2 mM l-glutamine, 100 U/100 µg/ml penicillin/streptomycin and 0.05 mM 2-mercaptoethanol (all from Sigma, St Louis, MO, USA). For differentiation and polarisation of THP-1 cells to macrophages, 25 ng/mL PMA, 20 ng/mL IFNγ, 100 ng/mL LPS, 20 ng/mL IL4 and 20 ng/mL IL13 (final concentrations) were used according to an established protocol with minor modifications14. The SRE Reporter HEK293 cell line was obtained from BPS Bioscience (San Diego, CA, USA) and used for monitoring the activity of the MAPK/ERK signalling pathway. The SRE Reporter HEK293 cell line contains a firefly luciferase gene under the control of Serum Response Element (SRE) responsive elements stably integrated into HEK293 cells, resulting in an ERK pathway-responsive reporter cell line. All cell lines were cultured at 37 °C in a humidified atmosphere containing 5% CO2. CD4+ T-cells from patients with SLE were purchased from Astarte Biologics, LLC (Bothell, WA, USA) and handled according to the manufacturer’s recommendations.
Materials
Mebendazole (MBZ), albendazole (ABZ), fenbendazole (FBZ), lipopolysaccharide (LPS), interferon gamma (IFNγ), IL4, IL13 and phorbol myristate acetate (PMA) were purchased from Sigma (St Louis, MO, USA). The ERK/MEK/Raf inhibitors used were purchased from Selleckchem (Houston, TX, USA). The compounds were kept as 10 mM stock solutions in dimethyl sulphoxide (DMSO, Sigma) or sterile water, and further diluted with culture medium (Sigma or ATCC) as needed.
Bioinformatic analysis using the LINCS L1000 platform
The drug-induced gene expression perturbations of MBZ were studied using the public LINCS Connectivity Map (CMap) resource (clue.io, formerly www.lincscloud.org)15,16 that contains a collection of hundreds-of-thousands of L1000 gene-expression profiles from cells grown in monolayer exposed to large numbers of small-molecule and genetic perturbagens. Since MBZ is present in the database, the gene expression profile can be compared with those of other drugs and perturbagens. Score and ranking were retrieved from the LINCS CMap database using default settings (best 4 cell lines). THP-1 gene expression data obtained using the L1000 assay (see below) were uploaded and analysed in a similar manner.
For the L1000 assay, drugs and tool compounds were transferred to monolayer plates using Echo Liquid Handler 550 (Labcyte Inc, Sunnyvale, CA). Following 1 h or 6 h incubation, the cell-culture medium was aspirated and cell lysis buffer (Genometry, Inc., Boston, MA, USA) added. After 30 min incubation at room temperature, cell samples were mixed. Homogeneous lysates were transferred to 384-well Nunc plates (Thermo Fisher Scientific, Waltham, MA, USA) and frozen at − 70 °C. Lysates were processed, and the resulting gene expression data subjected to a panel of quality control tests, performed by Genometry in their facility (www.genometry.com). The L1000 expression profiles delivered were scaled, normalized and log transformed, and denominated in the Affymetrix HG-U133A feature space.
Measurement of cytokines and phosphoproteins
The levels of cytokines and phosphoproteins were measured using the Luminex MAGPIX system and commercially available kits for various analytes (Bio-Rad, Hercules, CA, USA) and were performed as previously described11 according to the manufacturer’s instructions. The target of interest is bound to magnetic beads via antibodies, and detected using biotinylated antibodies with a fluorescent reporter. Briefly, in the cytokine assay the supernatant samples were incubated firstly with beads, secondly with detection antibody and finally with streptavidin-PE. Fluorescence was measured using the MAGPIX instrument (Bio-Rad) and concentration levels were determined using a fitted standard curve. For phosphoprotein assays, protein concentrations in the lysates were measured using a Micro BCA method (Thermo Fisher Scientific) in order to ensure equal amounts of samples in the assay before measuring using the same protocol as for the cytokine assay.
Measurement of tubulin polymerization
Tubulin polymerization from purified tubulin monomers was measured as increased fluorescence because of the incorporation of a fluorescent reporter into growing microtubules as previously described11. Reagents necessary for performing the assay were provided in the kit BK011 from Cytoskeleton (Denver, Colorado, USA) and fluorescence was measured at 1 min intervals for 60 min using a FLUOstar Omega (BMG Labtech GmbH, Offenburg, Germany).
Measurement of cell cycle
Cell cycle was assayed in THP-1 cells. These cells were plated in RPMI-1640 medium with 10% heat inactivated foetal bovine serum, 2 mM glutamine, 100 µg/ml streptomycin and 100 units/ml penicillin at 1 × 106 cells/well in 24-well plates. Test compounds were added to cells at 10 times the final concentration in medium and incubated at 37 °C 5% CO2 overnight. On the day of assay cells were permeabilized, the DNA stained with fluorescent dye (DAPI) and analysed according to the manufacturer’s instructions with “2-step Cell Cycle Assay” on a NucleoCounter NC-3000 (ChemoMetec A/S, Allerod, Denmark). Cellular fluorescence was quantified, and DNA content histograms were displayed. The phases of the cell cycle were gated and data collected.
Results
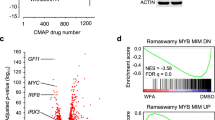
First, we performed a bioinformatic analysis to investigate whether the activation of ERK is unique to MBZ among tubulin binding agents (TBAs). We took advantage of the LINCS connectivity map database using the gene expression signatures for MBZ and a panel of well-known and clinically used TBAs as queries. The resulting drug-specific scores are based on the aggregated response from a panel of 6 non-haematological cell lines. MBZ showed strong negative correlations (− 99.7) to a predefined set of 8 MEK/ERK inhibitors in CMap which was not the case for the other TBAs showing correlations ranging between (+ 4 and + 77, Fig. 1a). These results indicate that MBZ but not the remaining TBAs strongly activates the MEK/ERK pathway.

Gene expression analysis of MBZ. Correlation of MBZ and a panel of TBAs with MEK inhibitors in the CMap database (a). Correlation of uploaded L1000 data (see material and methods) in response to 10 µM MBZ with PKC and MEK inhibitors (b). Effect of MBZ and other benzimidazoles on expression of a marker of ERK activity, DUSP6, using the L1000 platform (c).
Uploading our own MBZ induced gene expression L1000 signature from the THP-1 cell line also showed strong negative correlations with the MEK/ERK inhibitors (Fig. 1b). The highest positive correlations were observed for a set of PKC activators (Fig. 1b) which are known to induce ERK dependent THP-1 adhesion and differentiation17. The L1000 gene expression of DUSP6, a marker gene for ERK activation, dose dependently increased only in response to MBZ and to some extent flubendazole but no other benzimidazoles. (Fig. 1c). Finally, using the Reactome pathway analysis tool queried with the MBZ gene L1000 signature six of 25 top pathways enriched involved MAPK signalling (Supplementary Data).
Next, we employed a multiplex Luminex based assay to directly measure the effects of MBZ and the TBAs on MAPK phosphoprotein activity at 0.1, 1 and 10 µM using naive THP-1 cells as the model (Fig. 2). Again only MBZ increased MEK and ERK signalling with little or no effect observed for the other TBAs. MBZ also increased the activity of the ERK substrate P90RSK. Vinblastine and vincristine, on the other hand, increased primarily SAPK/JNK activity which is consistent with the literature13, whereas this was observed for MBZ only at the highest concentration (10 µM, Fig. 2). None of the TBAs, including MBZ, affected p38 phosphorylation.

Effect of MBZ and a panel of TBAs on MAPK phosphoprotein activity in naive THP-1 monocytes. In the last panel the effect of 10 µM MBZ and TBAs on IL1B release is shown. The experiments were repeated at least two times with similar results. MBZ mebendazole, FBZ fenbendazole, OBZ oxibendazole, ABZ albendazole.
When distal effects on cytokine release was measured only MBZ showed a significant effect on IL1B secretion. Very similar results were obtained in PMA differentiated THP-1 macrophages (Fig. 3). Interestingly in this model the effect of MBZ appeared potentiated causing ERK activation at lower concentrations. The above results in both THP-models were observed at concentrations of TBAs that affected tubulin polymerisation (Supplementary Fig. S1) and induced G2/M arrest (Supplementary Fig. S2).

Effect of MBZ and a panel of TBAs on MAPK phosphoprotein activity and IL1B release in PMA differentiated THP-1 macrophages. The experiments were repeated at least two times with similar results. MBZ mebendazole, FBZ fenbendazole, OBZ oxibendazole, ABZ albendazole.
The kinetics of MBZ induced ERK signalling appeared to be biphasic with one peak observed after 1 h followed by a decline and then increasing again at 6–24 h (Supplementary Fig. S3). Interestingly, in the THP-1 model MBZ differed from the classical M1 stimuli LPS/IFN, the latter increasing p38 signalling including downstream Hsp27 activity whereas MBZ primarily affected MEK/ERK phosphorylation (Supplementary Fig. S3). Increased ERK activity upon MBZ treatment was observed in THP-1 cells as well as in human PBMCs stimulated with anti-CD3 antibody and IL-2. The effect was completely abrogated by the MEK inhibitor U0126 (10 µM). Furthermore, MBZ increased ERK activity in CD4+ T-cells from SLE patients with known defective ERK signalling (Fig. 4). Finally, we employed a MAPK reporter HEK293 cell line and demonstrated that MBZ readily increased reporter activity even stronger than the positive control EGF (Fig. 5). Moreover, this activity was inhibited by selective RAF, MEK and ERK inhibitors (Fig. 5).

Effect of MBZ with or without MEK inhibitor U0126 on ERK activity in THP-1 monocytes (a), PBMC activated by antiCD3/IL2 (b) and CD4+ T-cells from patients with SLE (c,d).

Effect of MBZ and positive control EGF on ERK activity in MAPK reporter HEK293 cell line (a). In (b) the effect of MBZ with and without different ERK/MEK/Raf inhibitors is shown. The results are expressed as mean values + / − SEM for three independent experiments.
Discussion
In the present study we demonstrated that MBZ uniquely induce ERK activation in several cell types including those of non-haematological origin. Bioinformatic analysis has been used to identify pathogenetic mechanisms in diseases of different origins including immunoinflammatory diseases and cancer and helps to predict novel therapeutic targets as well as elucidate mode and mechanism of action18,19. Here we used a database of transcription drug response signatures, CMap, to compare MBZ to other TBAs. The striking negative correlation to MEK inhibitors in CMap is based on solid tumour cell lines only and was not observed for other compounds, including TBAs, in the database.
In the literature TBAs are long known to activate MAPK signalling but this effect has mostly been associated with JNK activation13 and TBAs have been reported to inhibit ERK in several cell systems20,21,22. This was confirmed in the present study in which only MBZ but no other TBAs, including other benzimidazoles, activated ERK to any significant extent. However, some of them indeed activated SAPK/JNK in accordance with the literature13. In the present study the tested concentrations of MBZ were relatively high (often 1–30 µM). However in most of the experimental models tested a clear effect on MEK/ERK activation was observed at 1 µM which is a plasma concentration achievable in the clinical setting23.
The reason for the MBZ selective ERK activation is still not clear but is apparently not only due to tubulin depolymerization per se. One potential explanation could involve specific interactions with the tubulin structure. MBZ is known to bind to the colchicine binding site of tubulin but this is also the case for other benzimidazoles24, thus providing no simple explanation for the MBZ induced selective ERK activation. However, MBZ has been demonstrated to inhibit DYRK1b at low nM concentrations which can lead to ERK activation25,26. Moreover, MBZ also potently inhibits BRAF8 which in cells with wild type RAF leads to paradoxical ERK activation27,28. Thus, RAF inhibitors selectively block ERK signalling in BRAF-mutant tumours but have the opposite effect in BRAF wild-type cells such as T-cells, where they cause hyper-activation of ERK signalling28. These additional pharmacodynamic effects may consequently contribute to the observed MBZ induced ERK activation. The potential mechanisms behind MBZ-induced ERK activation are depicted in Fig. 6.

Hypothesis explaining the difference between MBZ and other TBAs for MAPK activation. TBAs are known to activate MAPK signalling, however, mainly through SAP/JNK. MBZ but no other benzimidazoles activate ERK. Additionally, MBZ inhibits DYRK1b which can lead to ERK activation, as well as potently inhibits BRAF which in cells with wild type RAF leads to paradoxical ERK activation.
In cancer MAPK/ERK activation has mostly been associated with tumour growth promotion resulting from overactivity in RAS and RAF mutated signalling pathways12. However, in certain cell types and contexts ERK activation can, on the contrary, be a requirement for inducing cell death29,30. With respect to immune stimulation ERK is necessary for activating immune cells, both T-cells31 and macrophages32, and inducing immune cell mediated tumour cell killing33. For tumours with RAS or BRAF mutations with already maximal intrinsic ERK signalling, ERK mediated immune cell activation may shift the balance towards tumour suppression. However, for some tumour types growth promotion cannot be excluded.
In the immune-oncology setting studies have shown that engagement of PD-1 by PD-L1 inhibit the MEK/ERK MAPK pathway leading to suppression of T-cell activation and proliferation34,35. The effect of PD-1 on MEK/ERK and MAP kinases was found to be selective because PD-1 ligation could not inhibit the activation of JNK and p38 MAP kinases34. Also in macrophages signals from inhibitory receptors can downregulate the MEK/ERK pathway leading to inhibition of macrophage activation and function36. One example is the interaction of CD200 ligand (expressed on tumour cells) with CD200 receptors (CD200R) on myeloid cells which negatively regulates the activation of these cells secondary to MEK/ERK inhibition. CD200R expression is also associated with M2 polarisation of macrophages37. We speculate that MBZ may counteract these immune suppressive effects by restoring ERK activity. We have also previously shown that MBZ induces an M1 phenotype in human macrophage models and potentiates the anti-cancer activity of CD3/IL-2 activated peripheral blood mononuclear cells (PBMCs), the effect being attenuated by removal of CD14+ myeloid cells38.
However, ERK is not always a signal for immune stimulation and inflammation but can in certain cells and situations exert negative feedback on p38/JNK driven inflammation39,40. One explanation for this reciprocal feedback activity has been attributed to MAPK crosstalk. Both increased p38 and JNK activity can inhibit ERK through activation of PP2 and AP-1 transcription, respectively41. ERK activation, on the other hand, increases MKP1 protein stability and availability which in turn causes preferentially de-phosphorylation and inactivation of p38 and JNK39,40. The resulting decrease in p38 and JNK activity downregulates the pro-inflammatory response. ERK has also been shown to inhibit NF42,43. These potential mechanisms are schematically depicted in Supplementary Fig. S4.
Defective ERK signalling has been implicated in some autoimmune diseases including sarcoidosis44 and SLE45,46,47, with strongest evidence presented for the latter disease. For example, inhibition of MEK/ERK signalling by agents such as hydralazine and other MEK inhibitors can induce lupus and lupus-like autoimmune diseases45,47. Furthermore, decreased ERK activity in CD4+ T-cells obtained from SLE patients has been suggested to be a key factor in development of SLE by causing DNA hypomethylation, and consequent aberrant gene expression and disease manifestation45,46,47. The present study supports the potential utility of MBZ for increasing and normalising ERK activity in CD4+ cells from patients with SLE. Another postulated pathogenetic mechanism involves type I interferons released from plasmacytoid dendritic cells (PDC) believed to be a key initiating event in the development of autoimmune disease including SLE48. Indeed also in this case activation of the MEK/ERK pathway inhibits type I interferon release from PDCs49.
Given the mechanistic features suggested here, MBZ may be therapeutically useful also in autoimmune disease driven by Type I-interferons and/or p38 which may be ameliorated by ERK activation, such as systemic sclerosis, myositis, multiple sclerosis, Sjögren’s disease, rheumatoid arthritis and psoriasis48,50,51,52. However, it should be noted that for some of these diseases such as rheumatoid arthritis and multiple sclerosis there are conflicting reports demonstrating that inhibition of the MEK/ERK could ameliorate disease manifestation53,54 suggesting a more complex and different pathophysiology. For example, the pleiotropic proinflammatory cytokine macrophage migration inhibitory factor (MIF) which activates the MEK/ERK pathway is upregulated and centrally implicated in the pathogenesis of both multiple sclerosis and rheumatoid arthritis55,56,57. On the other hand, glatiramer acetate, an immunomodulating drug used for treatment of multiple sclerosis has been shown to activate MEK/ERK which potentially underlies the ability of glatiramer acetate to induce anti-inflammatory IL-1 receptor antagonist production58. Thus, the potential role of MEK/ERK signalling in autoimmune diseases indirectly suggested based on the association with type-I interferon and/or p38 signalling is clearly hypothetical and needs to be confirmed. Notably, only in SLE is there ample experimental evidence for defective ERK signalling being directly linked to disease pathophysiology45,46,47,49. Further supporting SLE as a potential target diagnosis for MBZ we have observed that MBZ significantly inhibits anti-dsDNA production and formation of immune complex deposits in the kidney without noticeable toxicity in the spontaneous NZB/W F1 mouse model of SLE (unpublished preliminary results).
Finally, MBZ has shown clinical activity in the diabetes setting59. In this clinical study MBZ increased insulin secretion and decreased plasma glucose levels in Type 1 and Type 2 diabetic patients59. Follow up experiments on isolated islets demonstrated that MBZ potentiated insulin release in the presence of stimulatory concentrations of glucose60. In the context of ERK activation recent studies may shed light on these observations demonstrating that ERK activation is associated with potentiation of glucose induced insulin secretion61,62.
In conclusion, in the present study we demonstrate that MBZ induced ERK activation is not mimicked by other tubulin active agents and occurs in both haematological and non-haematological cell types. This unique feature of MBZ is suggested to be utilised to treat conditions with defective ERK signalling.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding authors on reasonable request.
References
Sasaki, J.-I. et al. The anthelmintic drug mebendazole induces mitotic arrest and apoptosis by depolymerizing tubulin in non-small cell lung cancer cells. Mol. Cancer Ther. 1, 1201–1209s (2002).
Bai, R.-Y., Staedtke, V., Aprhys, C. M., Gallia, G. L. & Riggins, G. J. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro-oncology 13, 974–982 (2011).
Mukhopadhyay, T., Sasaki, J.-I., Ramesh, R. & Roth, J. A. Mebendazole elicits a potent antitumor effect on human cancer cell lines both in vitro and in vivo. Clin. Cancer Res. 8, 2963–2969 (2002).
Bai, R.-Y., Staedtke, V., Rudin, C. M., Bunz, F. & Riggins, G. J. Effective treatment of diverse medulloblastoma models with mebendazole and its impact on tumor angiogenesis. Neuro-oncology 17, 234–554 (2014).
Doudican, N., Rodriguez, A., Osman, I. & Orlow, S. J. Mebendazole induces apoptosis via Bcl-2 inactivation in chemoresistant melanoma cells. Mol. Cancer Res. 6, 1308–1315 (2008).
Doudican, N. A., Byron, S. A., Pollock, P. M. & Orlow, S. J. XIAP downregulation accompanies mebendazole growth inhibition in melanoma xenografts. Anticancer Drugs 24, 181–188 (2013).
Larsen, A. R. et al. Repurposing the antihelmintic mebendazole as a hedgehog inhibitor. Mol. Cancer Ther. 14, 3–13 (2014).
Nygren, P., Fryknäs, M., Agerup, B. & Larsson, R. Repositioning of the anthelmintic drug mebendazole for the treatment for colon cancer. J. Cancer Res. Clin. Oncol. 139, 2133–2140 (2013).
Nygren, P. & Larsson, R. Drug repositioning from bench to bedside: tumour remission by the antihelmintic drug mebendazole in refractory metastatic colon cancer. Acta Oncol. 53, 427–428 (2014).
Dobrosotskaya, I. Y., Hammer, G. D., Schteingart, D. E., Maturen, K. E. & Worden, F. P. Mebendazole monotherapy and long-term disease control in metastatic adrenocortical carcinoma. Endocr. Pract. 17, e59-62 (2011).
Blom, K. et al. The anticancer effect of mebendazole may be due to M1 monocyte/macrophage activation via ERK1/2 and TLR8-dependent inflammasome activation. Immunopharmacol. Immunotoxicol. 39, 199–210 (2017).
Kim, E. K. & Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405 (2010).
Fan, M. & Chambers, T. C. Role of mitogen-activated protein kinases in the response of tumor cells to chemotherapy. Drug Resist. Updates 4, 253–267 (2001).
Stewart, D. A., Yang, Y., Makowski, L. & Troester, M. A. Basal-like breast cancer cells induce phenotypic and genomic changes in macrophages. Mol. Cancer Res. 10, 727–738 (2012).
Subramanian, A. et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171, 1437-1452.e17 (2017).
Lamb, J. et al. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006).
Kurihara, Y. et al. αVβ3-integrin expression through ERK activation mediates cell attachment and is necessary for production of tumor necrosis factor alpha in monocytic THP-1 cells stimulated by phorbol myristate acetate. Cell. Immunol. 270, 25–31 (2011).
Kabadi, A., McDonnell, E., Frank, C. L. & Drowley, L. Applications of functional genomics for drug discovery. SLAS Discov. https://doi.org/10.1177/2472555220902092 (2020).
Lombardo, S. D. et al. Transcriptomic analysis reveals involvement of the macrophage migration inhibitory factor gene network in duchenne muscular dystrophy. Genes 10, 939–942 (2019).
Stone, A. A. & Chambers, T. C. Microtubule inhibitors elicit differential effects on MAP kinase (JNK, ERK, and p38) signaling pathways in human KB-3 carcinoma cells. Exp. Cell Res. 254, 110–119 (2000).
Brantley-Finley, C. et al. The JNK, ERK and p53 pathways play distinct roles in apoptosis mediated by the antitumor agents vinblastine, doxorubicin, and etoposide. Biochem. Pharmacol. 66, 459–469 (2003).
Fan, M. et al. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. J. Biol. Chem. 275, 29980–29985 (2000).
Braithwaite, P. A., Roberts, M. S., Allan, R. J. & Watson, T. R. Clinical pharmacokinetics of high dose mebendazole in patients treated for cystic hydatid disease. Eur. J. Clin. Pharmacol. 22, 161–169 (1982).
Lu, Y., Chen, J., Xiao, M., Li, W. & Miller, D. D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 29, 2943–2971 (2012).
Blom, K. et al. Mebendazole-induced M1 polarisation of THP-1 macrophages may involve DYRK1B inhibition. BMC Res. Notes 12, 234 (2019).
Gao, J. et al. Mirk/Dyrk1B mediates G0/G1 to S phase cell cycle progression and cell survival involving MAPK/ERK signaling in human cancer cells. Cancer Cell Int. 13, 2 (2013).
Poulikakos, P. I., Zhang, C., Bollag, G., Shokat, K. M. & Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430 (2010).
Callahan, M. K. et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol. Res. 2, 70–79 (2014).
Mebratu, Y. & Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer?. Cell Cycle 8, 1168–1175 (2009).
Cagnol, S. & Chambard, J.-C. ERK and cell death: mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS J. 277, 2–21 (2010).
Gaud, G., Lesourne, R. & Love, P. E. Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol. 18, 485–497 (2018).
Rao, K. M. MAP kinase activation in macrophages. J. Leukoc. Biol. 69, 3–10 (2001).
Radoja, S. et al. CD8(+) tumor-infiltrating T cells are deficient in perforin-mediated cytolytic activity due to defective microtubule-organizing center mobilization and lytic granule exocytosis. J. Immunol. 167, 5042–5051 (2001).
Boussiotis, V. A., Chatterjee, P. & Li, L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. 20, 265–271 (2014).
Dong, Y. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget 8, 2171–2186 (2017).
Ocaña-Guzman, R., Vázquez-Bolaños, L. & Sada-Ovalle, I. Receptors that inhibit macrophage activation: mechanisms and signals of regulation and tolerance. J. Immunol. Res. 2018, 8695157 (2018).
Koning, N. et al. Expression of the inhibitory CD200 receptor is associated with alternative macrophage activation. J. Innate Immun. 2, 195–200 (2010).
Rubin, J. et al. Mebendazole stimulates CD14+ myeloid cells to enhance T-cell activation and tumour cell killing. Oncotarget 9, 30805–30813 (2018).
Kondoh, K. Regulation of MAP kinases by MAP kinase phosphatases. Biochim. Biophys. Acta 1773, 1227–1237 (2007).
Toulouse, A. & Nolan, Y. M. A role for mitogen-activated protein kinase phosphatase 1 (MKP1) in neural cell development and survival. Neural Regen. Res. 10, 1748–1749 (2015).
Junttila, M. R., Li, S.-P. & Westermarck, J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 22, 954–965 (2008).
Maeng, Y.-S. et al. ERK is an anti-inflammatory signal that suppresses expression of NF-kappaB-dependent inflammatory genes by inhibiting IKK activity in endothelial cells. Cell Signal 18, 994–1005 (2006).
Carter, A. B. & Hunninghake, G. W. A constitutive active MEK –> ERK pathway negatively regulates NF-kappa B-dependent gene expression by modulating TATA-binding protein phosphorylation. J. Biol. Chem. 275, 27858–27864 (2000).
Rastogi, R. et al. Dysregulation of p38 and MKP-1 in response to NOD1/TLR4 stimulation in sarcoid bronchoalveolar cells. Am. J. Respir. Crit. Care Med. 183, 500–510 (2011).
Gorelik, G. & Richardson, B. Key role of ERK pathway signaling in lupus. Autoimmunity 43, 17–22 (2010).
Cedeño, S. et al. Defective activity of ERK-1 and ERK-2 mitogen-activated protein kinases in peripheral blood T lymphocytes from patients with systemic lupus erythematosus: potential role of altered coupling of Ras guanine nucleotide exchange factor hSos to adapter protein Grb2 in lupus T cells☆☆Supported by Grant S1–200000440 from Fondo Nacional de Ciencia, Innovación y Tecnologia (FONACIT), Ministerio de Ciencia y Tecnologia. Clin. Immunol. 106, 41–49 (2003).
Deng, C. et al. Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum. 48, 746–756 (2003).
Rönnblom, L. & Eloranta, M.-L. The interferon signature in autoimmune diseases. Curr. Opin. Rheumatol. 25, 248–253 (2013).
Janovec, V. et al. The MEK1/2-ERK pathway inhibits type I IFN production in plasmacytoid dendritic cells. Front. Immunol. 9, 364 (2018).
Crow, M. K. Type I interferon in organ-targeted autoimmune and inflammatory diseases. Arthritis Res. Ther. 12(Suppl 1), S5 (2010).
Saklatvala, J. The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr. Opin. Pharmacol. 4, 372–377 (2004).
Yao, Y. et al. Type I interferon: Potential therapeutic target for psoriasis?. PLoS ONE 3, e2737 (2008).
Brereton, C. F., Sutton, C. E., Lalor, S. J., Lavelle, E. C. & Mills, K. H. G. Inhibition of ERK MAPK suppresses IL-23- and IL-1-driven IL-17 production and attenuates autoimmune disease. J. Immunol. 183, 1715–1723 (2009).
Thiel, M. J. et al. Central role of the MEK/ERK MAP kinase pathway in a mouse model of rheumatoid arthritis: potential proinflammatory mechanisms. Arthritis Rheum. 56, 3347–3357 (2007).
Morand, E. F. & Leech, M. Macrophage migration inhibitory factor in rheumatoid arthritis. Front. Biosci. 10, 12–22 (2005).
Cavalli, E. et al. Upregulated expression of macrophage migration inhibitory factor, its analogue D-dopachrome tautomerase, and the CD44 receptor in peripheral CD4 T cells from clinically isolated syndrome patients with rapid conversion to clinical defined multiple sclerosis. Medicina 55, 667–672 (2019).
Fagone, P. et al. Contribution of the macrophage migration inhibitory factor superfamily of cytokines in the pathogenesis of preclinical and human multiple sclerosis: In silico and in vivo evidences. J. Neuroimmunol. 322, 46–56 (2018).
Carpintero, R. et al. Glatiramer acetate triggers PI3Kδ/Akt and MEK/ERK pathways to induce IL-1 receptor antagonist in human monocytes. Proc. Natl. Acad. Sci. USA 107, 17692–17697 (2010).
Caprio, S. et al. Improvement of metabolic control in diabetic patients during mebendazole administration: preliminary studies. Diabetologia 27, 52–55 (1984).
Owen, O. E. et al. Mebendazole and insulin secretion from isolated rat islets. Metab. Clin. Exp. 34, 567–570 (1985).
Youl, E. et al. Quercetin potentiates insulin secretion and protects INS-1 pancreatic β-cells against oxidative damage via the ERK1/2 pathway. Br. J. Pharmacol. 161, 799–814 (2010).
Bowe, J. E., Chander, A., Liu, B., Persaud, S. J. & Jones, P. M. The permissive effects of glucose on receptor-operated potentiation of insulin secretion from mouse islets: a role for ERK1/2 activation and cytoskeletal remodelling. Diabetologia 56, 783–791 (2013).
Acknowledgements
The study was supported by the Swedish Cancer Society, the Swedish Research Council and the Lions Cancer Research Fund. Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Contributions
Experimental design: R.L., K.B., P.N., M.F., A.L., V.P., C.A. Experimental work: K.B., M.B., M.J., L.L., J.R., T.S. Data analysis: C.A., M.B., R.L. Writing manuscript: R.L., C.A., P.N., A.L. Reviewing manuscript: All authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
R.L., P.N. and M.F. are co-founders and shareholders of Repos Pharma AB, a small Swedish research and development company dedicated to investigations of drug repositioning in the cancer area. AL is chairman of the board. The present work was funded only by academic grants. The remaining authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Andersson, C.R., Selvin, T., Blom, K. et al. Mebendazole is unique among tubulin-active drugs in activating the MEK–ERK pathway. Sci Rep 10, 13124 (2020). https://doi.org/10.1038/s41598-020-68986-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-68986-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
