
Abstract
Elevation of total cell-free DNA (cfDNA) in patients with preeclampsia is well-known; however, whether this change precedes the onset of symptoms remains inconclusive. Here, we conducted a nested case–control study to determine the elevation of cfDNA levels in women who subsequently developed preeclampsia. Methylated HYP2 (m-HYP2) levels were determined in 68 blood samples collected from women with hypertensive disorders of pregnancy, along with 136 control samples, using real-time quantitative PCR. The measured m-HYP2 levels were converted to multiples of the median (MoM) values for correction of maternal characteristics. The m-HYP2 levels and MoM values in patients with preeclampsia were significantly higher than in controls during the third trimester (P < 0.001, both), whereas those for women who subsequently developed preeclampsia did not differ during the second trimester. However, when patients with preeclampsia were divided based on the onset-time of preeclampsia or 10th percentile birth weight, both values were significantly higher in women who subsequently developed early-onset preeclampsia (P < 0.05, both) and preeclampsia with small-for-gestational-age (SGA) neonate (P < 0.01, both) than controls. These results suggested that total cfDNA levels could be used to predict early-onset preeclampsia or preeclampsia with SGA neonate.
Similar content being viewed by others


Introduction
Hypertensive disorders of pregnancy (HDP) are classified into four categories: chronic hypertension, preeclampsia (PE), PE superimposed on chronic hypertension, and gestational hypertension (GH). PE complicates 2–8% of pregnancies and is one of the leading causes of maternal and fetal morbidity and mortality1,2. In recent meta-analyses, aspirin prophylaxis was found to be associated with a significant risk reduction of PE in high-risk patients3,4. Therefore, early prediction would be important for proper management.
Many researchers have suggested diverse predictors, such as maternal characteristics (age, body mass index, nulliparity, multiple pregnancy, and previous history of PE), biophysical markers (uterine artery Doppler, blood pressure) or biochemical markers (pregnancy-associated plasma protein-A, vascular endothelial growth factor, placental growth factor, soluble fms-like tyrosine kinase 1, and soluble endoglin) for early prediction of PE5,6,7,8,9,10,11. Cell-free DNA (cfDNA) circulating in the maternal blood is also a candidate biomarker12.
It may be maternal or fetal in origin. Fetal cfDNA is shed from the syncytiotrophoblast as an apoptotic fragment during normal cell turnover, and released into maternal circulation. Although the mechanisms of PE are not completely understood, altered apoptosis is known to be involved in its pathogenesis13. Various authors have observed elevations in fetal cfDNA level, during the first and second trimesters, in patients who subsequently developed PE14,15,16, and suggested cfDNA as a predictive marker for early-onset PE or ‘any PE’17.
A poorly perfused placenta in patients with PE may release circulating factors into maternal circulation, causing damage to maternal vascular endothelial cells and leading to multi-system dysfunction17. The clinical syndrome of PE is a consequence of a wide systemic inflammatory response, and systemic inflammation is associated with the release of cfDNA into circulation18. Several studies have shown the amount of total cfDNA to be significantly elevated in patients with PE19,20,21. However, whether total cfDNA is elevated before symptom onset still remains unclear. A few previous studies had shown total cfDNA to be increased in patients with PE during the first or second trimester22,23,24, whereas a recent case–control study of a relatively large sample size demonstrated the total cfDNA levels to not be increased in the first trimester25.
The aim of this study was to determine whether the concentrations of total cfDNA in blood are increased in women during the second trimester of pregnancy who subsequently develop HDP during the third trimester. We used the methylated HYP2 gene as a total cfDNA marker to verify whether total cfDNA levels could be used to predict PE.
Results
Clinical characteristics of the study population
Clinical characteristics of the study population are presented in Table 1. There was no significant difference in nulliparity and gestational ages at the time of sampling across the groups (P > 0.05 for all). However, body mass index was significantly higher, and gestational age at delivery and birth weight were significantly lower in all patient groups compared to those in the control group (P < 0.05 for all). The maternal age of patient groups differed in the second trimester compared to that in the control group, but the difference was not observed in the third trimester.
Comparison of methylated HYP2 levels and multiples of the median (MoM) values between women with hypertensive disorders of pregnancy and controls
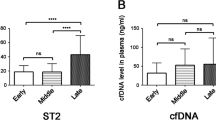
Comparison of methylated HYP2 levels in the maternal plasma between specific patient groups and controls is shown in Table 2. The methylated HYP2 levels and MoM values of the patients with PE were significantly higher than those of normal controls during the third trimester (P < 0.0001, both). In contrast, during the second trimester, the methylated HYP2 concentrations and MoM values of pregnant women who subsequently developed PE, and that of controls, were not significantly different. Nevertheless, when the patients were divided based on the onset time of PE, methylated HYP2 levels and MoM values were significantly higher in patients with early-onset PE than in control subjects (P = 0.042 and 0.044, respectively) (Table 3). Furthermore, when we divided the patients with PE based on the 10th percentile birth weight, both the median methylated HYP2 concentrations and MoM values were significantly higher in patients who subsequently developed PE, and having a small-for-gestational-age (SGA) neonate, than in patients who subsequently developed PE without SGA neonate (P = 0.032 and 0.034, respectively) and in control subjects (P = 0.008 and 0.009, respectively) (Table 4). However, these differences were not significant in patients who subsequently developed GH (Table 5). Additionally, we determined the relationship between total cfDNA levels and gestational age at sampling in patients and controls, according to onset-time of PE and accompanying SGA neonate, as shown in Fig. 1. The total cfDNA, in patients with early-onset PE and PE with SGA, was elevated early in the second trimester.

Relationship between total cell-free DNA levels and gestational age at sampling according to (a) onset-time of preeclampsia and (b) small-for-gestational-age neonate. GH gestational hypertension, PE preeclampsia, SGA small for gestational age.
Discussion
This study demonstrated that, during the second trimester, the concentrations of total cfDNA in patients who subsequently developed PE and GH were not significantly different from those in control subjects. However, the elevation became significant when the patient group was limited to early-onset PE or PE with SGA neonate.
Several studies have shown that fetal cfDNA is increased in patients who subsequently developed PE, and the elevation often occurs in the first trimester15. However, a few authors have reported the fetal fraction to be low in these patients, as observed during cfDNA screening for aneuploidy26,27. Rolnik et al. had indicated that the apparently discrepant results (lower fetal fraction as opposed to increase in absolute quantities of fetal cfDNA) may be due to a less-pronounced increase of fetal cfDNA than of maternal cfDNA, with a consequent reduction in fetal fraction26. In a previous study by our group, Kim et al.24 had shown that the combination of fetal cfDNA, total cfDNA, and pregnancy associated plasma protein-A can be a useful predictor for PE during the first trimester. In that study, we had demonstrated the total cfDNA levels to be significantly higher in patients who subsequently developed PE at 6–14 and 15–23 gestational weeks, using the HYP2 gene as a marker of total cfDNA. In contrast, Rolnik et al.28 had found a significant increase in the median total cfDNA measured at 11–13 gestational weeks in the early-onset PE group; however, the significance was not observed when the values were corrected for maternal characteristics and gestational age. Furthermore, neither the median total cfDNA nor the MoM values in the late-onset PE group differed from those in controls at 11–13 and 20–24 gestational weeks.
The proportion of early-onset PE is known to be approximately 20%, and around 30% of fetuses born to pregnant women with PE are below the 10th percentile birth weight29. Similarly, in our study, the proportion of early-onset PE was 20%, and that of PE with SGA neonate was 37% during the second trimester. Therefore, the low proportion of early-onset PE or PE with SGA neonate may explain the lack of significant differences. In contrast, the opposite result in previous studies may have been due to the higher proportion of such patients in the study population.
Early-onset PE is considered to be somewhat distinct from late-onset PE. The former is typically associated with placental dysfunction, reduction in placental volume, intrauterine growth restriction (IUGR), and adverse neonatal outcomes; In contrast, the latter is more often associated with normal placenta, normal fetal growth, and more favorable outcomes30. In this context, several authors have indicated PE as an etiologically heterogeneous disorder that occurs in at least two subsets, one with normal placental function and another involving placental dysfunction31,32.
There is a strong correlation between placental dysfunction and fetal growth restriction. IUGR is assigned to infants with a birth weight below 10th percentile for gestational age, having a pathologic restriction on fetal growth due to adverse genetic or environmental influences33. Therefore, SGA neonate complicated with PE can be regarded as IUGR. In a prospective study, Milosevic-Stevanovic et al. had shown that placental thickness and weight in patients with PE were significantly different depending on the presence or absence of IUGR. In histopathologic analysis, villous hypermaturity was more frequently observed in the placentas of patients with PE and IUGR34. Therefore, the elevation of total cfDNA levels in patients, who subsequently develop PE with IUGR, may be associated with hypermaturation of the placental villi.
The elevation of total cfDNA in patients with PE is thought to be associated with increased neutrophil extracellular trap (NET) production by their neutrophils. NET formation is a defense mechanism in which neutrophils are deployed as an alternative to phagocytosis35. Gupta et al. had found that a huge number of NETs was present in the intervillous space of preeclamptic placentae36. These NETs appear to be triggered by elevated release of placental micro-debris and may contribute to widespread systemic damage to the maternal endothelium37,38. NETs have been identified in both early- and late-onset PE39. However, it is currently unclear whether NETs appear prior to the onset of symptoms. Our results implied that, NETs may occur during the preclinical period, although they are limited in preeclamptic patients with placental dysfunction such as early-onset PE or PE with IUGR.
The elevation of total cfDNA may be associated with IUGR due to placental insufficiency regardless of PE. Crowley et al.40 had demonstrated that total cfDNA levels are significantly elevated in women with IUGR before 20 weeks of gestation, but not in women with PE. Thereafter, Al Nakib et al.41 showed that total cfDNA concentrations are significantly elevated in pregnant women with IUGR due to placental insufficiency, but not due to other causes of IUGR.
The pathophysiology of IUGR is similar to that of PE, and is associated with abnormal placentation, chronic utero-placental ischemia, increased trophoblast apoptosis, and enhanced maternal systemic inflammatory response42. In addition, both PE and IUGR promote endothelial cell dysfunction. Formanowicz et al. had reported that the sera collected from women with IUGR and IUGR with PE show a detrimental effect on endothelial cells, reducing their viability and proliferation, and generating oxidative stress owing to dysfunctional mitochondria43. Moreover, a few studies had demonstrated cfDNA to serve as an auxiliary biomarker of vascular endothelial dysfunction44,45. Therefore, the elevation of total cfDNA before symptom onset, in our patients with early-onset PE and PE with SGA neonate, may be associated with endothelial cell dysfunction. In women with a history of PE, maternal vascular dysfunction may persist for years46,47, and the risks of hypertension, cardiovascular disease, stroke, and end-stage renal disease may be increased later in life48,49,50. Yinon et al. had observed reduced flow-mediated dilatation and increased arterial stiffness, in women with previous early-onset PE and in women with previous IUGR without PE, 6–24 months postpartum. In contrast, women with a history of late-onset PE exhibited normal flow-mediated dilatation similar to the control subjects51. These findings can be explained by our results. We are not sure whether endothelial damage during pregnancy is the main cause of impaired maternal vascular function in postpartum women. However, a relatively longer period of endothelial cell dysfunction may worsen maternal vascular function in postpartum period, and increased total cfDNA level during the preclinical period, in patients with early-onset PE and PE with IUGR, may indicate the progression of endothelial cell dysfunction.
American College of Obstetrician and Gynecologists guidelines (2018) recommend that low-dose aspirin prophylaxis in women at high risk of PE should be initiated between 12 and 28 weeks of gestation and continued daily until delivery52. However, a few studies indicated that aspirin treatment reduces the risk of early-onset PE, but not term PE53,54. In this context, prediction of early-onset PE is important, since it may contribute to the identification of women who are most likely to respond to low-dose aspirin. In contrast, low-dose aspirin initiated after 16 weeks of gestation may not be as effective in reducing the risk of PE and fetal growth restriction3,53. Therefore, further studies on pregnant women before 16 weeks of gestation would be recommended.
In this study, we included only Korean pregnant women from a single center, and only non-smokers were included in the patient groups. Although this study has limitations its relatively small sample sizes, the homogeneity of our study population may compensate this weakness by minimizing influences from other causes, which can affect total cfDNA levels in maternal blood.
In conclusion, total cfDNA levels were significantly elevated in patients with PE during the third trimester regardless of the onset time of PE or whether the neonate was SGA. However, in the absence of symptoms during the second trimester, elevation of total cfDNA levels was observed only in patients with early-onset PE or PE and SGA neonate. In addition, total cfDNA levels in patients with PE and SGA neonate were significantly higher than in those with PE without SGA neonate. It supports the notion that PE with and without IUGR are two pathogenetically different entities. In future, well-designed studies would be required to confirm the elevation of total cfDNA, in patients with early-onset PE and in patients with IUGR due to placental insufficiency, before 16 weeks of gestation, and to identify the correlation with maternal vascular function in postpartum period. Simultaneously, based on our findings, efforts should continue toward better prediction of PE and IUGR during early pregnancy.
Methods
Study participants and samples
We performed a nested case–control study of women with singleton pregnancies who received routine prenatal care at the Department of Obstetrics and Gynecology at Cheil General Hospital between August 2010 and August 2014. This study was approved by the Institutional Review Board and Ethics Committee of Cheil General Hospital (#CGH-IRB-2013-54), and informed consent was obtained from all study participants prior to the study. All experiments were performed in accordance with the relevant guidelines and regulations. Maternal blood samples were prospectively collected when the participants underwent a routine blood test or were admitted for the management of hypertensive disorders of pregnancy at 15–19, 24–28, and 33–41 weeks according to our study protocol. We selected 68 patients, who were diagnosed with PE or GH in our hospital and delivered their baby in the third trimester, and 136 normal controls without medical or obstetric complications. PE was defined as hypertension (systolic blood pressure ≥ 140 mmHg and/or diastolic blood pressure ≥ 90 mmHg, twice, 4 h apart) and proteinuria (≥ 0.3 g/day urine collection and/or ≥ 1 + on dipstick testing) after 20 weeks of gestation. GH is a new-onset hypertension that occurs after 20 weeks of gestation without proteinuria. Early-onset PE was defined as PE diagnosed before 34 weeks of gestation, and late-onset PE was considered if it was diagnosed at or after 34 weeks. SGA was defined as birth weight below the 10th percentile55.
Laboratory analysis
DNA extraction, methylated DNA enrichment, and real-time quantitative PCR were performed as described in our previous study24. Maternal blood samples (10 mL) were collected in EDTA tubes and were immediately centrifuged at 1,600×g for 10 min at 4 °C. The supernatant plasma was re-centrifuged at 16,000×g for 10 min at 4 °C and aliquoted into 1 mL for circulating cfDNA extraction. Circulating cfDNA was extracted using the QIAamp DSP Virus Kit (Qiagen Hilden, Germany). The MethylMiner™ methylated DNA enrichment kit (Invitrogen, Carlsbad, CA., USA) with methyl-CpG binding domain (MBD) biotin protein was used to isolate methylated cfDNA from that extracted from maternal plasma. Finally, the isolated methylated cfDNA was concentrated using a DNA concentrator (Zymo Research Corp., Irvine, CA, USA) and then eluted in a final volume of 30 μL. Enrichment of methylated DNA enrichment was validated using control DNA (methylated and unmethylated DNA) included in the kit according to the manufacturer’s recommendations.
We measured the levels of total cfDNA by real-time quantitative PCR in all samples without failure of MBD capture. Quantification of the methylated HYP2 gene as an epigenetic marker of total cfDNA was performed in duplex reactions. Real-time quantitative PCR amplification was performed using the ABI 7500 Real Time System (Applied Biosystems, Foster City, CA, USA). Duplex reactions were prepared in a volume of 20 μL, using 5 μL of 4 × NEXpro qRT-PCR Master Mix (Geneslabs, Seongnam, Korea) and 6 μL of the methylated plasma DNA captured by MBD. Primers and probes were both used at a final concentration of 250 nM for HYP2. A standard curve using serial dilutions of single-stranded synthetic DNA oligonucleotides specific to the HYP2 amplicons (Bioneer, Daejeon, Korea) was employed. Each standard was amplified in triplicate and included in every PCR plate. All samples were amplified in triplicate and the final data reflected average of the results.
Comparison of methylated HYP2 levels between patients and controls
We converted the measured levels of methylated HYP2 to MoM values to correct for maternal characteristics, such as gestational age and maternal body mass index at the time of sampling, to increase the statistical power and compared the levels of methylated HYP2 and MoM values between patient groups (PE and GH) and controls. For further analysis, patients with PE were divided into subgroups according to the onset time of PE or diagnosis of SGA neonate, and the values of each group of patients (i.e. early-onset or late-onset PE, and PE with or without SGA neonate) were compared with those of controls.
Statistical analyses
Values are presented as frequencies (percentages) or medians (interquartile ranges), as appropriate. The three groups were compared using the Chi-square test or Fisher’s exact test for categorical variables, and the Kruskal–Wallis test were performed to compare continuous variables. If the Kruskal–Wallis test was significant, pair-wise comparisons of the three groups were performed using the Wilcoxon rank sum test with the step-up Bonferroni method. The MoM values of methylated HYP2 levels were calculated by dividing the expected methylated HYP2 levels by the actual measured methylated HYP2 levels. The expected methylated HYP2 level was calculated by quantile regression, which aimed at estimating the conditional median of dependent variable. In all tests, a threshold of P < 0.05 was defined as statistically significant. Statistical analyses were performed using SAS version 9.4 software (SAS, Inc., Cary, NC, USA; https://www.sas.com/) and R 3.4.1 (Vienna, Austria; https://www.R-project.org/).
References
Sibai, B. M. Diagnosis and management of gestational hypertension and preeclampsia. Obstet Gynecol. 102, 181–192 (2003).
Steegers, E. A., von Dadelszen, P., Duvekot, J. J. & Pijnenborg, R. Pre-eclampsia. Lancet 376, 631–644 (2010).
Roberge, S. et al. The role of aspirin dose on the prevention of preeclampsia and fetal growth restriction: systematic review and meta-analysis. Am. J. Obstet. Gynecol. 216, 110–120 (2017).
Meher, S., Duley, L., Hunter, K. & Askie, L. Antiplatelet therapy before or after 16 weeks’ gestation for preventing preeclampsia: an individual participant data meta-analysis. Am. J. Obstet. Gynecol. 216, 121–128 (2017).
Bartsch, E. et al. Clinical risk factors for pre-eclampsia determined in early pregnancy: systematic review and meta-analysis of large cohort studies. BMJ 353, i1753 (2016).
Velauthar, L. et al. First-trimester uterine artery Doppler and adverse pregnancy outcome: a meta-analysis involving 55,974 women. Ultrasound Obstet. Gynecol. 43, 500–507 (2014).
Mabuchi, A. et al. Significance of high-normal blood pressure during early second trimester for predicting the onset of hypertensive disorders in pregnancy. Hypertens. Pregnancy 35, 234–241 (2016).
Lim, J. H. et al. Effective prediction of preeclampsia by a combined ratio of angiogenesis-related factors. Obstet. Gynecol. 111, 1403–1409 (2008).
Lim, J. H. et al. Soluble endoglin and transforming growth factor-beta1 in women who subsequently developed preeclampsia. Prenat. Diagn. 29, 471–476 (2009).
Kleinrouweler, C. E. et al. Accuracy of circulating placental growth factor, vascular endothelial growth factor, soluble fms-like tyrosine kinase 1 and soluble endoglin in the prediction of pre-eclampsia: a systematic review and meta-analysis. BJOG 119, 778–787 (2012).
Poon, L. C. & Nicolaides, K. H. First-trimester maternal factors and biomarker screening for preeclampsia. Prenat. Diagn. 34, 618–627 (2014).
Levy, R. The role of apoptosis in preeclampsia. Isr. Med. Assoc. J. 7, 178–181 (2005).
Zhong, X. Y., Holzgreve, W. & Hahn, S. The concentrations of circulatory fetal DNA in maternal plasma are elevated prior to the onset of preeclampsia. Hypertens. Pregnancy 21, 77–83 (2002).
Farina, A. et al. Fetal DNA in maternal plasma as a screening variable for preeclampsia. A preliminary nonparametric analysis of detection rate in low-risk nonsymptomatic patients. Prenat. Diagn. 24, 83–86 (2004).
Sifakis, S., Zaravinos, A., Maiz, N., Spandidos, D. A. & Nicolaides, K. H. First-trimester maternal plasma cell-free fetal DNA and preeclampsia. Am. J. Obstet. Gynecol. 201(472), e1-7 (2009).
Kim, M. J. et al. Association of fetal-derived hypermethylated RASSF1A concentration in placenta-mediated pregnancy complications. Placenta 34, 57–61 (2013).
Contro, E., Bernabini, D. & Farina, A. Cell-free fetal DNA for the prediction of pre-eclampsia at the first and second trimesters: a systematic review and meta-analysis. Mol. Diagn. Ther. 21, 125–135 (2017).
van der Meer, A. J. et al. Systemic inflammation induces release of cell-free DNA from hematopoietic and parenchymal cells in mice and humans. Blood Adv. 3, 724–728 (2019).
Sekizawa, A. et al. Cell-free fetal DNA in the plasma of pregnant women with severe fetal growth restriction. Am. J. Obstet. Gynecol. 188, 480–484 (2003).
Lazar, L. et al. Relationship of circulating cell-free DNA levels to cell-free fetal DNA levels, clinical characteristics and laboratory parameters in preeclampsia. BMC Med. Genet. 10, 120 (2009).
Rafaeli-Yehudai, T. et al. Maternal total cell-free DNA in preeclampsia and fetal growth restriction: evidence of differences in maternal response to abnormal implantation. PLoS ONE 13, e0200360 (2018).
Salvianti, F. et al. Prospective evaluation of RASSF1A cell-free DNA as a biomarker of preeclampsia. Placenta 36, 996–1001 (2015).
Farina, A. et al. Total cell-free DNA (beta-globin gene) distribution in maternal plasma at the second trimester: a new prospective for preeclampsia screening. Prenat. Diagn. 24, 722–726 (2004).
Kim, S. Y. et al. Early prediction of hypertensive disorders of pregnancy using cell-free fetal DNA, cell-free total DNA, and biochemical markers. Fetal Diagn. Ther. 40, 255–262 (2016).
Silver, R. M. et al. Cell-free total and fetal DNA in first trimester maternal serum and subsequent development of preeclampsia. Am. J. Perinatol. 34, 191–198 (2017).
Rolnik, D. L., da Silva Costa, F., Lee, T. J., Schmid, M. & McLennan, A. C. Association between fetal fraction on cell-free DNA testing and first-trimester markers for pre-eclampsia. Ultrasound Obstet. Gynecol. 52, 722–727 (2018).
Genson, K. D. et al. Low fetal fraction of cell-free DNA predicts placental dysfunction and hypertensive disease in pregnancy. Pregnancy Hypertens. 16, 148–153 (2019).
Rolnik, D. L. et al. Maternal plasma cell-free DNA in the prediction of preeclampsia. Ultrasound Obstet. Gynecol. 45, 106–111 (2015).
Eskenazi, B., Fenster, L., Sydney, S. & Elkin, E. P. Fetal growth retardation in infants of multiparous and nulliparous women with preeclampsia. Am. J. Obstet. Gynecol. 169, 1112–1118 (1993).
Raymond, D. & Peterson, E. A critical review of early-onset and late-onset preeclampsia. Obstet. Gynecol. Surv. 66, 497–506 (2011).
Rasmussen, S. & Irgens, L. M. Fetal growth and body proportion in preeclampsia. Obstet. Gynecol. 101, 575–583 (2003).
Mayhew, T. M., Wijesekara, J., Baker, P. N. & Ong, S. S. Morphometric evidence that villous development and fetoplacental angiogenesis are compromised by intrauterine growth restriction but not by pre-eclampsia. Placenta 25, 829–833 (2004).
Wollmann, H. A. Intrauterine growth restriction: definition and etiology. Horm. Res. 49(Suppl 2), 1–6 (1998).
Milosevic-Stevanovic, J. et al. Preeclampsia with and without intrauterine growth restriction-two pathogenetically different entities?. Hypertens. Pregnancy 35, 573–582 (2016).
Branzk, N. et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 15, 1017–1025 (2014).
Gupta, A., Hasler, P., Gebhardt, S., Holzgreve, W. & Hahn, S. Occurrence of neutrophil extracellular DNA traps (NETs) in pre-eclampsia: a link with elevated levels of cell-free DNA?. Ann. N. Y. Acad. Sci. 1075, 118–122 (2006).
Goswami, D. et al. Syncytiotrophoblast microparticle shedding is a feature of early onset pre-eclampsia but not normotensive intrauterine growth restriction. Placenta 27, 56–61 (2006).
Messarli, M. et al. Fetomaternal interactions in pregnancies: placental microparticles activate peripheral blood monocytes. Placenta 31, 106–112 (2010).
Gupta, A. K., Gebhardt, S., Hillermann, R., Holzgreve, W. & Hahn, S. Analysis of plasma elastase levels in early and late onset preeclampsia. Arch. Gynecol. Obstet. 273, 239–242 (2006).
Crowley, A. et al. Free fetal DNA is not increased before 20 weeks in intrauterine growth restriction or pre-eclampsia. Prenat. Diagn. 27, 174–179 (2007).
Al Nakib, M. et al. Total and fetal cell-free DNA analysis in maternal blood as markers of placental insufficiency in intrauterine growth restriction. Fetal Diagn. Ther. 26, 24–28 (2009).
Ogge, G. et al. Leukocytes of pregnant women with small-for-gestational age neonates have a different phenotypic and metabolic activity from those of women with preeclampsia. Matern. Fetal Neonatal Med. 23, 476–487 (2010).
Formanowicz, D. et al. Preeclampsia with intrauterine growth restriction generates morphological changes in endothelial cells associated with mitochondrial swelling—an in vitro study. J. Clin. Med. 8, 1994 (2019).
Pokrywka, A. et al. The influence of hypoxic physical activity on cfDNA as a new marker of vascular inflammation. Arch. Med. Sci. 11, 1156–1163 (2015).
Borissoff, J. I. et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler. Thromb. Vasc Biol. 33, 2032–2040 (2013).
Agatisa, P. K. et al. Impairment of endothelial function in women with a history of preeclampsia: an indicator of cardiovascular risk. Am. J. Physiol. Heart Circ. Physiol. 286, H1389–H1393 (2004).
Germain, A. M. et al. Endothelial dysfunction: a link among preeclampsia, recurrent pregnancy loss, and future cardiovascular events?. Hypertension. 49, 90–95 (2007).
Vikse, B. E., Irgens, L. M., Leivestad, T., Skjaerven, R. & Iversen, B. M. Preeclampsia and the risk of end-stage renal disease. N. Engl. J. Med. 359, 800–809 (2008).
Wilson, B. J. et al. Hypertensive diseases of pregnancy and risk of hypertension and stroke in later life: results from cohort study. BMJ 326, 845 (2003).
Ray, J. G., Vermeulen, M. J., Schull, M. J. & Redelmeier, D. A. Cardiovascular health after maternal placental syndromes (CHAMPS): population-based retrospective cohort study. Lancet 366, 1797–1803 (2005).
Yinon, Y. et al. Vascular dysfunction in women with a history of preeclampsia and intrauterine growth restriction: insights into future vascular risk. Circulation 122, 1846–1853 (2010).
ACOG. ACOG committee opinion no. 743: low-dose aspirin use during pregnancy. Obstet. Gynecol. 132, e44–e52 (2018).
Roberge, S., Bujold, E. & Nicolaides, K. H. Aspirin for the prevention of preterm and term preeclampsia: systematic review and meta-analysis. Am. J. Obstet. Gynecol. 218, 287–293 (2018).
Atallah, A. et al. Aspirin and preeclampsia. Presse Med. 48, 34–45 (2019).
Lim, J. S. et al. New Korean reference for birth weight by gestational age and sex: data from the Korean Statistical Information Service (2008–2012). Ann. Pediatr. Endocrinol. Metab. 19, 146–153 (2014).
Acknowledgements
This work was funded by a Grant (HI17C0778) from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea.
Author information
Authors and Affiliations
Contributions
D.W.K. contributed to the conception and design of the study, analysis and interpretation of data, and drafting of the article. S.Y.K. and H.J.K. helped with the performance of experiments and analysis of data. J.H.L. revised the manuscript for critically important intellectual content. Y.H.K. made substantial contributions to the concept and design of the study and gave final approval of the version to be published. H.M.R. collected samples, made substantial contributions to the concept and design, and gave final approval of the version to be published.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kwak, D.W., Kim, S.Y., Kim, H.J. et al. Maternal total cell-free DNA in preeclampsia with and without intrauterine growth restriction. Sci Rep 10, 11848 (2020). https://doi.org/10.1038/s41598-020-68842-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-68842-1
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
