
Abstract
Breast cancer (BC), as one of the leading causes of death among women, comprises several subtypes with controversial and poor prognosis. Considering the TNM (tumor, lymph node, metastasis) based classification for staging of breast cancer, it is essential to diagnose the disease at early stages. The present study aims to take advantage of the systems biology approach on genome wide gene expression profiling datasets to identify the potential biomarkers involved at stage I, stage II, stage III, and stage IV as well as in the integrated group. Three HER2-negative breast cancer microarray datasets were retrieved from the GEO database, including normal, stage I, stage II, stage III, and stage IV samples. Additionally, one dataset was also extracted to test the developed predictive models trained on the three datasets. The analysis of gene expression profiles to identify differentially expressed genes (DEGs) was performed after preprocessing and normalization of data. Then, statistically significant prioritized DEGs were used to construct protein–protein interaction networks for the stages for module analysis and biomarker identification. Furthermore, the prioritized DEGs were used to determine the involved GO enrichment and KEGG signaling pathways at various stages of the breast cancer. The recurrence survival rate analysis of the identified gene biomarkers was conducted based on Kaplan–Meier methodology. Furthermore, the identified genes were validated not only by using several classification models but also through screening the experimental literature reports on the target genes. Fourteen (21 genes), nine (17 genes), eight (10 genes), four (7 genes), and six (8 genes) gene modules (total of 53 unique genes out of 63 genes with involving those with the same connectivity degree) were identified for stage I, stage II, stage III, stage IV, and the integrated group. Moreover, SMC4, FN1, FOS, JUN, and KIF11 and RACGAP1 genes with the highest connectivity degrees were in module 1 for abovementioned stages, respectively. The biological processes, cellular components, and molecular functions were demonstrated for outcomes of GO analysis and KEGG pathway assessment. Additionally, the Kaplan–Meier analysis revealed that 33 genes were found to be significant while considering the recurrence-free survival rate as an alternative to overall survival rate. Furthermore, the machine learning calcification models show good performance on the determined biomarkers. Moreover, the literature reports have confirmed all of the identified gene biomarkers for breast cancer. According to the literature evidence, the identified hub genes are highly correlated with HER2-negative breast cancer. The 53-mRNA signature might be a potential gene set for TNM based stages as well as possible therapeutics with potentially good performance in predicting and managing recurrence-free survival rates at stages I, II, III, and IV as well as in the integrated group. Moreover, the identified genes for the TNM-based stages can also be used as mRNA profile signatures to determine the current stage of the breast cancer.
Similar content being viewed by others

Introduction
Breast cancer (BC) is one of the most common health threatening problems among women in the world, leading to death of those patients with BC1. It has been reported in 2019 that the incidence and mortality of breast cancer worldwide are 24.2% and 15.0%, respectively, deserving more attention from healthcare systems and policy-makers1. To clinically classify the status of breast cancer, the American Joint Committee on Cancer (AJCC) has announced eight editions on the Tumor-Node-Metastasis (TNM)-based staging of breast cancer, specifically for treatment and prognosis2,3. Since more than 50% of the affected patients were died, increasing the survival rate of these patients is highly important by determining the stage of the disease. The earlier the identification of the stage, the more superior the survival rate. To increase the therapeutic efficiency and consider the molecular portrait differences in BC along with their different clinical outcomes4, breast cancer can be classified into six main subtypes, including normal-like, luminal A, luminal B, HER2-positive, basal-like, and claudin-low5; the classification has also been confirmed by the Cancer Genome Atlas (TCGA) program6.
It has been frequently reported that the human epidermal growth factor receptor (HER) family (i.e., HER-1, HER-2, HER-3, and HER-4) plays a pivotal role in various cancers7. Among them, HER-2 (known as HER-2/neu gene), as an oncogene with 1,255 amino acids and 185kD transmembrane glycoprotein with tyrosine kinase activity, is located at chromosome 177,8. Moreover, HER-2/neu gene makes breast cancer classified as HER2-positive and HER2-negative9. In 15–30% of patients with invasive breast carcinomas, an overexpression or amplification of HER2 has been identified7,10.
It is worth mentioning that is not effective for HER2-negative. Although, endocrine therapy is the target of chemotherapy, there are no successful reports for survival rates of these types of patients in the literature11. Moreover, several traditional diagnostic approaches such as mammography, magnetic resonance imaging (MRI), ultrasound, computerized tomography (CT), positron emission tomography (PET), and biopsy have been studied in breast cancer diagnosis12.
Nowadays, molecular biomarkers have been proposed to provide more efficiency in the prognosis and diagnosis of cancers in deficiency of traditional cancer tests. Additionally, the biomarkers are now regularly utilized to better understand the development of the tumors13. Hence, owing to the large number of stored microarray gene expression profiles by several genomics laboratories in the most publicly available database websites such as National Center for Biotechnology Information (NCBI), their analyses by various bioinformatics and systems biology analyses are essential4. Finally, these biomarkers will be helpful in personalizing the treatments for each patient with their special stage of the disease4. Considering the HER2-targeted therapy, there are still no predictive biomarkers validated for the prognosis and diagnosis of the stages of breast cancer14,15.
Consequently, the aim of the current study is to identify the potential biomarkers in breast cancer at stages I, II, III, IV as well as in the integrated group simultaneously regarded as one. To reach this aim, three microarray gene expression profiling datasets have been included to identify the differentially expressed genes (DEGs). By prioritizing those DEGs, their cellular and molecular functions will be further analyzed. Then, the involved GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) signaling pathways will be studied. Moreover, the protein–protein interaction network for all stages are developed based on the STRING database, and the significant hub genes are identified by clustering algorithm from which the gene biomarkers will later be determined based on their higher connectivity degrees. Finally, the Kaplan–Meier analysis tool was used to assess recurrence-free survival rates of the identified gene biomarkers.
Materials and methods
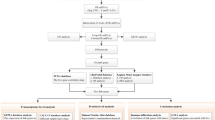
Figure 1 presents the summarization of the flowchart diagram of the approach to satisfy the research question.

Flowchart of the current research approach step by step to achieve the final validated gene biomarkers in terms of recurrence free survival in HER2-negative breast cancer.
Data sources
All the datasets used in this study were retrieved from the NCBI GEO database (i.e., https://www.ncbi.nlm.nih.gov/geo/). The platform and file type of the breast cancer microarray datasets were GPL96 [HG-U133A] Affymetrix Human Genome U133A Array and CEL files, respectively. To cover the aim of this study, GSE124647, GSE129551, and GSE124646 were used as train set including 140 biopsy samples from metastatic patients with stage IV breast cancer, 147 samples from patients with stages I, II, III, and IV breast cancer, and 10 normal samples (0 percent cancer) out of 100 samples, respectively. Moreover, GSE15852 (i.e., includes 43 normal, 8 grade 1 ~ stage I, 23 grade 2 ~ stage II, and 12 grade 3 ~ stage III samples) was used as a test set for external validation.
Data preprocessing and identification of differentially expressed genes (DEGs)
The BRB-ArrayTools (v4.6.0, stable version), an excel graphical user interface (GUI) for communicating with R (v 3.5.1) programming environment developed by Dr. Richard Simon and the BRB-ArrayTools Development Team, was used for all stages of preprocessing (i.e., data import, data filtering, and normalization), gene annotation using “hthgu133a.db” R annotation package16 and identification of DEGs. During the data import phase, Microarray Suite version 5.0 (MAS 5.0) algorithm was utilized, and then spot filtering, quantile normalization, and gene filtering (gene exclusion criteria of fold change ≤ 2 with expression data values less than %20) were carried out. Next, class comparison between groups of arrays in terms of their label classification was performed to identify the differentially expressed genes (DEGs) by enabling the two options, including univariate permutation tests and restricting gene list based on the fold change threshold with their default values (i.e., 10,000 and 2, respectively). All of the identified DEGs were stored for the next stage (i.e., prioritization of DEGs) as test group. Furthermore, the volcano plot and box plot of the imported data were demonstrated for each stage versus the normal samples.
Prioritization for DEGs
To prioritize identified DEGs from the previous section using the evidence of the literature, GeneCards17 and ToPPGene18 websites were used, respectively. The GeneCards database site (i.e., https://genecards.org) was used to extract the literature evidence on reported genes (denoted by the train group) for a specific disease by using approximately 150 web sources and the keywords. For this purpose, the used keywords included < “breast cancer” + ”stage I” > , < “breast cancer” + ”stage II” > , < “breast cancer” + ”stage III” > , < “breast cancer” + ”stage IV” > , as well as inclusion of the results of all four stages. Then, the ToPPGene website (i.e., https://toppgene.cchmc.org), which used the functional annotation and protein interactions to prioritize the imported gene list, was used to order the test group of genes based on the train group to determine the most significant DEGs in all stages of breast cancer with the p-value less than 0.05. Moreover, the ToPPGene website uses the similarity scores of the train group based on fuzzy and Pearson correlation measurement values to score and rank the test group.
Gene ontology, pathway and functional enrichment analyses of prioritized DEGs
To determine the biological and molecular functional processes of the prioritized gene list as well as their significant enriched pathways, the online tool provided in the DAVID v. 6.8 (Database for Annotation, Visualization, and Integrated Discovery) website (i.e., https://david.abcc.ncifcrf.gov/summary.jsp)19,20 was applied. This website took the advantages of the gene ontology (GO) annotation analysis and the Kyoto Encyclopedia of Genes and Genomes (KEGG) to cover the required properties. Moreover, the results with the p-value ≤ 0.05 were considered significant.
Protein–protein interaction (PPI) network construction
The protein–protein interaction network among prioritized DEGs was constructed by the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING database ver. 11 plugin21 for Cytoscape v.3.7.122). The current STRING database (since January 19, 2019) contains 24,584,628 proteins from 5,090 organisms with 3,123,056,667 interactions. Moreover, the STRING database is experimentally dependent on BIND, DIP, GRID, HPRD, IntAct, MINT, and PID, and the cumulative information is extracted from curated websites Biocarta, BioCyc, GO, KEGG, and Reactome21. During the gene list import using the Cytoscape software, the confidence score cutoff value was set as 0.4 for PPI network construction and visualization. In the PPI network, the involved proteins are denoted by nodes, and their corresponding protein–protein interactions are presented as edges. To further investigate the PPI network of each of the breast cancer stages, the module (hub gene) analysis was performed using ClusterOne v.1.0 cytoscape plugin23 with its default values. Then, the significant modules with the p-value ≤ 0.05 were retrieved for biomarker identification. A protein with the highest connectivity degree in each candidate module will be considered a biomarker.
Validation of gene biomarkers
To validate the identified gene biomarkers for each stage, three validation approaches were considered. These include (i) the Kaplan–Meier (KM) plotter tool, (ii) classification model development and validation, and (iii) literature search for the identified gene biomarkers.
Kaplan–Meier plotter tool
To further validate the prognostic value of the gene biomarkers obtained from the hub genes of five groups, the free online Kaplan–Meier (KM) plotter tool was used24,25. Using the KM plotter tool, a meta-analysis based approach on thirty-five separate datasets was presented to assess the gene biomarkers in terms of various survival rates such as relapse free survival (RFS) and overall survival (OS). However, it has been reported that there is no significant difference between recurrence or relapse or disease free survival and overall survival rates26,27. To this end, the relapse free survival (RFS) (n = 3,955) was used by restricting the analysis to only HER2 (ERBB2) considering the HER2 nature of the three abovementioned datasets. Moreover, to generate high-resolution images, an option, namely “Generate high resolution TIFF file” was enabled before drawing the Kaplan–Meier plot and then, their p-values were recorded for target biomarkers. Additionally, by analyzing the RFS rate, the clinical outcomes of a disease would be measured if the time to death of the patient would be observed rather than validating the prognostic value of the gene biomarkers at particular stages of a disease.
Classification model development and validation
To validate the prognostic value of the identified biomarkers for a specific disease, a non-linear classification model was developed. For this purpose, nine classification models in Orange 3.22.0, including support vector machine, k-nearest neighbors, stochastic gradient descent, random forest, artificial neural network, Naïve Bayes, logistic regression, CN2 rule inducer, and adaboost were considered28. Furthermore, cross-validated accuracy (CA), precision (positive predictive value), recall (sensitivity), F1 score (a harmonic mean of sensitivity), and AUC (area under curve) were assessed using validation criteria such as k-fold cross-validation (k = 5, 10), LOOV (leave-one-out validation) as well as testing the model on train and test sets. Overall, the developed models would be validated both internally and externally.
Literature screening for potential genes
Another way of validating the identified genes was carried out based on the frequent appearance of the reported genes through experimental wet-labs of the literature investigations for the disease.
Results
Data preprocessing
The numbers of genes remained after applying the filtering criteria at stages I (normal:10, stage I:20), II (normal:10, stage II:80), III (normal:10, stage III:15), IV (normal:10, stage IV:141), and in the integrated group (normal:10, all samples at stage I, II, III, and IV:256) were 1,873, 2,034, 2,016, 2,279, and 2,471, respectively. Among the filtered genes, 832 (341 downregulated genes and 491 upregulated genes), 836 (392 downregulated genes and 444 upregulated genes), 980 (444 downregulated genes and 536 upregulated genes), 731 (455 downregulated genes and 276 upregulated genes), and 735 (464 downregulated genes and 271 upregulated genes) DEGs were identified using the two-sample t-test for the order of the abovementioned stages.
Prioritization of DEGs
After searching the GeneCards database for the specified breast cancer terms, 2,264, 1,611, 1,856, 855, and 6,586 DEGs for stages I, II, III, IV, and the integrated group were extracted and exported as a .csv file and were set as training datasets for five groups, separately. Moreover, the identified DEGs for five groups from BRB-ArrayTools were set as test datasets. Then, the ToppGene database ranked the input test datasets based on training datasets in five groups separately for each stage. Considering the threshold of the p-value < 0.05, the numbers of the selected DEGs for the above order of stages were 287, 339, 365, 347, and 224 that could play an important role in five specified stages of breast cancer. Among those DEGs identified for stage I, 131 genes were downregulated and 156 genes were upregulated. The values of downregulated and upregulated genes for stages II, III, IV and all stage were 174 and 165, 176 and 189, 218 and 129 as well as 134 and 90, respectively. Table 1 presents the list of the top 10 upregulated and downregulated genes ranked for all stages considering their low p-values.
GO enrichment and KEGG pathway analysis
The output of the DAVID bioinformatics tool provides diverse biological and functional analyses on the prioritized genes in five groups. These include biological processes (BP), cellular components (CC), and molecular functions (MF) for GO analysis as well as the KEGG pathway assessment. Considering stage I, several biological processes (e.g., reactive oxygen species metabolic process, hemopoiesis), cellular components (e.g., proteinaceous extracellular matrix, extracellular exosome), molecular functions (e.g., actin binding, ATP binding), and KEGG pathways (e.g., Influenza A, Tyrosine metabolism) are mainly enriched by DEGs (Fig. 2a). Moreover, the DEGs at stage II are associated with extracellular matrix organization and cellular response to fibroblast growth factor stimulus in terms of BP, with extracellular exosome and proteinaceous extracellular matrix in terms of CC, with protein binding and actin binding in terms of MF as well as focal adhesion and ECM-receptor interaction in terms of KEGG pathways (Fig. 2b). The key genes at stage III are enriched in BP related to the positive regulation of the apoptotic process and extracellular matrix organization, in CC related to extracellular exosome and cytosol, in MF related to protein binding and ATP binding, and in KEGG pathways related to Tyrosine metabolism and TNF signaling pathway (Fig. 2c). Additionally, at stage IV, extracellular matrix organization, extracellular exosome, protein binding, and focal adhesion are the most statistically significant enrichments in BP, CC, MF groups and KEGG pathways (Fig. 2d). The GO analysis results of the integrated group show that DEGs in groups BP, CC, MF are significantly enriched in complement activation, extracellular exosome, and calcium ion binding. Furthermore, the KEGG pathways analysis for all stages reveals that complement and coagulation cascades and Staphylococcus aureus infection are significantly enriched by prioritized DEGs (Fig. 2e).

The biological processes (BP), cellular components (CC), and molecular functions (MF) for GO analysis as well as the KEGG pathway assessment for (a) stage I, (b) stage II, (c) stage III, (d) stage IV, and (e) Integrated group.
PPI network analysis and hub genes identification
Using the Cytoscape and STRING database plugin, PPI networks are constructed for five groups (i.e., stage I (284 nodes and 512 edges), stage II (338 nodes and 1,263 edges), stage III (363 nodes and 1,170 edges), stage IV (346 nodes and 1909 edges), and the integrated group (221 nodes and 519 edges)). Among genes with higher interconnectivity within the constructed PPI networks of five groups, SMC4 (degree = 24, downregulated), FN1 (degree = 50, downregulated), FOS (degree = 42, upregulated), JUN (degree = 69, downregulated), and KIF11 and RACGAP1 (degree = 27, upregulated) for stage I, stage II, stage III, stage IV, and all stages, respectively, have the highest connectivity degrees in their PPI networks.
The significant outcomes for the ClusterOne module analysis in Cytoscape (p-value < 0.05) reveal 14, 9, 8, 4, and 6 protein modules for stages I, II, III, IV, and the integrated group, respectively.
Verification of central gene biomarkers
KM plotter tool
According to the visualization and numerical results obtained from the KM plotter and analysis tool, it has been revealed that 33 out of 53 potential biomarkers have a statistical significant association with the recurrence of free survival for five groups in HER2 breast cancer. Table 2 lists the characteristics of each of 53 genes in terms of their stages, gene symbol and expression, and overall p-value.
Performance of nine classifiers
The classification prediction results of all nine non-linear models (i.e., AUC, CA, F1 score, precision, and recall parameters) were investigated. In the k-fold cross-validation procedure to keep and possibly increase the stability of the models within the folds, the stratification sampling is used. Except, the performance of the models on the test set, almost all of the machine learning classifiers are trained and cross-validated at the highest values while considering the five-fold cross validation, ten-fold cross validation, stratified shuffle sampling trained on 66% of data, leave one out validation, and trained and tested on the whole dataset. Once the trained model is tested on the test set, the performance results for stages I, II, and III show that naïve Bayes, random forest, and naïve Bayes outperform the other classifiers with 0.87, 0.83, and 0.89 AUC values, respectively. The results are indicative of the fact that the computational classification models are capable of validating the identified genes from the systems biology approach for several stages of breast cancer.
Literature screening for identified genes
The other tactic commonly used in the systems biology related studies for validating the identified genes from a specific computational methodology is to gather the required evidence from the literature reports on a specific determined gene in a known disease (i.e., breast cancer). To this end, searching results present that all of the fifty three genes are found to be responsible for cell proliferation, growth, motility, and development at several stages of breast cancer disease. The next section discusses detailed information on these genes (Table 2).
Discussion
Breast cancer as a heterogeneous disease and the most common invasive cancer is the second leading cause of mortality among women globally29. During the last thirty years, the trend of mortality rate for breast cancer in developed countries has been dramatically decreased; however, the condition for low-income countries has no significant changes30. The success in the mortality rate reduction of breast cancer in high-income countries is mostly owing to the improved treatment and early stage diagnosis as well as the appropriate selection and administration of therapies30. This will be followed by prolonging RFS and OS without complications29.
In this research, three microarray datasets, including stages I, II, III, IV, and the integrated group, were used, preprocessed, normalized and analyzed from which the significant DEGs for five groups were identified. After that, they were ranked based on the literature involved genes in breast cancer and selected based on the statistical significant p-value < 0.05. Then, GO and KEGG pathways analyses as well as PPI network construction were performed. The biological processes (BP), cellular components (CC), and molecular functions (MF) were also assessed for enrichment pathways. Moreover, the PPI network analysis using the STRING database revealed several effective hub genes for five groups separately. The significant gene biomarkers with the highest connectivity degree within the hub genes were selected. The validation of the obtained gene biomarkers in terms of recurrence free survival rate in HER2 was statistically carried out by Kaplan–Meier plotter tool with p-values less than 0.05. Moreover, the internal and external validation procedures revealed that the machine learning classification models specifically those developed based on naïve Bayes and random forest by employing various biomarkers at several stages were successful in differentiating between stages and normal samples with good predictive power. Finally, in Table 2, the available evidences collected from the experimental literature reported for breast cancer has been retrieved and listed according to the identified gene biomarkers. Additionally, some of the identified biomarkers were found to be common among different TNM stages. For example, IRF7 was the significant biomarker for stages I, II, and III; and, ACTA2 biomarker was found to have an increasing expression across stages II and III.
According to the outcomes of the current study, we identified a signature of potential biomarkers for BC stages to specifically diagnose breast cancer at developed stages as well as very early stages. These biomarkers could potentially be the target of wet-lab researchers for future investigations. The mathematical models developed for BC prediction and diagnosis at various stages showed significantly high and reasonable performance in clinical outcomes employing the identified biomarkers. It is worth noting that the current study is conducted for the first time that studied the high throughput gene profiling datasets for four stages of BC as well as its integrated stage. Finally, the strong point of the study relied on the three validation methodologies, however, the Kaplan–Meier analysis did not find some of the biomarkers statistically significant.
The systems biology approach could enlighten the path for wet-lab investigators in rapid identification of stages in patients with BC. Moreover, the developed non-linear models could be utilized in prediction procedure after the gene expression values for target biomarkers are determined through experimental tests. The workflow of the current study could be applied for other future microarray studies in terms of involving and investigating the stages of the diseases. Furthermore, the identified biomarkers along with their involved signaling pathways could be beneficial for drug design and discovery agents considering various disease stages and hence, the disease could be controlled, managed and treated at very early stages.
Any researches specifically those carried out on systems biology approaches will have limitations and it seems to be normal. Due to the computational nature of these studies, there will remain gaps between the wet- and dry-labs for further validating the results. The experimental and clinical literature studies do only report on the genes involved in BC disease without stating their stages. The lack of available sufficient microarray datasets in the repository databases investigating the stages of BC made us consider the stages and grades of BC equivalent for the validation process.
During the last decades, extensive genome-wide association studies and next generation sequencing techniques were conducted and applied to identify the potent biomarkers using bioinformatics and experimental approaches for various diseases such as Parkinson’s disease and prostate cancer considering the exponential growth of Big Data generation in the field31,32,33,34,35. For future researches, it is useful to investigate the genome-based studies in a centralized manner to provide the datasets in further details in terms of being more specific at the disease stages and the follow-up procedures. Moreover, owing to the large generation of genome datasets, handling and managing them computationally and experimentally are still of many researches’ interest in the world. Therefore, close cooperation among systems biologists, bioinformaticians, and biologists is required in to identify potential biomarkers and their involvement in signaling pathways. In other words, understanding the functions of the target signaling pathways in specific diseases is highly important in accelerating the development of new experimental drugs and diagnostics, paving the ways for personalized medicine and improving translational sciences32,36,37.
Conclusions
In this study, three HER2-negative breast cancer datasets were analyzed to identify differentially expressed genes and construct protein–protein interaction networks as well as GO enrichment and KEGG pathway analyses for the TNM-based staging system. The results indicate that a 53-gene signature is responsible for breast cancer prognosis at various stages. The identified gene signature could be further utilized in personalizing medicine for individuals with breast cancer. The identified PPI modules significantly involved at different stages of breast cancer show a different number of connectivity ranging from 1 to 69. The interesting finding noticeable in the results is that the lower number of interactions within hub genes is not correlated with the importance of genes as potential biomarkers. For example, module 5 with only three genes and two connections shows significant expression (downregulation) in the integrated group. Her2-negative breast cancer was further confirmed by the literature reports. Moreover, the Kaplan–Meier tool for assessing the recurrence-free survival rate is not a measure to exclude a biomarker based only on its statistical significant p-value. For instance, in Table 2, there are 20 genes identified to be non-significant in the RFS rate assessment evaluated by the KM tool. However, for example, IRF7 identified as a biomarker for almost all groups has not been significantly related to the RFS rate. However, according to the literature, IRF7 is significantly correlated with breast cancer development. Therefore, non-significant p-value in the KM assessment does not decrease the importance of an identified biomarker. The outcomes of this research have paved the way to evaluate the status of breast cancer development in terms of the TNM-based staging system. All of the identified DEGs were involved in breast cancer as confirmed by the evidence available in the literature derived solely from experimental studies. What is missing from the clinical data in the literature is the staging of the condition, which now can be answered using the panel of gene biomarkers proposed in this study.
References
Ferlay, J. et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 144, 1941–1953. https://doi.org/10.1002/ijc.31937 (2019).
Koh, J. & Kim, M. J. Introduction of a new staging system of breast cancer for radiologists: an emphasis on the prognostic stage. Korean J Radiol 20, 69–82. https://doi.org/10.3348/kjr.2018.0231 (2019).
Cserni, G., Chmielik, E., Cserni, B. & Tot, T. The new TNM-based staging of breast cancer. Virch. Arch. 472, 697–703. https://doi.org/10.1007/s00428-018-2301-9 (2018).
Yang, Y. et al. Identification of LCN1 as a potential biomarker for breast cancer by bioinformatic analysis. DNA Cell Biol. https://doi.org/10.1089/dna.2019.4843 (2019).
Deng, J. L., Xu, Y. H. & Wang, G. Identification of potential crucial genes and key pathways in breast cancer using bioinformatic analysis. Front. Genet. 10, 695. https://doi.org/10.3389/fgene.2019.00695 (2019).
Tomczak, K., Czerwinska, P. & Wiznerowicz, M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp. Oncol. 19, A68-77. https://doi.org/10.5114/wo.2014.47136 (2015).
Iqbal, N. & Iqbal, N. Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol. Biol. Int. 2014, 852748. https://doi.org/10.1155/2014/852748 (2014).
Wu, Y. et al. Circulating HER-2 mRNA in the peripheral blood as a potential diagnostic and prognostic biomarker in females with breast cancer. Oncol Lett. 16, 3726–3734. https://doi.org/10.3892/ol.2018.9091 (2018).
Ramachandran, S. & Badve, S. S. Milestones in the discovery of HER2 proto-oncogene and trastuzumab (Herceptin). Breast Cancer Res. Treat. 112, 522–533 (2008).
Ross, J. S. et al. The HER-2 receptor and breast cancer: ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist 14, 320–368. https://doi.org/10.1634/theoncologist.2008-0230 (2009).
Gonzalez, A. et al. A definition for aggressive disease in patients with HER-2 negative metastatic breast cancer: an expert consensus of the Spanish Society of Medical Oncology (SEOM). Clin. Transl. Oncol. 19, 616–624. https://doi.org/10.1007/s12094-016-1571-4 (2017).
Wang, L. Early diagnosis of breast cancer. Sensors. https://doi.org/10.3390/s17071572 (2017).
Hequet, D. et al. Prospective, multicenter French study evaluating the clinical impact of the Breast Cancer Intrinsic Subtype-Prosigna(R) Test in the management of early-stage breast cancers. PLoS ONE 12, e0185753. https://doi.org/10.1371/journal.pone.0185753 (2017).
Ades, F. et al. Luminal B breast cancer: molecular characterization, clinical management, and future perspectives. J. Clin. Oncol. 32, 2794–2803. https://doi.org/10.1200/jco.2013.54.1870 (2014).
Pan, C. et al. KLP-PI: a new prognostic index for luminal B HER-2-negative breast cancer. Hum. Cell 32, 172–184. https://doi.org/10.1007/s13577-018-00229-x (2019).
Carlson, M. hthgu133a.db: Affymetrix HT Human Genome U133 Array Plate Set annotation data (chip hthgu133a). R package version 3.2.3 (2016).
Stelzer, G. et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Current protocols in bioinformatics 54, 31–33. https://doi.org/10.1002/cpbi.5 (2016).
Chen, J., Bardes, E. E., Aronow, B. J. & Jegga, A. G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37, W305-311. https://doi.org/10.1093/nar/gkp427 (2009).
da Huang, W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. https://doi.org/10.1093/nar/gkn923 (2009).
da Huang, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. https://doi.org/10.1038/nprot.2008.211 (2009).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607-d613. https://doi.org/10.1093/nar/gky1131 (2019).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. https://doi.org/10.1101/gr.1239303 (2003).
Nepusz, T., Yu, H. & Paccanaro, A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 9, 471. https://doi.org/10.1038/nmeth.1938 (2012).
Gyorffy, B. et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 123, 725–731. https://doi.org/10.1007/s10549-009-0674-9 (2010).
Qi, L. et al. MicroRNAs associated with lung squamous cell carcinoma: New prognostic biomarkers and therapeutic targets. J. Cell. Biochem. https://doi.org/10.1002/jcb.29216 (2019).
Buzdar, A. U. et al. Disease-free and overall survival among patients with operable HER2-positive breast cancer treated with sequential vs concurrent chemotherapy: the ACOSOG Z1041 (Alliance) randomized clinical trial. JAMA Oncol. 5, 45–50. https://doi.org/10.1001/jamaoncol.2018.3691 (2019).
Punt, C. J. et al. Endpoints in adjuvant treatment trials: a systematic review of the literature in colon cancer and proposed definitions for future trials. J. Natl Cancer Inst. 99, 998–1003. https://doi.org/10.1093/jnci/djm024 (2007).
Demšar, J. et al. Orange: data mining toolbox in python. J. Mach. Learn. Res. 14, 2349–2353 (2013).
Guler, E. N. Gene expression profiling in breast cancer and its effect on therapy selection in early-stage breast cancer. Eur. J. Breast Health 13, 168–174. https://doi.org/10.5152/ejbh.2017.3636 (2017).
Carioli, G. et al. Trends and predictions to 2020 in breast cancer mortality: Americas and Australasia. Breast (Edinburgh, Scotland) 37, 163–169. https://doi.org/10.1016/j.breast.2017.12.004 (2018).
Qi, X., Yu, C., Wang, Y., Lin, Y. & Shen, B. Network vulnerability-based and knowledge-guided identification of microRNA biomarkers indicating platinum resistance in high-grade serous ovarian cancer. Clin. Transl. Med. 8, 1–11 (2019).
Shang, J. et al. Evaluation and comparison of multiple aligners for next-generation sequencing data analysis. BioMed Res. Int. 1, 4. https://doi.org/10.1155/2014/309650 (2014).
Shen, B. et al. Translational informatics for Parkinson’s disease: from big biomedical data to small actionable alterations. Genom. Proteom. Bioinform. https://doi.org/10.1016/j.gpb.2018.10.007 (2019).
Shen, L. et al. Knowledge-guided bioinformatics model for identifying autism spectrum disorder diagnostic MicroRNA biomarkers. Sci. Rep. 6, 39663 (2016).
Zhu, J. et al. Screening key microRNAs for castration-resistant prostate cancer based on miRNA/mRNA functional synergistic network. Oncotarget 6, 43819 (2015).
Chen, J. et al. Long non-coding RNAs in urologic malignancies: functional roles and clinical translation. J. Cancer 7, 1842 (2016).
Yang, Y., Chen, B., Tan, G., Vihinen, M. & Shen, B. Structure-based prediction of the effects of a missense variant on protein stability. Amino Acids 44, 847–855 (2013).
Wang, Y. et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet (London, England) 365, 671–679. https://doi.org/10.1016/s0140-6736(05)17947-1 (2005).
Einav, U. et al. Gene expression analysis reveals a strong signature of an interferon-induced pathway in childhood lymphoblastic leukemia as well as in breast and ovarian cancer. Oncogene 24, 6367–6375. https://doi.org/10.1038/sj.onc.1208797 (2005).
Wang, Z. et al. Periostin promotes immunosuppressive premetastatic niche formation to facilitate breast tumour metastasis. J. Pathol. 239, 484–495. https://doi.org/10.1002/path.4747 (2016).
Budczies, J. et al. Comparative metabolomics of estrogen receptor positive and estrogen receptor negative breast cancer: alterations in glutamine and beta-alanine metabolism. J. Proteom. 94, 279–288. https://doi.org/10.1016/j.jprot.2013.10.002 (2013).
Zang, H., Li, N., Pan, Y. & Hao, J. Identification of upstream transcription factors (TFs) for expression signature genes in breast cancer. Gynecol. Endocrinol. 33, 193–198. https://doi.org/10.1080/09513590.2016.1239253 (2017).
Ivanov, S. V. et al. Diagnostic SOX10 gene signatures in salivary adenoid cystic and breast basal-like carcinomas. Br. J. Cancer 109, 444–451. https://doi.org/10.1038/bjc.2013.326 (2013).
Panaccione, A., Guo, Y., Yarbrough, W. G. & Ivanov, S. V. Expression profiling of clinical specimens supports the existence of neural progenitor-like stem cells in basal breast cancers. Clin. Breast Cancer 17, 298-306.e297. https://doi.org/10.1016/j.clbc.2017.01.007 (2017).
Cyr-Depauw, C. et al. Chordin-like 1 suppresses bone morphogenetic protein 4-induced breast cancer cell migration and invasion. Mol. Cell. Biol. 36, 1509–1525. https://doi.org/10.1128/mcb.00600-15 (2016).
Yang, C. et al. The integrin alpha(v)beta(3–5) ligand MFG-E8 is a p63/p73 target gene in triple-negative breast cancers but exhibits suppressive functions in ER(+) and erbB2(+) breast cancers. Cancer Res 71, 937–945. https://doi.org/10.1158/0008-5472.CAN-10-1471 (2011).
Carrascosa, C. et al. MFG-E8/lactadherin regulates cyclins D1/D3 expression and enhances the tumorigenic potential of mammary epithelial cells. Oncogene 31, 1521–1532. https://doi.org/10.1038/onc.2011.356 (2012).
Isaya, G. Mitochondrial iron-sulfur cluster dysfunction in neurodegenerative disease. Front. Pharmacol. 5, 29. https://doi.org/10.3389/fphar.2014.00029 (2014).
Kalyuga, M. et al. ELF5 suppresses estrogen sensitivity and underpins the acquisition of antiestrogen resistance in luminal breast cancer. PLoS Biol. 10, e1001461. https://doi.org/10.1371/journal.pbio.1001461 (2012).
Chakrabarti, R. et al. Elf5 inhibits the epithelial-mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nat. Cell Biol. 14, 1212–1222. https://doi.org/10.1038/ncb2607 (2012).
Scribner, K. C., Behbod, F. & Porter, W. W. Regulation of DCIS to invasive breast cancer progression by Singleminded-2s (SIM2s). Oncogene 32, 2631–2639. https://doi.org/10.1038/onc.2012.286 (2013).
Yang, F. et al. Co-expression networks revealed potential core lncRNAs in the triple-negative breast cancer. Gene 591, 471–477. https://doi.org/10.1016/j.gene.2016.07.002 (2016).
Touraine, P. et al. Increased expression of prolactin receptor gene assessed by quantitative polymerase chain reaction in human breast tumors versus normal breast tissues. J. Clin. Endocrinol. Metab. 83, 667–674. https://doi.org/10.1210/jcem.83.2.4564 (1998).
Clevenger, C. V., Furth, P. A., Hankinson, S. E. & Schuler, L. A. The role of prolactin in mammary carcinoma. Endocr. Rev. 24, 1–27. https://doi.org/10.1210/er.2001-0036 (2003).
Graichen, R. et al. The growth hormone-binding protein is a location-dependent cytokine receptor transcriptional enhancer. J. Biol. Chem. 278, 6346–6354. https://doi.org/10.1074/jbc.M207546200 (2003).
Sun, M. et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc. Natl. Acad. Sci. 110, 9920–9925 (2013).
Chai, F. et al. Systematically identify key genes in inflammatory and non-inflammatory breast cancer. Gene 575, 600–614 (2016).
Wu, C. et al. Mechanosensitive PPAP2B regulates endothelial responses to atherorelevant hemodynamic forces. Circ. Res. 117, e41–e53 (2015).
Westcott, J. M. et al. An epigenetically distinct breast cancer cell subpopulation promotes collective invasion. J. Clin. Investig. 125, 1927–1943. https://doi.org/10.1172/jci77767 (2015).
Benhaj, K., Akcali, K. C. & Ozturk, M. Redundant expression of canonical Wnt ligands in human breast cancer cell lines. Oncol. Rep. 15, 701–707 (2006).
Russo, J., Moral, R., Balogh, G. A., Mailo, D. & Russo, I. H. The protective role of pregnancy in breast cancer. Breast Cancer Res. 7, 131 (2005).
Dong, H., Claffey, K. P., Brocke, S. & Epstein, P. M. Expression of phosphodiesterase 6 (PDE6) in human breast cancer cells. SpringerPlus 2, 680 (2013).
Paine-Saunders, S., Viviano, B. L. & Saunders, S. GPC6, a Novel Member of the Glypican Gene Family, Encodes a Product Structurally Related to GPC4 and Is Colocalized withGPC5on Human Chromosome 13. Genomics 57, 455–458 (1999).
Liu, Y. et al. Isoflavones in soy flour diet have different effects on whole-genome expression patterns than purified isoflavone mix in human MCF-7 breast tumors in ovariectomized athymic nude mice. Mol. Nutr. Food Res. 59, 1419–1430 (2015).
Fernandez, S. V. et al. Expression and DNA methylation changes in human breast epithelial cells after bisphenol A exposure. Int. J. Oncol. 41, 369–377 (2012).
Fan, S. H. et al. CERS2 suppresses tumor cell invasion and is associated with decreased V-ATPase and MMP-2/MMP-9 activities in breast cancer. J. Cell. Biochem. 116, 502–513 (2015).
Erez-Roman, R., Pienik, R. & Futerman, A. H. Increased ceramide synthase 2 and 6 mRNA levels in breast cancer tissues and correlation with sphingosine kinase expression. Biochem. Biophys. Res. Commun. 391, 219–223 (2010).
Pan, H. et al. Cloning, mapping, and characterization of a human homologue of the yeast longevity assurance gene LAG1. Genomics 77, 58–64 (2001).
Schengrund, C.-L. Gangliosides: glycosphingolipids essential for normal neural development and function. Trends Biochem. Sci. 40, 397–406. https://doi.org/10.1016/j.tibs.2015.03.007 (2015).
Kuo, W.-H. et al. Molecular characteristics and metastasis predictor genes of triple-negative breast cancer: a clinical study of triple-negative breast carcinomas. PLoS ONE 7, e45831 (2012).
Ruckhäberle, E. et al. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res. Treat. 112, 41–52 (2008).
Che, J., Huang, Y., Xu, C. & Zhang, P. Increased ceramide production sensitizes breast cancer cell response to chemotherapy. Cancer Chemother. Pharmacol. 79, 933–941 (2017).
Milde-Langosch, K. et al. Prognostic relevance of glycosylation-associated genes in breast cancer. Breast Cancer Res. Treat. 145, 295–305 (2014).
Moini, J. Epidemiology of Diabetes 57–73 (Elsevier, Amsterdam, 2019).
Nath, A. & Chan, C. Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci. Rep. 6, 18669 (2016).
Malvia, S. et al. Study of gene expression profiles of breast cancers in Indian women. Sci. Rep. 9, 10018 (2019).
Bickel, P. E., Tansey, J. T. & Welte, M. A. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochem. Biophys. Acta. 1791, 419–440. https://doi.org/10.1016/j.bbalip.2009.04.002 (2009).
Kim, S., Lee, Y. & Koo, J. S. Differential expression of lipid metabolism-related proteins in different breast cancer subtypes. PLoS ONE 10, e0119473 (2015).
Cefan Zhou, M. W. et al. Prognostic significance of PLIN1 expression in human breast cancer. Oncotarget 7, 54488 (2016).
Qian, X. et al. CCNB2 overexpression is a poor prognostic biomarker in Chinese NSCLC patients. Biomed. Pharmacother. 74, 222–227 (2015).
Prat, A. et al. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 18, 123–133 (2013).
Miller, W. et al. Microarray analysis of sequential tumour biopsies from patients receiving neoadjuvant therapy is able to distinguish sub-populations of breast cancers with differential response to the aromatase inhibitor, letrozole. Breast Cancer Res. Treat. 88, 3139 (2004).
Walker, G. et al. Estrogen-regulated gene expression predicts response to endocrine therapy in patients with ovarian cancer. Gynecol. Oncol. 106, 461–468 (2007).
Sava, G. P. et al. Common variation at 12q24.13 (OAS3) influences chronic lymphocytic leukemia risk. Leukemia 29, 748–751. https://doi.org/10.1038/leu.2014.311 (2015).
Callari, M. et al. Subtype-dependent prognostic relevance of an interferon-induced pathway metagene in node-negative breast cancer. Mol. Oncol. 8, 1278–1289. https://doi.org/10.1016/j.molonc.2014.04.010 (2014).
Tsai, M.-H. et al. Gene expression profiling of breast, prostate, and glioma cells following single versus fractionated doses of radiation. Cancer Res 67, 3845–3852 (2007).
Jeong, G. et al. A Kelch domain-containing KLHDC7B and a long non-coding RNA ST8SIA6-AS1 act oppositely on breast cancer cell proliferation via the interferon signaling pathway. Sci. Rep. 8, 12922 (2018).
Larson, P. S. et al. CDKN1C/p57 kip2 is a candidate tumor suppressor gene in human breast cancer. BMC Cancer 8, 68 (2008).
Yang, X. et al. CDKN1C (p57KIP2) is a direct target of EZH2 and suppressed by multiple epigenetic mechanisms in breast cancer cells. PLoS ONE 4, e5011 (2009).
Huang, J. M. & Kim, J. DNA methylation analysis of the mammalian PEG3 imprinted domain. Gene 442, 18–25 (2009).
Harrison, K. et al. Breast cancer risk and imprinting methylation in blood. Clin. Epigenet. 7, 92 (2015).
Schauwecker, S. M., Kim, J. J., Licht, J. D. & Clevenger, C. V. Histone H1 and chromosomal protein HMGN2 regulate prolactin-induced STAT5 transcription factor recruitment and function in breast cancer cells. J. Biol. Chem. 292, 2237–2254 (2017).
Moon, H.-G. et al. Prognostic and functional importance of the engraftment-associated genes in the patient-derived xenograft models of triple-negative breast cancers. Breast Cancer Res. Treat. 154, 13–22 (2015).
Alsner, J., Rødningen, O. K. & Overgaard, J. Differential gene expression before and after ionizing radiation of subcutaneous fibroblasts identifies breast cancer patients resistant to radiation-induced fibrosis. Radiother. Oncol. 83, 261–266 (2007).
Casey, T. et al. Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast Cancer Res. Treat. 114, 47–62 (2009).
He, H. et al. Low expression of SLC22A18 predicts poor survival outcome in patients with breast cancer after surgery. Cancer Epidemiol. 35, 279–285 (2011).
Xu, J., Zhang, W., Tang, L., Chen, W. & Guan, X. Epithelial-mesenchymal transition induced PAI-1 is associated with prognosis of triple-negative breast cancer patients. Gene 670, 7–14 (2018).
Tomaskovic-Crook, E., Thompson, E. W. & Thiery, J. P. Epithelial to mesenchymal transition and breast cancer. Breast Cancer Res. 11, 213 (2009).
Frankel, L. B. et al. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J. Biol. Chem. 283, 1026–1033 (2008).
Jeon, M. et al. Dimerization of EGFR and HER2 induces breast cancer cell motility through STAT1-dependent ACTA2 induction. Oncotarget 8, 50570–50581. https://doi.org/10.18632/oncotarget.10843 (2017).
Miaskowski, C. et al. Lymphatic and angiogenic candidate genes predict the development of secondary lymphedema following breast cancer surgery. PLoS ONE 8, e60164 (2013).
Garg, A. D., De Ruysscher, D. & Agostinis, P. Immunological metagene signatures derived from immunogenic cancer cell death associate with improved survival of patients with lung, breast or ovarian malignancies: A large-scale meta-analysis. Oncoimmunology 5, e1069938 (2016).
El Roz, A., Bard, J.-M., Huvelin, J.-M. & Nazih, H. LXR agonists and ABCG1-dependent cholesterol efflux in MCF-7 breast cancer cells: relation to proliferation and apoptosis. Anticancer Res. 32, 3007–3013 (2012).
Schmitz, G., Langmann, T. & Heimerl, S. Role of ABCG1 and other ABCG family members in lipid metabolism. J. Lipid Res. 42, 1513–1520 (2001).
Ciriello, G. et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 163, 506–519 (2015).
Kraus, M. R. C. et al. Two mutations in human BICC1 resulting in Wnt pathway hyperactivity associated with cystic renal dysplasia. Hum. Mutat. 33, 86–90 (2012).
Sahay, D. et al. The LPA1/ZEB1/miR-21-activation pathway regulates metastasis in basal breast cancer. Oncotarget 6, 20604 (2015).
Barok, M. et al. Characterization of a novel, trastuzumab resistant human breast cancer cell line. Front. Biosci. (Elite Ed) 2, 627–640 (2010).
LeVan, T. D. et al. Genetic variants in circadian rhythm genes and self-reported sleep quality in women with breast cancer. J/ Circadian Rhythms 17, 6 (2019).
Chandrashekar, D. S. et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 19, 649–658. https://doi.org/10.1016/j.neo.2017.05.002 (2017).
Abdel-Fatah, T. M. et al. Genomic and protein expression analysis reveals flap endonuclease 1 (FEN1) as a key biomarker in breast and ovarian cancer. Mol. Oncol. 8, 1326–1338 (2014).
Lv, Z. et al. Association of functional FEN1 genetic variants and haplotypes and breast cancer risk. Gene 538, 42–45 (2014).
Chen, B. et al. Curcumin inhibits proliferation of breast cancer cells through Nrf2-mediated down-regulation of Fen1 expression. J. Steroid Biochem. Mol. Biol. 143, 11–18 (2014).
Lilla, C., Koehler, T., Kropp, S., Wang-Gohrke, S. & Chang-Claude, J. Alcohol dehydrogenase 1B (ADH1B) genotype, alcohol consumption and breast cancer risk by age 50 years in a German case–control study. Br. J. Cancer 92, 2039 (2005).
Terry, M. B. et al. Alcohol metabolism, alcohol intake, and breast cancer risk: a sister-set analysis using the Breast Cancer Family Registry. Breast Cancer Res. Treat. 106, 281–288 (2007).
Visvanathan, K. et al. Alcohol dehydrogenase genetic polymorphisms, low-to-moderate alcohol consumption, and risk of breast cancer. Alcoholism 31, 467–476 (2007).
Kan He, W. L. et al. The stromal genome heterogeneity between breast and prostate tumors revealed by a comparative transcriptomic analysis. Oncotarget 6, 8687 (2015).
Myal, Y., Leygue, E. & Blanchard, A. A. Claudin 1 in breast tumorigenesis: revelation of a possible novel “claudin high” subset of breast cancers. BioMed Res. Int. https://doi.org/10.1155/2010/956897 (2010).
Blockhuys, S. & Wittung-Stafshede, P. Roles of copper-binding proteins in breast cancer. Int. J. Mol. Sci. 18, 871 (2017).
Denoyer, D., Masaldan, S., La Fontaine, S. & Cater, M. A. Targeting copper in cancer therapy:‘Copper That Cancer’. Metallomics 7, 1459–1476 (2015).
Cheng, Y. et al. Fibulin 1 is downregulated through promoter hypermethylation in gastric cancer. Br. J. Cancer 99, 2083 (2008).
Bardin, A. et al. Transcriptional and posttranscriptional regulation of fibulin-1 by estrogens leads to differential induction of messenger ribonucleic acid variants in ovarian and breast cancer cells. Endocrinology 146, 760–768 (2005).
Hodgkinson, V. C. et al. Pilot and feasibility study: comparative proteomic analysis by 2-DE MALDI TOF/TOF MS reveals 14-3-3 proteins as putative biomarkers of response to neoadjuvant chemotherapy in ER-positive breast cancer. Journal of Proteomics 75, 2745–2752 (2012).
Xiang, Y.-J. et al. Screening for candidate genes related to breast cancer with cDNA microarray analysis. Chronic Dis. Transl. Med. 1, 65–72 (2015).
Vazquez-Martin, A. et al. Metformin regulates breast cancer stem cello ntogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle 9, 3831–3838 (2010).
Hill, V. K. et al. Genome-wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res 71, 2988–2999 (2011).
Loss, L. A. et al. Prediction of epigenetically regulated genes in breast cancer cell lines. BMC Bioinform. 11, 305 (2010).
Ma, X.-J., Dahiya, S., Richardson, E., Erlander, M. & Sgroi, D. C. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res. 11, R7 (2009).
Mackay, A. et al. Molecular response to aromatase inhibitor treatment in primary breast cancer. Breast Cancer Res. 9, R37 (2007).
Chen, L. et al. High levels of nucleolar spindle-associated protein and reduced levels of BRCA1 expression predict poor prognosis in triple-negative breast cancer. PLoS ONE 10, e0140572 (2015).
Iyer, J., Moghe, S., Furukawa, M. & Tsai, M.-Y. What’s Nu (SAP) in mitosis and cancer?. Cell. Signal. 23, 991–998 (2011).
Pongor, L. et al. A genome-wide approach to link genotype to clinical outcome by utilizing next generation sequencing and gene chip data of 6,697 breast cancer patients. Genome medicine 7, 104 (2015).
Karousou, E. et al. Collagen VI and hyaluronan: the common role in breast cancer. BioMed Res. Int. 1, 4. https://doi.org/10.1155/2014/606458 (2014).
Nayak, S. R. et al. A role for histone H2B variants in endocrine-resistant breast cancer. Hormones Cancer 6, 214–224 (2015).
Jiang, M. et al. KIF11 is required for proliferation and self-renewal of docetaxel resistant triple negative breast cancer cells. Oncotarget 8, 92106 (2017).
Lucanus, A. & Yip, G. Kinesin superfamily: roles in breast cancer, patient prognosis and therapeutics. Oncogene 37, 833–838 (2018).
Pors, K. & Moreb, J. S. Aldehyde dehydrogenases in cancer: an opportunity for biomarker and drug development?. Drug Discov. Today 19, 1953–1963 (2014).
van den Hoogen, C. et al. The aldehyde dehydrogenase enzyme 7A1 is functionally involved in prostate cancer bone metastasis. Clin. Exp. Metas. 28, 615–625. https://doi.org/10.1007/s10585-011-9395-7 (2011).
Zhang, Q., Liang, Z., Gao, Y., Teng, M. & Niu, L. Differentially expressed mitochondrial genes in breast cancer cells: potential new targets for anti-cancer therapies. Gene 596, 45–52 (2017).
Sansregret, L. & Nepveu, A. Gene signatures of genomic instability as prognostic tools for breast cancer. Future Oncol. 7, 591–594 (2011).
Acknowledgements
The authors would like to thank the Research Office of Tabriz University of Medical Sciences and acknowledge the Biotechnology Research Center for providing financial support under Grant No. 64904.
Author information
Authors and Affiliations
Contributions
B.S. and S.D. contributed to the design and implementation of the research. E.A. and S.A. worked out the numerical calculations and outcomes for the experiment. All authors (E.A., S.A., B.S. and S.D.) discussed and aided in interpreting the results and contributed to the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Amjad, E., Asnaashari, S., Sokouti, B. et al. Systems biology comprehensive analysis on breast cancer for identification of key gene modules and genes associated with TNM-based clinical stages. Sci Rep 10, 10816 (2020). https://doi.org/10.1038/s41598-020-67643-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-67643-w
This article is cited by
-
In-vivo studies of targeted and localized cancer drug release from microporous poly-di-methyl-siloxane (PDMS) devices for the treatment of triple negative breast cancer
Scientific Reports (2024)
-
Synergistic effects of flavonoids and paclitaxel in cancer treatment: a systematic review
Cancer Cell International (2023)
-
A systematic review of anti-cancer roles and mechanisms of kaempferol as a natural compound
Cancer Cell International (2022)
-
Impact of Gene Biomarker Discovery Tools Based on Protein–Protein Interaction and Machine Learning on Performance of Artificial Intelligence Models in Predicting Clinical Stages of Breast Cancer
Interdisciplinary Sciences: Computational Life Sciences (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
