
Abstract
Extreme overproduction of gratuitous proteins can overload cellular protein production resources, leading to growth defects, a phenomenon known as the protein burden/cost effect. Genetic screening in the budding yeast Saccharomyces cerevisiae has isolated several dubious ORFs whose deletions mitigated the protein burden effect, but individual characterization thereof has yet to be delineated. We found that deletion of the YJL175W ORF yielded an N-terminal deletion of Swi3, a subunit of the SWI/SNF chromatin remodeling complex, and partial loss of function of Swi3. The deletion mutant showed a reduction in transcription of genes encoding highly expressed, secreted proteins and an overall reduction in translation. Mutations in the chromatin remodeling complex could thus mitigate the protein burden effect, likely by reallocating residual cellular resources used to overproduce proteins. This cellular state might also be related to cancer cells, as they frequently harbor mutations in the SWI/SNF complex.
Similar content being viewed by others


Introduction
Expression levels of intracellular proteins are tightly controlled to maintain organism capacity for proliferation and survival, and an excess of proteins can cause cellular dysfunction1,2,3. Potentially, any harmless protein inhibits cell growth when it is extremely overproduced, because it depletes cellular protein production resources. This phenomenon is known as the protein burden/cost effect3,4,5,6. The protein burden effect was initially observed as growth defects of bacterial cells overexpressing gratuitous proteins7, and later analyzed in yeast as well2,4,6,8. The protein burden effect is triggered by the cost of gene expression upon overexpression of gratuitous proteins; the overexpression overloads cellular transcription and translation resources4,9. Because the protein burden effect is triggered by the massive overexpression of unneeded proteins, cancer cells, where an increase in the chromosome numbers is common10,11, should be under the condition of the protein burden effect. While the protein burden effect initially appears to be a simple phenomenon, little is known about the physiological conditions and cellular responses triggered by the protein burden effect.
Extreme overexpression of fluorescent proteins such as GFP and RFP are thought to trigger this effect4,6,8,12. To clarify the physiology of cells suffering from protein burden effect, we recently conducted genetic profiling13. Upon isolating a series of deletion and temperature-sensitive mutants harboring genetic interactions affecting the overproduction of GFP (GFP-op) in the budding yeast Saccharomyces cerevisiae, we found that the deletion of certain uncharacterized ORFs mitigated growth defects triggered by GFP-op. However, the molecular details underpinning alleviation of the protein burden effect in these mutants remained unclear.
The SWI/SNF complex is a chromatin remodeling complex that remodels nucleosomes and changes chromatin structure by using the hydrolysis energy of ATP14. The SWI/SNF complex is evolutionarily conserved in eukaryotes15. In humans, the complex is also known as BAF or PBAF complex15. The SWI/SNF complex is composed of 12 subunits in budding yeast and 11–15 subunits in human16,17. The SWI/SNF complex has Swi2/Snf2 as the ATPase component, Swi3 and Snf5 as the core subunits, and several other accessory subunits. 20% of all human cancers have mutations in the SWI/SNF complex subunits18. For instance, 98% of Rhabdoid tumors and 20–40% of Familial schwannomatosis have a homozygous deletion or truncating mutation in SNF519. However, the underlying mechanism relating to the mutations in the SWI/SNF complex and cancer physiology is still unclear.
In this study, we characterized the yeast deletion mutants in which the growth defects triggered by GFP-op are mitigated, and revealed that one of the deletion mutants unexpectedly created an N-terminal deletion of SWI3, a component of the SWI/SNF complex, and a reduction in transcription levels of certain genes. We thereby suggest that transcriptional alterations may free up ribosomes to accept ectopically expressed mRNA for translation and mitigate the protein burden effect.
Results
Deletion of YJL175W ORF mitigates GFP-op-triggered growth defects
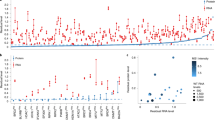
Overproduction of GFP (yEGFP3) under the control of strong TDH3 promoter from a multicopy plasmid pTOW40836 causes growth defects, probably due to the protein burden effect6 (Fig. 1C). We recently performed a systematic screening of deletion mutants and temperature-sensitive mutants in which GFP-op triggered growth defects were aggravated or mitigated13. We performed the synthetic genetic array analysis20 to obtain genetic interaction scores between GFP-op and the mutants. The genetic interaction score indicates how much the growth of GFP-op in a mutant differs from what is expected from each of the growth of GFP-op in the wild type and the growth of the vector control in the mutant. If the score is negative (namely the mutant negatively interacts with GFP-op), the growth defect triggered by GFP-op is aggravated. While if the score is positive (namely the mutant positively interacts with GFP-op), the growth defect is mitigated.

Deletion of YJL175W mitigates GFP-op-triggered growth defects. (A) Dubious ORFs of whose deletions showed positive interactions with GFP-op overlapping other ORFs. Gray and blue arrows show dubious ORFs, and the verified ORFs overlapped with them, respectively. (B) Genetic interaction scores of indicated mutants with GFP-op. For each mutant, the scores from two independent experiments are shown. Data were obtained from Kintaka et al.13. Genetic interaction score was calculated from colony size differences among control strains. NA: not analyzed. (C,D) Max growth rates (orange bars) and max GFP fluorescence levels (green boxes) of wild type (WT) and indicated mutants grown in the synthetic medium (−Leu/Ura). (C) The max growth rate and max GFP fluorescence p-values. (D) The max GFP fluorescence p-values. (E) Growth curves of WT and the YJL175W deletion mutant with empty vector in synthetic medium (−Leu/Ura). (F) Growth curves and GFP fluorescence of WT and the YJL175W deletion mutant upon GFP-op in synthetic medium (−Leu/Ura).
The screening isolated 100 mutants positively interact with GFP-op13, and the mutants contained the deletions of four dubious ORFs; YGL024W, YGL218W, YJL175W, and YKL053W. These ORFs overlap with and are located on opposite strands from the verified genes PGD1, MDM34, SWI3, and ASK1 and are thus unlikely to encode functional proteins (Saccharomyces Genome Database, Fig. 1A). Figure 1B shows the genetic interaction scores between GFP-op and the indicated mutants. All ykl053wΔ and TS mutants of ASK1 (ask1-2 and ask1-3) demonstrated positive interactions, suggesting that ykl053wΔ disrupts the function of the ASK1 gene. Conversely, although ygl218wΔ and yjl175wΔ yielded positive interactions, mdm34Δ and swi3Δ did not. This suggests that the deletions of YGL218W and YJL175W result in different consequences due to loss of function of MDM34 and SWI3.
We then analyzed growth rates and GFP levels in liquid media to confirm the positive interactions between GFP-op and deletion mutants of three dubious ORFs. Among the three deletion mutants, only yjl175wΔ showed a significantly higher growth rate (p = 0.002; Fig. 1C); yjl175wΔ also presented with higher GFP levels than wild type (WT) cells (p = 0.0001; Fig. 1C). This phenotype was not observed in swi3Δ cells wherein GFP levels were lower than those in WT cells (p = 0.01; Fig. 1D), indicating that yjl175wΔ does not cause loss of function by SWI3. Figure 1E,F show the growth curves and GFP expression levels of yjl175wΔ and WT cells, and the dramatic decrease in growth rate triggered by GFP-op in WT was not observed in these cells. We thus focused on YJL175W-SWI3 for subsequent analysis.
Deletion of YJL175W leads to partial loss of function of Swi3
We then performed transcriptome (RNA-seq) analyses to elucidate the consequences of YJL175W deletion. We first analyzed the transcripts expressed at the YJL175W-SWI3 locus (Fig. 2A). Although the deletion of YJL175W removes the 5′ region of SWI3 (Figs. S1 and S2), partial SWI3 transcripts were still expressed, with an estimated expression level at about 63% of WT. The end of the transcript (dotted line, Fig. 2A) suggests that the deletion of YJL175W produced a truncated Swi3 lacking its N-terminal 193 amino acid (Fig, S2). Swi3 is a subunit of the SWI/SNF chromatin remodeling complex21, and known functional domains of Swi3 are located at the C-terminus22,23; the truncated Swi3 contains all three domains, SWIRM, SANT, and LZ (Fig. 2A), suggesting that a truncated Swi3 retains some function. Western blot analysis validated the expression of truncated Swi3 in yjl175wΔ cells (Fig. 2B).

Deletion of YJL175W creates partial loss of function of Swi3. (A) RNA-seq reads mapped to the YJL175W-SWI3 locus in the wild type (WT) and yjl175w∆ transcripts displayed using IGV (2.4.9). Corresponding locations of Swi3 domains and YJL175W are also shown. Dotted line represents the predicted transcript end of truncated SWI3 in yjl175w∆. Expression levels of the SWI3 transcripts (TPM) in each cell are also shown. (B) Truncated form of Swi3 expressed in yjl175w∆ cells. The TAP-tag was fused to the C-terminus of SWI3 in WT and yjl175w∆, and Swi3-TAP was then detected by Western blot. Total cellular protein content is also shown. The full-length gel and blot images are shown in Fig. S5. (C) Relationship between expression changes of transcripts upon SWI3 and YJL175W deletion. Red dots show transcripts with a false discovery rate (FDR) < 0.05 and log2 FC > |1|. (D,E) Expression changes of transcripts of transcription factors under swi3∆ and yjl175w∆. Only transcription factors known to be affected by the SWI3 deletion16 are shown. In E, average log2 expression changes of increased and decreased genes under swi3∆ and yjl175w∆ are shown. (F) Distributions of expression changes of transcripts in swi3∆ and yjl175w∆. The sample number is represented by n and the Pearson correlation coefficiency by r. Expression change is shown as log2 FC over WT.
To further assess the effects of YJL175W deletion, we next compared the transcriptional profiles of swi3Δ16 and yjl175Δ (Supplementary data set). Complete transcriptional changes associated with SWI3 and YJL175W deletions were weakly correlated (r = 0.37) (dots, Fig. 2C), a correlation that increased (r = 0.68) when transcripts with expressions significantly changed by yjl175wΔ were compared (red dots, Fig. 2C). The previous study demonstrated that the expression levels of mRNAs encoding transcription factors were significantly altered in swi3∆ cells16, and indeed, the YJL175W deletion presented with expression changes in transcription factors similar to those associated with SWI3 deletion (r = 0.61; Fig. 2D,E). These results suggest that the YJL175W deletion resulted in a similar transcriptional change as the deletion of SWI3. However, the transcriptional change range was much wider in swi3Δ cells (standard deviation (SD) = 0.86) than that in yjl175wΔ cells (SD = 0.63; Fig. 2F), indicating that the deletion of YJL175W resulted in less pronounced transcriptional changes than the deletion of SWI3. This difference in transcriptional changes was also observed for the expression of transcription factors; although changes in the transcription factors were equally distributed in swi3∆ cells, the transcriptional decrease was far greater than the increase in yjl175∆ cells (Fig. 2E).
We thereby concluded that the YJL175W deletion caused partial loss of function of SWI3, particularly related to the activation of transcription of Swi3 targets, as the underlying cause of the difference between the phenotypes of swi3∆ and yjl175w∆ shown in Fig. 1.
Transcriptional consequences of N-terminal deletion of Swi3
We then analyzed the transcriptional profile of yjl175wΔ in more detail to further delineate the transcriptional consequences of the N-terminal deletion of Swi3 (swi3ΔN) created by the deletion of YJL175W. Figure 3A shows variations in transcripts between WT and yjl175w∆ cells. The number of decreased genes (230) was about 3.4 times higher than the number of increased genes (67). Overall alterations of expression levels of these transcripts upon yjl175w∆ are shown in Fig. 3B. The decreased 230 genes constituted 4.58% of the total transcripts in WT cells and were decreased to 1.18% in yjl175w∆. The increased 67 transcripts constituted only 0.14% of total WT transcripts and were increased to only 0.45% in yjl175w∆ (Fig. 3B). This asymmetrical distribution, showing more decreased transcripts than increased ones (Fig. 3C), suggests an overall reduction of transcription in yjl175w∆ cells compared with that in WT cells. Of note, however, these ratios do not necessarily reflect intracellular mRNA ratios, as the number of each transcript is normalized to the number of total transcripts.

Transcriptional consequences of deletion of YJL175W. (A) Comparison of transcript levels between WT and yjl175w∆. Blue and red dots indicate significantly increased transcripts (FDR < 0.05 and log2 FC > 1) and decreased transcripts (FDR < 0.05 and log2 FC < − 1), respectively. Numbers of increased and decreased genes are also shown. (B) Distributions of all, 67 increased, and 230 decreased transcripts upon YJL175W deletion; their proportions in WT and in yjl175w∆ transcripts are also shown as the percentage. The percentage was calculated as follows: (the sum of TPMs of 67 increased or 230 decreased transcripts)/(the sum of TPMs of all 6401 genes) × 100. (C) Alteration of transcripts upon deletion of YJL175W. The transcript percentage was calculated as in (B). (D) Expression levels of transcripts coding for secreted proteins (secretome) in WT and yjl175w∆. Purple dots show transcripts of secretome proteins. The secretome protein list (1088 proteins) was obtained from Costa et al.28. (E,F) Proportions of the numbers of genes (E) and sums of transcripts (F) of secretome proteins in the 230 genes significantly decreased upon YJL175W deletion. Transcript sum is calculated by the sum of the TPMs of contained transcripts. (G) Expression levels of the top 100 highly expressed genes in WT and yjl175w∆. Five representative genes with expression levels significantly reduced upon YJL175W deletion are shown. Genes with asterisks showed a greater than twofold decrease upon YJL175W deletion. The adjusted p-values of the expression changes of indicated genes were <2.1E-30 (Table S3) (H). Expression levels of transcripts of essential genes in WT and yjl175w∆. Dark blue dots represent transcripts of essential genes. Essential gene list (1274 genes) was obtained from Giaever et al.29. Expression change is shown as log10 TPM.
We then performed gene ontology (GO) enrichment analysis to reveal processes and genes affected in the yjl175w∆ cells (Tables 1, 2 and S1). The set of 67 increased genes was enriched with GOs related to methionine biosynthetic processes, likely associated with an increase in the transcription factors MET28 and MET32 (Fig. 2E)24,25. The set of 230 decreased genes was enriched with GOs related to transposon activity, which is likely associated with a decrease in the transcription factors TYE7 and TEC1 (Fig. 2E)26,27. Decreased genes were significantly enriched with GO genes for secreted proteins28 (Table 2, GO0071944: cell periphery); in fact, 77 out of the 230 decreased genes encoded secretory proteins (Fig. 3D,E), a level that was significantly higher than expected (p = 1.62E–26). Transcripts of secreted proteins accounted for more than 60% of the decreased transcripts, whereas transposon-related transcripts accounted for only 0.2% (Fig. 3F), suggesting that those secreted proteins were highly expressed. Figure 3G shows the expression levels of the top 100 highly expressed genes in WT. Specific reductions in yjl175w∆ transcripts encoding secretory proteins, such as CWP2, MFA2, SED1, NCW1, and MFA1, were observed, and no transcript increased more than twofold. The decreased secretory genes were not essential for viability29, and the yjl175w∆ cells did not present with reductions in growth under normal growth conditions (Fig. 1E). We thereby speculated that the YJL175W deletion might only affect the transcription of non-essential genes. In fact, essential genes made up just 5% (15 genes) of the 297 genes with expression levels altered by more than twofold (Fig. 3H), which was far fewer than expected (p = 1.82E–15).
We thus concluded that the swi3ΔN mutant generated by the deletion of YJL175W caused a selective transcriptional reduction of genes encoding highly expressed, non-essential secreted proteins.
N-terminal deletion of Swi3 leads to change in translation status
Given that swi3ΔN (yjl175w∆) was shown to mitigate the protein burden effect, we speculated that translation status in the mutant cells affected the ectopic expression of proteins therein. We thus analyzed the polysome profiles of WT and yjl175w∆ (Fig. 4A) by calculating polysome/monosome ratios (Fig. 4B) to represent translation efficacy in cells. In the vector control, the polysome/monosome ratio was significantly lower in yjl175w∆ than that in WT; that is, the number of translating ribosomes was lower in yjl175w∆. This result suggests that the reduction in transcription of some genes in yjl175w∆ might lead to a reduction in overall translation, which might free up ribosomes used to produce foreign proteins. Interestingly, the difference in polysome/monosome ratio between WT and yjl175w∆ cells did not occur under GFP-op conditions (Fig. 4B), suggesting that under such conditions, the same number of ribosomes is engaged in translation in yjl175w∆ as that in WT, leading to the higher GFP levels observed in the mutant.

Translational consequence upon deletion of YJL175W. (A) Polysome profiling of WT and yjl175w∆ cells under normal (vector) and GFP-op conditions. Predicted monosome and polysome peaks are shown. (B) Ratios of polysome to monosome in indicated conditions. (C,D) Conceptual pie chart models interpreting how translational changes observed in yjl175w∆ mitigated growth defects upon GFP-op. These charts show the hypothetical allocations of ribosomes in indicated conditions, but their percentages are not based on the real data. See the main text for a detailed explanation of these charts.
Discussion
In this study, we showed that the deletion of YJL175W unexpectedly led to a partial loss of function of Swi3 (swi3∆N) and mitigated growth defects triggered by GFP-op, a condition implicated in the protein burden effect. We believe that YJL175W is a misannotated ORF because there is no homologous protein even in closely-rerated Saccharomyces species (data not shown), and no transcript corresponding to the YJL175W locus was detected (Fig. S3). Therefore, the phenotype of the YJL175W deletion should be solely created by the truncation of Swi3 accidentally created by the YJL175W deletion. A substantial portion of transcription was reduced upon swi3∆N via the selective reduction of transcripts encoding highly expressed, secreted proteins (Fig. 3). This transcriptional reduction also led to a reduction in total protein translation (Fig. 4A,B). Cellular conditions created by the YJL175W deletion were speculated in accordance with these results. Figure 4C,D show conceptual pie chart models explaining such conditions. In normal conditions, ribosomes are used to translate both proteins that are and are not required for growth (Fig. 4C, WT). In yjl175w∆ cells, the number of transcripts encoding proteins not required for growth was lower, and consequently, ribosomes tasked with translating those transcripts were freed up to perform other translations (Fig. 4C, yjl175w∆). In GFP-op, ribosomes are used to translate GFP, leading to a reduction in the number of ribosomes translating proteins required for growth followed by growth defects (Fig. 4D, WT). Free ribosomes created by yjl175w∆ reinstates extra ribosomes for GFP translation, avoiding a reduction in the translation of proteins required for growth (Fig. 4D, yjl175w∆) and mitigating growth defects.
Although yjl175w∆ cells have an increased capacity to produce exogenic proteins, this phenomenon is associated with an apparent trade-off. These cells are sensitive to stressors, such as high temperature, alkaline pH, and many chemicals (Saccharomyces Genome Database), perhaps due to a transcriptional reduction of stress tolerance-related genes. The selective reduction of transcription not required for normal growth, but required for stress responses, likely affords additional resources for the production of exogenic proteins. In yjl175w∆ cells, total resources for protein production were unchanged compared with those in WT cells; however, the allocation of these resources was found to be altered.
Swi3 is a subunit of the SWI/SNF chromatin remodeling complex that regulates transcription by remodeling chromosomes. The deletion of this subunit thereby causes significant changes in the transcriptional profiles of many genes16 (Fig. 2). The N-terminal deletion of Swi3 created by yjl175w∆ leaves three important domains associated with Swi3 function, and transcriptional change via yjl175w∆ is less pronounced than that via swi3∆ (Fig. 2C). Interestingly, the N-terminal deletion does not result in milder transcriptional change but instead yields a different transcriptional profile from that of swi3∆, such as opposing directional changes observed in the transcripts of ribosomal proteins (Fig. S4). This finding indicates that the N-terminus of Swi3 might influence a specific set of genes.
A hallmark of cancer cells is an increase in chromosome number that triggers a massive overproduction of proteins10,11. Cancer cells must thereby evolve to overcome the protein burden effect. At least 20% of all human cancers contain mutations in the SWI/SNF complex including Swi3 homolog BAF155/SMARCC1 and BAF170/SMARCC218,30,31. Loss of expression or the C-terminal truncation of BAF155 is associated with the proliferation of human cancer cell lines32. While mutations of Swi3 homologs found in cancer cells are not entirely same as the N-terminal deletion of Swi3 described here in yeast, it might create a similar situation to that of evolved cancer cells, that is, mutations in the SWI/SNF complex associated with an extensive transcriptional reduction and mitigation of the protein burden effect, facilitating rapid growth.
Materials and Methods
Strains used in this study
Strains used in this study are listed in Table 3. The yjl175w∆ deletion cassette was generated by PCR using the genome of yjl175w∆ strain in the yeast knockout collection (Horizon) as a template with the primers 5′-CGGCCGCTCTAGAACTAGTGGATCCGATGGAATTTCTTTGTAAACGCA-3′ and 5′-ATTGGGTACCGGGCCCCCCCTCGAGGCCCAAAAACGTATCTCTGCTTA. The cassette was cloned into the pRS413 vector via gap-repair cloning method in yeast. The C-terminal coding region of truncated SWI3 in the plasmid was fused in-frame to the TAP tag generated by PCR using a TAP collection strain as a template (Dharmacon) and gap-repair cloning in yeast. PCR reactions for yjl175w∆-TAP cassette were performed with the primers 5′-GATGGAATTTCTTTGTAAACGCATT-3′ and 5′-GCCCAAAAACGTATCTCTGCTTAAA-3′. BY4741 was transformed with the yjl175w∆-TAP cassette by homologous recombination and selected on yeast extract/peptone/dextrose (YPD) plates containing 200 µg/mL G418. Insertion of the cassette was verified by genomic PCR with primers external to the integration locus, 5′-GACCGTTCCCAGTTAAGGTCGA-3′ and 5′-CGCTGCCAATGCTGAAGTATGT-3′.
Growth conditions and yeast transformation
Yeast culture and transformation were performed as previously described33. YPD medium was used for yeast culture. Synthetic complete (SC) medium lacking uracil (–Ura) or leucine and uracil (−Leu/Ura) was used to culture yeast cells harboring plasmids. Strains used in this study are listed in Table 1. BY4741 was used as WT control. GFP is overproduced under control of the TDH3 promoter on the 2-µm plasmid pTOW4083634. pTOW40836 was used as empty vector control.
RNA-seq analysis
Sample preparation for RNA-seq was performed as described in Takasaki et al.35. BY4741 and yjl175w∆ were grown in SC − Ura at 30 °C and sampled during the logarithmic growth phase. Cells were collected by centrifugation at 10,000 g for 10 min. Total RNA from 0.5 g of collected yeast cell samples was extracted using a FastRNA Pro Soil-Direct Kit (Qbiogene) according to the manufacturer’s instructions. Samples were treated with DNase to remove genomic DNA from total RNA with Recombinant DNaseI (RNase-free) (TaKaRa). After ethanol precipitation, the purified total RNA was stored at −80 °C until use in subsequent experiments. RNA purity and concentrations were estimated with a NanoDrop spectrophotometer (Thermo Fisher Scientific). RNA quality and quantity were estimated with a Bioanalyzer (Agilent, Tokyo, Japan). cDNA from purified mRNA was synthesized using a cDNA Synthesis Kit (TaKaRa) according to defined protocols. Briefly, first, poly(A) RNA was reverse-transcribed with an oligo (dT)-T7 primer containing a T7 promoter sequence; this was used to synthesize double-stranded cDNA. cDNA templates were then transcribed in vitro with T7 RNA polymerase (TaKaRa), yielding large amounts of antisense RNA (aRNA). Finally, aRNA was further reverse-transcribed to cDNA with a biotinated oligo (dT) primer for next generation sequencing. Sequencing of the synthesized cDNA was performed by paired-end sequencing on an Illumina Hiseq2000 sequencing system provided by the Hokkaido System Science Co., Ltd. We analyzed three biological replicates for each strain.
Transcriptome data analysis
RNA-seq data of Swi3 knockout and WT strains (GEO, ID: 302174480, 302174481, 30174486, and 302174487)16 were downloaded from the SRA database. Our RNA-seq data of deletion of YJL175W and WT are available from the SRA database (SRA, ID: SRR10848971 and SRR10848972). All RNA-seq data, including ours and Dutta’s, were aligned to the Ensembl R64-1-1 genome using HISAT2 version 2.1.0 with gene annotations from Ensembl R64-1-1. HISAT2 options were -p 8 –dta. Ensembl R64-1-1 was obtained from iGenomes by Illumina, Inc. Visualizations of mapped fragments were conducted with an Integrative Genome Viewer (2.4.9). Assembly and estimation of transcript abundances were performed with HTSeq version 0.11.1. Downstream analysis was conducted using Python (3.6.8). Transcripts per kilobase millions (TPMs) were calculated according to a previously described method36. Calculated TPM values are provided in Tables S2 and S3.
TAP-tag western blot
Detection of TAP-tag protein by Western blot was performed as described in Ishikawa et al.37. Yeast strains were aerobically cultured at 30 °C in 2 mL of YPD medium. Optical density at 660 nm (OD660) was measured, and units of 1 OD660 were harvested during the log phase. Cells were treated with 1 mL of 0.2 N NaOH for 5 min at room temperature and then suspended in 2× NuPAGE LDS sample buffer (Invitrogen) and heated to 70 °C for 10 min. Protein lysate in the supernatant was labeled with EzLabel FluoroNeo (ATTO) and subjected to polyacrylamide gel electrophoresis with lithium dodecyl sulfate (SDS-PAGE), followed by Western blot with PAP (Sigma-Aldrich) (1:2000) and peroxidase-conjugated secondary antibody (Nichirei Biosciences) (1:1000). We used a NuPAGE 4–12% Bis-Tris Gel (Invitrogen) for SDS-PAGE and an iBlot Transfer Stack PVDF membrane (Invitrogen) for Western blot. Chemiluminescence was induced by SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) and detected on a LAS-4000 image analyzer (Fujifilm) using ImageQuant LAS 4000 (GE Healthcare).
Polysome profiling
Frozen yeast cells were mixed with frozen droplets of 600 µL lysis buffer (20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol, 100 µg/mL cycloheximide, and 1% Triton X-100) and lysed with Multi-beads Shocker (Yasui Kikai). Lysates were treated with 25 U of TURBO DNase (Thermo Fisher Scientific) and cleared by centrifugation at 20,000 g for 10 min at 4 °C. RNA concentration in the lysate was measured with a Qubit RNA BR Assay Kit (Thermo Fisher Scientific). Sucrose gradients (10–50% sucrose in lysis buffer without Triton X-100) were prepared in 14 × 95 mm open-top Polyclear centrifuge tubes (SETON) using a Gradient Station (BioComp). Lysates containing 20 µg RNA were loaded on top of the sucrose gradients and centrifuged at 35,300 rpm for 2.5 h at 4 °C using a rotor P40ST (Hitachi Koki). After ultra-centrifugation, the absorbance at 254 nm was measured continuously on a Bio-mini UV monitor (ATTO).
Statistical analysis
Where appropriate, values are expressed as mean ± SD. Statistical analyses of RNA-seq date from three biological replicates were conducted using the Benjamini–Hochberg method, and a false discovery rate <0.05 was considered statically significant38. Statistical analyses with more than two groups were performed by Bonferroni correction. An adjusted p-value of <0.05 was considered statistically significant. All data are representative of multiple repeated experiments.
References
Sopko, R. et al. Mapping pathways and phenotypes by systematic gene overexpression. Mol. Cell 21, 319–330 (2006).
Makanae, K., Kintaka, R., Makino, T., Kitano, H. & Moriya, H. Identification of dosage-sensitive genes in Saccharomyces cerevisiae using the genetic tug-of-war method. Genome Res. 23, 300–311 (2013).
Moriya, H. Quantitative nature of overexpression experiments. Mol. Biol. Cell 26, 3932–3939 (2015).
Kafri, M., Metzl-Raz, E., Jona, G. & Barkai, N. The Cost of Protein Production. Cell Rep 14, 22–31 (2016).
Snoep, J. L., Yomano, L. P., Westerhoff, H. V. & Ingram, L. O. Protein burden in Zymomonas mobilis: Negative flux and growth control due to overproduction of glycolytic enzymes. Microbiology 141, 2329–2337 (1995).
Eguchi, Y. et al. Estimating the protein burden limit of yeast cells by measuring the expression limits of glycolytic proteins. Elife 7, 1–23 (2018).
Kurland, C. G. & Dong, H. Bacterial growth inhibition by overproduction of protein. Mol. Microbiol. 21, 1–4 (1996).
Farkas, Z. et al. Hsp70-associated chaperones have a critical role in buffering protein production costs. Elife 7, 1–23 (2018).
Frumkin, I. et al. Gene Architectures that Minimize Cost of Gene Expression. Mol. Cell 65, 142–153 (2017).
Tang, Y. C. & Amon, A. Gene copy-number alterations: A cost-benefit analysis. Cell 152, 394–405 (2013).
Ben-David, U. & Amon, A. Context is everything: aneuploidy in cancer. Nat. Rev. Genet 21, 44–62 (2020).
Kintaka, R., Makanae, K. & Moriya, H. Cellular growth defects triggered by an overload of protein localization processes. Sci. Rep 6, 1–11 (2016).
Kintaka, R. et al. Genetic Profiling of Protein Burden and Nuclear Export Overload. bioRxiv 2020.02.25.962068 (2020).
Kassabov, S. R., Zhang, B., Persinger, J. & Bartholomew, B. SWI/SNF unwraps, slides, and rewraps the nucleosome. Mol. Cell 11, 391–403 (2003).
Phelan, M. L., Sif, S., Narlikar, G. J. & Kingston, R. E. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol. Cell 3, 247–253 (1999).
Dutta, A. et al. Composition and Function of Mutant Swi/Snf Complexes. Cell Rep 18, 2124–2134 (2017).
Helming, K. C., Wang, X. & Roberts, C. W. M. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell 26, 309–317 (2014).
Kadoch, C. et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 45, 592–601 (2013).
Wilson, B. G. & Roberts, C. W. M. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 11, 481–492 (2011).
Baryshnikova, A. et al. Synthetic genetic array (SGA) analysis in Saccharomyces cerevisiae and schizosaccharomyces pombe. Methods in Enzymology 470, (Elsevier Inc., 2010).
Peterson, C. L. & Herskowitz, I. Characterization of the yeast SWI1, SWI2, and SWI3 genes, which encode a global activator of transcription. Cell 68, 573–583 (1992).
Boyer, L. A. et al. Essential role for the SANT domain in the functioning of multiple chromatin remodeling enzymes. Mol. Cell 10, 935–942 (2002).
Da, G. et al. Structure and function of the SWIRM domain, a conserved protein module found in chromatin regulatory complexes. Proc. Natl. Acad. Sci. USA 103, 2057–2062 (2006).
Kuras, L., Cherest, H., Surdin-Kerjan, Y. & Thomas, D. A heteromeric complex containing the centromere binding factor 1 and two basic leucine zipper factors, Met4 and Met28, mediates the transcription activation of yeast sulfur metabolism. EMBO J. 15, 2519–2529 (1996).
Blaiseau, P. L., Isnard, A. D., Surdin-Kerjan, Y. & Thomas, D. Met31p and Met32p, two related zinc finger proteins, are involved in transcriptional regulation of yeast sulfur amino acid metabolism. Mol. Cell. Biol. 17, 3640–3648 (1997).
Servant, G. et al. Tye7 regulates yeast Ty1 retrotransposon sense and antisense transcription in response to adenylic nucleotides stress. Nucleic Acids Res. 40, 5271–5282 (2012).
Laloux, I., Dubois, E., Dewerchin, M. & Jacobs, E. TEC1, a gene involved in the activation of Ty1 and Ty1-mediated gene expression in Saccharomyces cerevisiae: cloning and molecular analysis. Mol. Cell. Biol. 10, 3541–3550 (1990).
Costa, E. A., Subramanian, K., Nunnari, J. & Weissman, J. S. Defining the physiological role of SRP in protein-targeting efficiency and specificity. Science (80−) 359, 689–692 (2018).
Giaever, G. et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 387–391 (2002).
Biegel, J. A., Busse, T. M. & Weissman, B. E. SWI/SNF chromatin remodeling complexes and cancer. Am. J. Med. Genet. Part C Semin. Med. Genet 166, 350–366 (2014).
Shain, A. H. & Pollack, J. R. The Spectrum of SWI/SNF Mutations, Ubiquitous in Human Cancers. PLoS One 8 (2013).
del Bove, J. et al. Identification of a core member of the SWI/SNF complex, BAF155/SMARCC1, as a human tumor suppressor gene. Epigenetics 6, 1444–1453 (2011).
Amberg, D. C. & Strathern, J. N. Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual. (CSHL press, 2005).
Moriya, H., Makanae, K., Watanabe, K., Chino, A. & Shimizu-Yoshida, Y. Robustness analysis of cellular systems using the genetic tug-of-war method. Mol. Biosyst. 8, 2513–2522 (2012).
Takasaki, K. et al. Discovery of Glycoside Hydrolase Enzymes in an Avicel-Adapted Forest Soil Fungal Community by a Metatranscriptomic Approach. Plos One 8 (2013).
Li, B., Ruotti, V., Stewart, R. M., Thomson, J. A. & Dewey, C. N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26, 493–500 (2009).
Ishikawa, K., Makanae, K., Iwasaki, S., Ingolia, N. T. & Moriya, H. Post-Translational Dosage Compensation Buffers Genetic Perturbations to Stoichiometry of Protein Complexes. PLoS Genet. 13, 1–22 (2017).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing Yoav Benjamini; Yosef Hochberg Journal of the Royal Statistical Society. Series B (Methodological), Vol. 57, No. 1. (1995), pp. J. R. Stat. Soc. 57, 289–300 (1995).
Brachman, C. B. et al. Designer Deletion Strains derived from Saccharomyces cerevisiae S288C: a Useful set of Strains and Plasmids for PCR-mediated Gene Disruption and Other Applications. Yeast 14, 115–132 (1998).
Winzeler, E. A. et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science (80–) 285, 901–906 (1999).
Ghaemmaghami, S. et al. Global analysis of protein expression in yeast. Nature 425, 737–41 (2003).
Acknowledgements
We thank the members of the Moriya lab for advice and helpful discussions. This work was partly supported by JSPS KAKENHI grant numbers 15KK0258, 17H03618, and 18K19300. This paper is partly based on results obtained from a project commissioned by the New Energy and Industrial Technology Development Organization (NEDO). The funding agencies were not involved in study design, data collection and analysis, decision to publish, or manuscript preparation.
Author information
Authors and Affiliations
Contributions
N.S., Y.E. and H.M. designed the study; N.S., Y.E., R.K., K.M., Y.S., S.I., M.K. and N.K. performed experiments and analyzed the data; N.S. and H.M. prepared figures and wrote the manuscript. M.K., Y.E., Y.S. and S.I. reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saeki, N., Eguchi, Y., Kintaka, R. et al. N-terminal deletion of Swi3 created by the deletion of a dubious ORF YJL175W mitigates protein burden effect in S. cerevisiae. Sci Rep 10, 9500 (2020). https://doi.org/10.1038/s41598-020-66307-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66307-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
