
Abstract
In this paper, applying the density scaling idea to an associated liquid 4-methyl-2-pentanol used as an example, we identify different pressure-volume-temperature ranges within which molecular dynamics is dominated by either complex H-bonded networks most probably leading to supramolecular structures or non-specific intermolecular interactions like van der Waals forces. In this way, we show that the density scaling law for molecular dynamics near the glass transition provides a sensitive tool to detect thermodynamic regions characterized by intermolecular interactions of different type and complexity for a given material in the wide pressure-volume-temperature domain even if its typical form with constant scaling exponent is not obeyed. Moreover, we quantify the observed decoupling between dielectric and mechanical relaxations of the material in the density scaling regime. The suggested methods of analyses and their interpretations open new prospects for formulating models based on proper effective intermolecular potentials describing physicochemical phenomena near the glass transition.
Similar content being viewed by others

Introduction
Investigations of intra- and intermolecular interactions characterizing different molecular structures have held a tremendous amount of attention of many researchers since the 19th century. However, there are still unsatisfactorily explored issues in this extensive field. A commonly known challenge constitutes supramolecular structures and their specific interactions in various thermodynamic phases. This matter becomes especially difficult to investigate if we do not limit our study to the ambient pressure conditions, but extend it over a wide temperature-pressure range to result in relatively large variations in the density domain. In this paper, we present a novel approach to detect regions within which supramolecular structures characterized by complex specific interactions are expected in the thermodynamic space.
Since the beginning of this century, a very promising idea of the density scaling of molecular dynamics in viscous liquids has been successfully developed to make progress in the search for a complete and commonly accepted theory of the glass transition and related phenomena1,2,3,4. According to this idea, a dynamic quantity Y (e.g., viscosity, structural relaxation time or segmental relaxation time in case of polymers) measured at different temperatures T and pressures p along different isobars and isotherms can be scaled onto one master curve well described by a function f of the single variable Γ as follows
where \(\Gamma ={\rho }^{\gamma }/T\) with the density ρ defined as the inverse specific volume \({V}^{-1}\) and the scaling exponent γ which is a material constant independent of thermodynamic conditions according to the typical viewpoint at the beginning of this idea development5. A significance of Eq. (1) relies on the theoretical grounds for its scaling exponent γ related to an exponent of the dominant repulsive part (\(\approx 3\gamma \)) of the effective short-range intermolecular potential derived from the well-known Lennard-Jones potential, which have been satisfactorily verified by computer simulations6,7,8,9,10. Therefore, the power density scaling (PDS) law expressed by Eq. (1) gives a tempting opportunity to study macroscopic material properties by using their underlying intermolecular potential as well as to evaluate parameters of the relevant intermolecular potential based on macroscopic quantities measured for various materials.
It should be noted that the scaling law given by Eq. (1) was called thermodynamical scaling by Casalini and Roland5. This name has been widely used by them and other authors, but almost simultaneously, another term has been proposed, i.e., scaling with a power law in density11. Later, this term has been popularized as density scaling or density scaling law by the Roskilde group, which significantly contributed to understand theoretical grounds for Eq. (1), starting from finding a strong correlation between instantaneous average virial and potential energy in simple simulations models6,7,8. According to the latter approach and earlier publications4, in this paper, we prefer to use the term power density scaling law, which invokes the power form of the density scaling function implemented in Eq. (1), and additionally enables to informatively specify whether the scaling exponent of the PDS law is invariant or not.
In many cases of materials belonging to different material groups (such as van der Waals liquids and polymer melts, but also ionic liquids, some phases of liquid crystals, and metallic glass formers), it has been experimentally validated that the PDS law is obeyed when \(\gamma =const\) for a given material at least to a good approximation12. Thus, the PDS law bears hallmarks of universality, becoming a convenient tool to gain a better insight into complex physicochemical phenomena occurring mainly near the glass transition, but also in the normal liquid state13 and some liquid crystal phases14,15.
Excluding results of molecular dynamics simulations in mainly isotropic simple models, which do not seem to be representative for real materials usually consisted of anisotropic molecules, a few van der Waals liquids16,17 and several metals17,18 have been reported to possess scaling exponents γ dependent on state points or only density, but these cases are still hotly debated19,20,21. However, undoubted examples of the deviation from the PDS law with the invariant scaling exponent γ for a given material have been found among associated liquids22,23,24,25,26. Then, a question arises whether the PDS law with the scaling exponent γ dependent on thermodynamic conditions for a given material may usefully inform us about some properties of the material? To answer this question, in this paper, we perform the density scaling based analysis of supercooled monohydroxy alcohol, 4-methyl-2-pentanol (4M2P) as an example of associated liquids, and consequently provide a handy tool to identify the range of thermodynamic variables (p-V-T), within which complex hydrogen-bonded networks should be formed.
Results and Discussion
To collect an experimental data set covering a wide thermodynamic range, we have performed extensive measurements of dielectric relaxations and viscosity at ambient and elevated pressure as well as the speed of sound under high pressure, density and specific isobaric heat capacity at atmospheric pressure for 4M2P. Details of the measurement techniques have been described elsewhere27,28,29,30,31 and their brief description including experimental ranges for 4M2P is presented in Methods.
It is worth noting that monohydroxy alcohols with hydroxyl group located at or close to the terminal position of the carbon chain constitute an important group of associated liquids. For these materials, chain-like H-bonded structures prevail32. Motions of these transient chains are source of the so-called Debye-like relaxation observed mainly in the dielectric spectra of the alcohols like e.g. 1-propanol or 2-ethyl-1-hexanol (2E1H)33,34. This process (exponentially broadened at elevated pressure) is slower and decoupled from the structural relaxation, although it also exhibits a non-Arrhenius temperature dependence of the relaxation times, \({\tau }_{D}\)32. In case of 4M2P, we have also observed the same kind of dielectric relaxation (see Methods herein and Supplementary Fig. S1) along 16 isotherms and 1 isobar at ambient pressure (see Supplementary Fig. S2). It should be noted that comparison of the Debye-like relaxations on the dielectric loss spectra of 2E1H and 4M2P reveals virtually the same intensity of these processes (see Supplementary Fig. S3). It was shown35 that for primary alcohols the amplitudes of the Debye-like mode are very similar. This indicates the same architecture and population of H-bonded structures, in that case dominated by chain-like associates. On the other hand, for alcohols with hydroxyl group located at non-terminal position on the alkyl chain, e.g. 5-methyl-3-heptanol and 4-methyl-3-heptanol, the intensity of the Debye-like process decrease36,37. This is the consequence of increasing number of ring-like entities in the liquid. Since the amplitude of the Debye-like mode in 2E1H is the same as for 4M2P it can be concluded that for the latter alcohol H-bonded structures are dominated by the transient chains. With pressurization the intensity of the Debye-like mode increases, however this increase is smaller than in the material isobarically cooled at ambient pressure (see Supplementary, inset to Fig. S4). This difference is reflected by changes of dielectric strength, Δε, of the relaxation during isobaric cooling and isothermal pressurization (see Supplementary Fig. S4). Although for both thermodynamic paths Δε increases with increasing τ but for isothermal results this increase is much lower. It has to be noticed that in case of total decomposition of H-bonded structures responsible for the Debye-like process during pressurization we will expect a gradual disappearance of this relaxation. Though this is not the case here and it can be concluded that although the compressive forces modify the properties of Debye-like mode markedly, they definitely do not destroy the chain-like associated structures that are the source of this process.
To attempt to scale the Debye-like relaxation times in terms of Eq. (1), the experimental dielectric data collected in the T-p domain should be transformed to the T-V or T-ρ domain. To do that we have parametrized the dependence V(T,p) by fitting the pVT measurement data to the commonly known Tait equation of state38 to find values of its parameters shown in Methods. Then, the isothermal and isobaric dependences \({\tau }_{D}(V)\) have been established (Supplementary Fig. S5).
A convenient criterion for the density scaling expressed by Eq. (1) with \(\gamma =const\) can be formulated by the following linear equation39,
where the parameter C may depend only on the dynamic quantity Y and the scaling exponent γ is a constant slope of the log-log dependence of T vs ρ (or \({V}^{-1}\)) to a good approximation. Then, the PDS law with the invariant scaling exponent γ is obeyed for a given material.
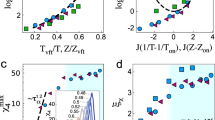
We have applied the PDS criterion to the experimental Debye-like relaxation times of 4M2P at \({\tau }_{D}=const\) selected every half decade in the range of \({\tau }_{D}\) between 0.1μs and 0.1 s. As can be seen in Fig. 1(a), Eq. (2) with \(\gamma =const\) is not met in the entire analysis range, which includes the temperature-density regions of both the invariant and state-point dependent scaling exponent γ, where the latter region has been shaded. From the thermodynamic region of the linear dependences of \({\tau }_{D}\) described by Eq. (2), we have established a constant value of the scaling exponent, \(\gamma =2.50\pm 0.02\), which is relatively small and similar to earlier reported values of γ for some H-bonded liquids (e.g. sorbitol)5 that satisfied the PDS law with \(\gamma =const\) to a good approximation. To better recognize the thermodynamic region of state-point dependent scaling exponent γ, we have moved horizontally the isochrones shown in Fig. 1(a) to the isochrone at \({\tau }_{D}=0.1{\rm{s}}\), which is linear in the analysis range. In this way, we have illustrated the deviation from the PDS law with \(\gamma =const\) in Fig. 1(b). Moreover, in the inset in Fig. 1(b), we have reconverted the T-ρ domain to the T-p domain of the analysis based on Eq. (2), showing for convenience the T-p region of state-point dependent scaling exponent γ.

(a) Log-log plot of temperature vs. inverse volume at different constant Debye-like relaxation times. (b) Determination of density scaling exponent by horizontal shifts of the isochrones shown in panel (a) onto the isochrone at τD = 0.1 s. The shaded areas in both panels represent the pVT domain within which the density scaling with γ = const is not satisfied.
All the analyses based on the PDS criterion (Eq. (2)), which are presented in Fig. 1, give evidence that the Debye-like relaxation times of 4M2P conform to the PDS law (Eq. (1)) with \(\gamma =const\) at \({\tau }_{D} > 1\,{\rm{ms}}\) (excluding almost all points at ambient pressure), while at \({\tau }_{D}\le 1\,{\rm{ms}}\) only above some limiting values of pressure. The limiting values of p systematically increase (from 0.1 MPa to 650 MPa) with increasing values of temperature (from 160 K to 273 K). Correspondingly, the entire isotherms of \({\tau }_{D}\) between 283 K and 323 K can be scaled according to Eq. (1) with \(\gamma =2.5\), while other isotherms of \({\tau }_{D}\) measured at temperatures between 203 K and 273 K satisfy the PDS law with this scaling exponent γ above some value of \({\tau }_{D}\) separately established for each of them (Fig. 2(a)). Along the demarcation line between the invariant and state-point dependent scaling exponent γ for the low-temperature isotherms, the values of \({\tau }_{D}\) increases with decreasing isotherm temperature from \({\tau }_{D}=2\,{\rm{\mu }}{\rm{s}}\) at T = 273 K and reaches a plateau \({\tau }_{D}\approx 1\,{\rm{ms}}\) at \({\rm{T}}\le 233{\rm{K}}\). This analysis leads to no density scaling with \(\gamma =2.5\), which is presented in Fig. 2(b) for the values of \({\tau }_{D}\) below the demarcation line in case of the low-temperature isotherms. It is worth noting that Casalini and Ransom have recently shown40 that the scaling exponent gamma increases with increasing pressure, tending to a constant value in the high pressure limit in case of a few hydrogen bonded materials. Our results obtained for 4M2P reveal a more complex dependence of the scaling exponent on thermodynamic conditions, because we have found that the pressure dependence of the scaling exponent for 4M2P strongly changes with varying temperature. That is, as already mentioned, an increase in temperature is related to a considerable increase in the pressure limit above which the \(T{V}^{\gamma }\)–scaling is satisfied with \(\gamma =2.5\) by the Debye-like relaxation times for 4M2P (see the inset in Fig. 1(b)).

(a) Density scaling plot with the exponent γ = 2.5 after excluding parts of isotherms at low temperatures and short relaxation times according to the analysis shown in Fig. 1. The atmospheric isobar does not match the scaling pattern satisfied by isotherms at high temperatures and selected parts of isotherms at low temperatures. (b) Density scaling plot with the exponent γ = 2.5, including the atmospheric isobar and isotherms at low temperatures; such a density scaling is invalid for isotherms at low temperatures and short relaxation times.
The analyses of the thermodynamic evolution of the Debye-like relaxation times \({\tau }_{D}\) depicted in Figs. 1 and 2, which are respectively based on the PDS criterion (Eq. (2)), and then on the PDS law (Eq. (1)), show that the density scaling behavior of 4M2P is more complex than those earlier observed for associated liquids. For instance, in case of the already mentioned monohydroxy alcohol 2E1H, Fragiadakis et al.41 reported that the thermodynamic scaling is generally not valid, and later Pawlus et al.25 have shown that the PDS law with a constant \(\gamma =1.8\) can be, however, successfully applied to the Debye-like relaxation times \({\tau }_{D}\), but only up to the pressure of ca. 0.5 GPa, above which the scaling exponent γ linearly increases with \({\tau }_{D}\). Thus, it is uniquely defined by the time scale of H-bonded structures rearrangements, which are reflected in the Debye-like dielectric process of 2E1H. An associated liquid N,N-diethyl-meta-toluamide (DEET), widely used as an insect repellent, reveals a more complicated dependence of the scaling exponent γ on the structural relaxation time τ described by a cubic function in the entire range of the density scaling based analysis. However, the monohydroxy alcohol 4M2P considered herein is characterized by two separate thermodynamic regions, within which the PDS law with \(\gamma =const\) is either valid or broken, where the scaling exponent γ in the latter one cannot be described by any function only of the time scale \({\tau }_{D}\) as can be deduced from Fig. 1(a). Moreover, the scaling exponent γ cannot be also any function only of density, which was earlier suggested for two supercooled van der Waals liquids measured in the wide T-p range16, because it has not been possible to superimpose the nonlinear parts of isochrones in Fig. 1(b). It means that the scaling exponent γ for 4M2P is state-point dependent in the thermodynamic region where the PDS law is broken for this material, i.e., it varies when temperature and pressure changes.
In case of associated liquids, the deviations from the PDS law with \(\gamma =const\) are most likely caused by forming complex H-bonded networks. Earlier investigations of the dynamics of H bonds under high pressure42,43 showed that the increasing pressure rather results in the increase in the degree of H bonds (i.e., in their strength or/and number) at least up to 1 GPa. Seemingly, the found T-p range of the state-point dependent scaling exponent γ for 4M2P follows a different pattern of behavior, because the limit pressure between the invariant and state-point dependent γ considerably changes with varying temperature (see the inset in Fig. 1(b)). However, the mentioned earlier reports on H bonds under high pressure were actually based on an analysis of the liquid fragility determined at different glass transition temperatures Tg at different values of pressure, where Tg is defined in some arbitrarily chosen isochronal conditions defined by a constant structural relaxation time. In this context, it should be noted that the isochronal compression is accompanied with an increase in temperature. This experimental fact was used to explain the increase in the degree of H bonds of propylene glycol oligomers up to ca. 1 GPa due to decreasing intermolecular distances under compression, and the decrease in the degree of H bonds at \(p > 1\,{\rm{GPa}}\) pressure due to an increasing role of temperature fluctuations in molecular dynamics of those associated liquids. If we inspect the isochrones at \({\tau }_{D}\le 1\,{\rm{ms}}\) (see Fig. 1(a)), we can observe a very similar behavior in case of 4M2P. Along these isochrones, the thermodynamic ranges of the state-dependent scaling exponent γ are located at low temperatures and densities, i.e., at low temperatures and pressures, while the PDS law with \(\gamma =2.5\) is obeyed above some values of temperature and density (and above some corresponding value of p), which depend on the time scale of isochrone. Thus, the complex density scaling of the Debye-like relaxation times of 4M2P observed at \({\tau }_{D}\le 1\,{\rm{ms}}\) can be explained by changes in the degree of H bonds due to competing effects of compression and temperature fluctuations on the H-bonded network dynamics similarly as reported for propylene glycol oligomers near the glass transition42,43. Analogically, the validity of the PDS law with \(\gamma =2.5\) for 4M2P above \({\tau }_{D}=1\,{\rm{ms}}\) at each considered temperature, excluding pressures near the ambient conditions, suggests that the temperature fluctuations dominate the dynamics of H bonds of 4M2P at these time scales even at low temperatures. It shows that the PDS criterion (Eq. (2)) is a very sensitive tool to detect the thermodynamic regions of complex specific interactions like strong or/and numerous hydrogen bonds (if γ varies) and typical molecular interactions like van der Waals forces (if γ is invariant) for a given material.
Finally, we have performed the density scaling based analysis of viscosity η measured for 4M2P along an atmospheric isobar and a few isotherms up to \(p=1\,{\rm{GPa}}\) in order to compare its results with the previous one for \({\tau }_{D}\). This study has been motivated by the well-known difficulties in experimental explorations of the structural dielectric relaxation in monohydroxy alcohols.
For such materials, including 4M2P, the structural dielectric loss peak is usually covered by the dominating Debye-like dielectric loss peak at sufficiently high pressures due to a higher sensitivity of the structural process than the Debye-like relaxation to the isothermal compression35. All the viscosity data measured for 4M2P and shown as functions of T, p, and V in Supplementary Figs. S6 and S7 available has turned out to satisfy the PDS law (Eq. (1)) with the same scaling exponent \(\gamma =2.5\) as that for \({\tau }_{D}\) (Fig. 3). It suggests that the same effective intermolecular potential might be relevant to the mechanical relaxation and the Debye-like dielectric relaxation in case of 4M2P at least in the thermodynamic region of the invariant scaling exponent γ for \({\tau }_{D}\). Nevertheless, the Debye-like relaxation times and viscosity data superimpose on two different scaling curves, which indicate a decoupling between \({\tau }_{D}\) and η for this material. To quantify this decoupling, we apply the Avramov model version44,45,46 that conforms to the PDS law with \(\gamma =const\) (Eq. (1)), which can be formulated for a dynamic quantity, \(Y={Y}_{0}\exp [{(A{\rho }^{\gamma }/T)}^{D}]\), where Y0, A, D, and γ are its parameters. By fitting separately the Debye-like relaxation times determined in the region of the invariant scaling exponent γ and all the measured viscosity data to the Avramov model, we have confirmed the value \(\gamma =2.50\pm 0.02\) for 4M2P in case of \({\tau }_{D}\) and η, and respectively for these quantities, we have found different values of its other fitting parameter, \({D}_{{\tau }_{D}}=5.11\pm 0.02\) and \({D}_{\eta }=2.96\pm 0.02\). By dividing the Avramov equations applied to \({\tau }_{D}\) and η with the same value of γ, one can show that a decoupling between these quantities can be quantified by the ratio \({D}_{{\tau }_{D}}/{D}_{\eta }\) different from unity as it is for 4M2P \(({D}_{{\tau }_{D}}/{D}_{\eta }\approx 1.73)\). The decoupling between η and \({\tau }_{D}\) accompanied with the same scaling exponent γ for these quantities is expected to result in the same exponent of the repulsive part of the effective intermolecular potentials relevant to the mechanical and Debye-like dielectric relaxations of 4M2P, but other parameters of the effective potentials may differ. Whereas the thermodynamic region of the state-point dependent scaling exponent γ observed only for \({\tau }_{D}\) shows that the dielectric spectroscopy enables to probe selectively the complex dynamics of H-bonded networks, which seems to be covered by other mechanical relaxation modes or incompletely contribute to the structural mechanical relaxation.

Comparison of the density scaling of viscosity and Debye-like relaxation time with γ = 2.5, including all viscosity data and only isothermal Debye-like relaxation times shown in Fig. 2(a). This scaling pattern is satisfied by all examined viscosity data, but the master plot for η does not match that for τ, because there is a decoupling between η and τD, which can be quantified by different values Dη and \({D}_{{t}_{D}}\) found from fitting temperature-volume dependences of η and τD to the Avramov model.
Summary and Conclusions
In summary, it should be emphasized that the density scaling criterion (Eq. (2)) has turned out to be a powerful tool to investigate relevant effective intermolecular potentials based on measurements of macroscopic dynamic quantities and vice versa independently of the validity of the PDS law (Eq. (1)) with \(\gamma =const\). The density scaling criterion applied to detect thermodynamic regions within which molecular dynamics is dominated by either typical or complex specific intermolecular interactions, depending on the invariant or varying values of γ for a given material, should considerably facilitate to work out intermolecular potentials to properly and effectively model various physical phenomena near the glass transition. Additionally, the suggested comparative analysis of the density scaling functions for different dynamic quantities for a given material enables to identify possible differences in the thermodynamic evolution of these quantities in the wide pVT range and may give hints not only on the exponent of the repulsive part of the effective intermolecular potential but also on its other parameters. Our findings open new prospects for gaining a better insight into possible changes in the character and complexity of intermolecular interactions in various materials, not only associated liquids, depending on thermodynamic conditions.
Methods
Broadband dielectric spectroscopy (BDS) extensive studies of 4M2P have been conducted at ambient and elevated pressure. In ambient conditions, the BDS system equipped with the Alpha impedance analyzer and Quatro Cryosystem by Novocontrol GmBH have been employed in the frequency range from 10μHz up to 3 MHz at temperatures from 150 K up to 323 K. High pressure isothermal dielectric measurements have been performed in the pressure range from 0.1 MPa up to 1.8 GPa (varying depending on isotherms) and in the temperature range from 203 K up o 323 K, using two pressure systems described elsewhere27. Similarly to other monohydroxy alcohols with hydroxyl group located at the terminal position of the carbon chain (see e.g. refs. 32,33), we have observed the so-called Debye-like relaxation at elevated pressure (or the Debye process at ambient pressure) in case of 4M2P (see Supplementary Fig. S1), which has been measured along 16 isotherms and 1 atmospheric isobar. It should be noted that whereas at ambient pressure two relaxations can be discerned and the dominating one has an exponential shape (see the panel (a) in Supplementary Fig. S1), with compression both processes approach and only a single stretched Debye-like relaxation is observed. The relaxation times for this dominating process were estimated from the maximum of the relaxation curve (see black arrows in Supplementary Fig. S1). The obtained dependences of the Debye-like relaxation time \({\tau }_{D}\) on temperature at ambient pressure and pressure at constant temperatures are shown respectively in the panels (a) and (b) of Supplementary Fig. S2.
The 4-methyl-2-pentanol (min. 0.99 mass fraction, water content less than 2·10−4 mass fraction) was from Alfa Aesar. Based on the experimental speed of sound under high pressures, density and specific isobaric heat capacity at atmospheric pressure, the pρT data and related quantities were estimated for pressures up to 100 MPa at temperatures from 293.15 to 318.15 K by the acoustic method. The details concerning the experimental and estimation methods can be found in the previous works30,31. The measurement temperature-volume dependence of the specific volume V(T,p), where \(V={\rho }^{-1}\), has been parametrized by fitting the pVT experimental data to the commonly known Tait equation of state38, \(V(T,p)=({A}_{0}+{A}_{2}T+{A}_{2}{T}^{2})\{1-C\,\mathrm{ln}\,[1+p/({b}_{0}\exp (\,-\,{b}_{1}T))]\}\). The following values of the Tait equation parameters have been obtained for 4M2P, A0 = (1.2157 ± 0.0002) cm3/g, A1 = (1.14 ± 0.01)∙10−3cm3/(g ◦C), A2 = (2.87 ± 0.11)∙10−6cm3/(g °C2), C = 0.0837 ± 0.0002, b0 = (103.8 ± 0.4) MPa, and b1 = (7.56 ± 0.05)∙10−3◦C−1. Then, the isobaric and isothermal dependences \({\tau }_{D}(V)\) have been established (see Supplementary Fig. S5).
The viscosity has been measured in falling cylinder viscometers28,29 to pressure of 1 GPa at three temperatures, 298 K, 308 K, and 323 K. A linear variable differential transformer monitors the sinker position rather than detecting the end of fall by electrical contact. Estimated uncertainties are 3% for viscosity, 0.5 °C for temperature and the greater of 1 MPa and 0.4% for pressure. The obtained dependences of viscosity η on temperature at ambient pressure and pressure at constant temperatures are shown in the panels (a) and (b) of Supplementary Fig. S6, respectively. Using the aforementioned parametrized dependence V(T,p), the isobaric and isothermal dependences of viscosity have been expressed by a function of volume (see Supplementary Fig. S7).
References
Roland, C. M., Hensel-Bielowka, S., Paluch, M. & Casalini, R. Supercooled dynamics of glass-forming liquids and polymers under hydrostatic pressure. Rep. Prog. Phys. 68, 1405–1478 (2005).
Floudas, G., Paluch, M., Grzybowski, A. & Ngai, K. L. Origin of Glass Formation in Molecular Dynamics of Glass-Forming Systems: Effects of Pressure. (Series: Advances in Dielectrics, Series ed. Kremer, F.), ch. 2, pp. 39–65 (Springer, Berlin Heidelberg 2011).
Roland, C. M. Viscoelastic behavior of rubbery materials. (Oxford University Press, 2011).
Grzybowski, A. & Paluch, M. Universality of Density Scaling in The Scaling of Relaxation Processes, (eds. Kremer, F. & Loidl, A.), ch. 4, pp. 77–119 (Springer Nature, 2018).
Casalini, R. & Roland, C. M. Thermodynamical scaling of the glass transition dynamics. Phys. Rev. E 69, 062501 (2004).
Pedersen, U. R., Bailey, N. P., Schrøder, T. B. & Dyre, J. C. Strong Pressure-Energy Correlations in van der Waals Liquids. Phys. Rev. Lett. 100, 015701 (2008).
Bailey, N. P., Pedersen, U. R., Gnan, N., Schrøder, T. B. & Dyre, J. C. Pressure-energy correlations in liquids. I. Results from computer simulations. J. Chem. Phys. 129, 184507 (2008).
Bailey, N. P., Pedersen, U. R., Gnan, N., Schrøder, T. B. & Dyre, J. C. Pressure-energy correlations in liquids. II. Analysis and consequences. J. Chem. Phys 129, 184508 (2008).
Coslovich, D. & Roland, C. M. Thermodynamic Scaling of Diffusion in Supercooled Lennard-Jones Liquids. J. Phys. Chem. B 112, 1329–1332 (2008).
Coslovich, D. & Roland, C. M. Pressure-energy correlations and thermodynamic scaling in viscous Lennard-Jones liquids. J. Chem. Phys. 130, 014508 (2009).
Dreyfus, C., Le Grand, A., Gapinski, J., Steffen, W. & Patkowsk, A. Scaling the α-relaxation time of supercooled fragileorganic liquids. Eur. Phys. J B42, 309–319 (2004).
Wojnarowska, Z. & Paluch, M. High-Pressure Dielectric Spectroscopy for Studying the Charge Transfer in Ionic Liquids and Solids in Dielectric Properties of Ionic Liquids. (ed. Paluch, M.), (Series: Advances in Dielectrics, Series ed. Kremer, F.), ch. 4, pp. 73–113 (Springer, Berlin Heidelberg 2016).
Thoms, E., Grzybowski, A., Pawlus, S. & Paluch, M. Breakdown of the Simple Arrhenius Law in the Normal Liquid State. J. Chem. Phys. Lett. 9, 1783–1787 (2018).
Urban, S. High Pressure Investigations of Liquid Crystals in Handbook of Liquid Crystals, (eds. Goodby, J. W. et al.), vol. 2, ch. 15, pp. 443–486 (Wiley-VCH, Weinheim 2014).
Grzybowski, A., Urban, S., Mroz, S. & Paluch, M. Activation volume of selected liquid crystals in the density scaling regime. Sci. Rep 7, 42174 (2017).
Bøhling, L. et al. Scaling of viscous dynamics in simple liquids:theory, simulation and experiment. New J. Phys. 14, 113035 (2012).
Sanz, A. et al. Experimental Evidence for a State-Point-Dependent Density-Scaling Exponent of Liquid Dynamics. Phys. Rev. Lett. 122, 055501 (2019).
Hummel, F., Kresse, G., Dyre, J. C. & Pedersen, U. R. Hidden scale invariance of metals. Phys. Rev. B 92, 174116 (2015).
Casalini., R. & Roland, C. M. The “anomalous” dynamics of decahyroisoquinoline revisited. J. Chem. Phys. 144, 024502 (2016).
Casalini, R., Bair, S. & Roland, C. M. Density scaling and decoupling in o-terphenyl, salol, and dibutyphthalate. J. Chem. Phys. 145, 064502 (2016).
Ransom, T. C., Casalini, R., Fragiadakis, D., Holt, A. P. & Roland, C. M. Comment on “Experimental Evidence for a State-Point-Dependent Density-Scaling Exponent of Liquid Dynamics”. Phys. Rev. Lett. 123, 189601 (2019).
Grzybowski, A., Grzybowska, K., Zioło, J. & Paluch, M. Correlations between isobaric and isochoric fragilities and thermodynamical scaling exponent for glass-forming liquids. Phys. Rev. E 74, 041503 (2006).
Grzybowski, A., Grzybowska, K., Zioło, J. & Paluch, M. Reply to “Comment on ‘Correlations between isobaric and isochoric fragilities and thermodynamical scaling exponent for glass-forming liquids’. Phys. Rev. E 76, 013502 (2007).
Pronin, A. A. et al. Pressure-induced change in the relaxation dynamics of glycerol. JETP Letters 92, 479–483 (2010).
Pawlus, S., Paluch, M. & Grzybowski, A. Communication: Thermodynamic scaling of the Debye process in primary alcohols. J. Chem. Phys. 134, 041103 (2011).
Paluch, M. et al. General rules prospected for the liquid fragility in various material groups and different thermodynamic conditions. J. Chem. Phys. 141, 134507 (2014).
Paluch, M. et al. Test of the Einstein-Debye Relation in Supercooled Dibutylphthalate at Pressures up to 1.4 GPa. Phys. Rev. Lett. 90, 175702 (2003).
Bair, S. & Qureshi, F. Accurate Measurements of Pressure-Viscosity Behavior in Lubricants. Tribology Transactions 45, 390–396 (2002).
Bair, S. High pressure rheology for quantitative elastohydrodynamics: Second Edition; Elsevier: Amsterdam 2019.
Jężak, S., Dzida, M. & Zorębski, M. High pressure physicochemical properties of 2-methylfuran and 2,5-dimethylfuran – second generation biofuels. Fuel 184, 334–343 (2016).
Dzida, M. & Góralski, P. Excess molar heat capacities for (decan-1-ol + n-heptane) at temperatures from (290 to 318) K. Experimental results and theoretical description using the ERAS model. J. Chem. Thermodyn. 38, 962–969 (2006).
Böhmer, R., Gainaru, C. & Richert, R. Structure and dynamics of monohydroxy alcohols –Milestones towards their microscopic understanding, 100 years after Debye. Physics Reports 545, 125–195 (2014).
Hansen, C., Stickel, F., Berger, T., Richert, R. & Fischer, E. W. Dynamics of glass-forming liquids. III. Comparing the dielectric α- and β-relaxation of 1-propanol and o-terphenyl. J. Chem. Phys. 107, 1086–1093 (1997).
Huth, H., Wang, L.-M., Schick, C. H. & Richert, R. Comparing calorimetric and dielectric polarization modes in viscous 2-ethyl-1-hexanol. J. Chem. Phys. 126, 104503 (2007).
Wikarek, M., Pawlus, S., Tripathy, S. N., Szulc, A. & Paluch, M. How Different Molecular Architectures Influence the Dynamics of H-Bonded Structures in Glass-Forming Monohydroxy Alcohols. J. Phys. Chem. B 120, 5744–5752 (2016).
Singh, L. P. & Richert, R. Watching Hydrogen-Bonded Structures in an Alcohol Convert from Rings to Chains. Phys. Rev. Lett. 109, 167802 (2012).
Gainaru, C. et al. Oscillatory shear and high-pressure dielectric study of 5-methyl-3-heptanol. Colloid Polym. Sci. 292, 1913 (2014).
Tait, P. G. Physics and Chemistry of the Voyage of H. M. S. Challenger. vol. 2, part 4, pp. 941–951 (HMSO, London 1888).
Paluch, M., Grzybowska, K. & Grzybowski, A. Effect of high pressure on the relaxation dynamics of glass-forming liquids. J. Phys.: Condens. Matter 19, 205117 (2007).
Casalini, R. & Ransom, T. C. On the experimental determination of the repulsive component of the potential from high pressure measurements: What is special about twelve? J. Chem. Phys. 151, 194504 (2019).
Fragiadakis, D., Roland, C. M. & Casalini, R. Insights on the origin of the Debye process in monoalcohols from dielectricspectroscopy under extreme pressure conditions. J. Chem. Phys. 132, 144505 (2010).
Grzybowska, K., Pawlus, S., Mierzwa, M., Paluch, M. & Ngai, K. L. Changes of relaxation dynamics of a hydrogen-bonded glass former after removal of the hydrogen bonds. J. Chem. Phys. 125, 144507 (2006).
Grzybowska, K. et al. Dynamic Crossover of Water Relaxation in Aqueous Mixtures: Effect of Pressure. J. Phys. Chem. Lett. 1, 1170–1175 (2010).
Casalini, R., Mohanty, U. & Roland, C. M. Thermodynamic interpretation of the scaling of the dynamics of supercooled liquids. J. Chem. Phys. 125, 014505 (2006).
Grzybowski, A., Paluch, M., Grzybowska, K. & Haracz, S. Communication: Relationships between Intermolecular potential, thermodynamics, and dynamic scaling in viscous systems. J. Chem. Phys. 133, 161101 (2010).
Paluch, M. et al. A Relationship between Intermolecular Potential, Thermodynamics, and Dynamic Scaling for a Supercooled Ionic Liquid. J. Phys. Chem. Lett. 1, 987–992 (2010).
Acknowledgements
The current work was realized due to the financial support of the project No. UMO-2015/17/B/ST3/01221 by the National Science Centre, Poland
Author information
Authors and Affiliations
Contributions
S.P., A.G. and M.P. formulated the reported interpretation of the density scaling idea and wrote the manuscript. S.P., S.K. and M.W. measured dielectric data and performed standard analyses of the dielectric data. S.B. measured viscosity data and performed standard analyses of the viscosity data. M.D. and P.G. experimentally obtained pressure-volume-temperature data. A.G. preformed density scaling based analyses of experimental data, the results of which have been reported in the manuscript. All authors discussed and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pawlus, S., Grzybowski, A., Kołodziej, S. et al. Density Scaling Based Detection of Thermodynamic Regions of Complex Intermolecular Interactions Characterizing Supramolecular Structures. Sci Rep 10, 9316 (2020). https://doi.org/10.1038/s41598-020-66244-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66244-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
