
Abstract
Trioza erytreae is the main vector for ‘Candidatus Liberibacter africanus’, the causative agent of African Citrus Greening disease. The insect is widespread in Africa, and has recently disseminated to Southwestern Europe. This study aimed at generating reference mitogenome sequences for T. erytreae, as a background for future genetic diversity surveys. Complete mitochondrial sequences of three specimens collected in Ethiopia, Uganda and South Africa were recovered using Ion Torrent technology. The mitogenomes of T. erytreae from Uganda and Ethiopia were highly similar, and distinct from that found in South Africa. The phylogeographic structure of T. erytreae was assessed using genetic clustering and pairwise distances, based on a dataset of public COI sequences recorded as T. erytreae. The dataset revealed ten haplotypes with strong phylogeographic structure in Africa and Europe. Three haplotypes found in Kenya on Clausena anisata belonged to pairs separated by distances as high as 11.2%, and were basal to all other sequences. These results indicate that not all sequences identified as T. erytreae belong to the same species, and that some degree of specificity with different plant hosts is likely to exist. This study provides new baseline information on the diversity of T. erytreae, with potential implications for the epidemiology of African Citrus Greening disease.
Similar content being viewed by others

INTRODUCTION
Citrus is one of the most important fruit crops globally, with an average annual production of over 73 million metric tons (MT)1. Numerous citrus species are commercially cultivated worldwide, including sweet orange (Citrus sinensis Osbeck), lemon (Citrus limon [L.] Burm. f.), lime (Citrus aurantifolia [Cristm.] Swingle), grapefruit (Citrus paradisi Macfad), and mandarin (Citrus reticulata Blanco). Sweet orange is the most popular citrus, and accounts for more than half of total world production1.
Currently, the leading citrus producers in Africa are Egypt and South Africa, with an average of 1.4 million MT of oranges per year in South Africa1. In spite of high market demand and great agricultural potential for citrus in Africa, production has been declining2,3. For example, the total yield of oranges has gradually decreased from 205,895 hg/ha in 2014 to 198,710 hg/ha in 2017, as well as the total yield of lemons and limes, from 148,431 hg/ha to 135,435 hg/ha, in the same period 1. Numerous biological factors constrain citrus production in Africa, including important insect pests (aphids, whiteflies and triozids), and viral and bacterial diseases (Citrus Tristeza and Citrus Greening Disease).
The African citrus triozid, Trioza erytreae (Del Guercio) (Hemiptera: Triozidae), is one of the most damaging citrus pests in Africa2,4. Trioza erytreae was first recorded in 1929 in South Africa5, and it is widely distributed on the continent. The insect has been reported in 19 out of the 54 African countries: Angola, Kenya, Ethiopia, Eritrea, Madagascar, Malawi, Mauritius, La Réunion, South Africa, Sudan, Swaziland, St. Helen, Tanzania, Uganda, Zambia, Democratic Republic of the Congo, Rwanda, Comoros, and Cameroon6. Outside Africa, T. erytreae was first detected in Madeira (Portugal) in 19947, and it has now spread to the southwest of the Iberian Peninsula through northern Spain and Portugal8.
Trioza erytreae causes damage to citrus plants by larval feeding, which results in notching and curling of the leaves. In addition, the production of honeydew by the insect promotes the growth of sooty mould on the plant, decreasing its productivity. Although the negative impact of larval feeding is significant, the most injurious activity of the insect results from its role as the vector for ‘Candidatus Liberibacter africanus’ (Laf), a phloem-limited bacterium responsible for the African Citrus Greening disease (ACG)9. In Eastern and Southern Africa, ACG is reported to have high impact on citrus production, especially in the highlands, causing yield losses of 25% to 100%10,11. While T. erytreae is the primary vector for Laf12, the insect is also able to transmit another citrus pathogen – ‘Candidatus Liberibacter asiaticus’ (Las) - under experimental conditions13. Las is associated with the severe Huanglongbing (HLB) disease, normally transmitted by Diaphorina citri in Asia, and North and South America14,15,16. In Africa, Las was reported for the first time in Ethiopia in 2009, in the absence of D. citri, its primary vector17. Recent reports of T. erytreae field populations carrying Las in the Ethiopian highlands, in the absence of D. citri, support the possibility that the insect is able to transmit the pathogen under natural conditions. Las was also reported on citrus in Uganda18 and Tanzania19, but this finding was due to a misidentification, and the bacteria detected were most likely ‘Candidatus Liberibacter africanus spp. clausenae’ (LafCl)20. Therefore, Las has not yet been confirmed in Uganda or Tanzania. LafCl has only been detected on Clausena anisata, an African member of the family Rutaceae21, and has not been found on citrus in South Africa22. The confirmed presence of Las in Africa, along with the widespread occurrence of T. erytreae and its ability to transmit the pathogen, contribute to a scenario of significant threat to citrus production on the continent.
As the control of T. erytreae is directly linked to the management of ACG, understanding the bioecology of the triozid is relevant for its management and control23,24. The genetic diversity and phylogeographic structure of T. erytreae are largely unknown, except for a study which focused on the generation of DNA barcodes for species identification in the genus Trioza25, and a recent work aimed at testing the host range of Tamarixia dryi, a parasitoid wasp associated with T. erytreae in South Africa26.
The objective of the present study was to establish a background for the assessment of the genetic diversity and phylogeography of T. erytreae in Africa, as the species is present in a wide range of bioclimatic regions and different plant hosts, across large geographical distances. For that purpose, we sequenced and compared the complete mitogenomes of three individuals collected in Ethiopia, South Africa and Uganda. The phylogenetic position of T. erytreae within the family Triozidae was recovered in the context of publicly available mitogenome sequences. The intraspecific haplotype structure and the genetic diversity of T. erytreae were assessed using a compilation of publicly available COI sequences.
Results and Discussion
The complete mitochondrial genomes of three adult specimens of T. erytreae collected from citrus plants in Ethiopia (TE-ETH), Uganda (TE-UG) and South Africa (TE-SA) were sequenced with three objectives: 1) to generate reference mitogenomes for future studies, 2) to gain insights into the phylogeographic structure of the species in Africa, and 3) to assess the phylogenetic position of the new mitogenomes within the family Triozidae, using publicly available complete sequences.
Mitogenomics
The Ion Torrent run resulted in an average of 22.5 million reads for each sample with average read length of 175 bp. The de novo assembly generated a contig with 133,872 reads for TE-ETH, a contig with 135,931 reads for TE-UG, and a contig with 31,073 reads for TE-SA. The average sequence coverage was 2,861x for TE-ETH, 2,651x for TE-UG, and 37,460x for TE-SA. A complete mitogenome sequence for T. erytreae was available on Genbank (NC_038142.1), and could potentially be used for the mapping and assembly of the new NGS data. However, the sequence contained an unusual, non-annotated 1,689 bp region between ND2 and tRNATrp, additionally to the annotated AT-rich region (control region). Animal mitogenomes are compact, and do not generally contain large non-coding intergenic regions apart from the control region. Therefore, the new mitogenomes were recovered using de novo assembly and reference-based assembly. The mitogenomes recovered using the de novo assembly did not have the unannotated 1,689 bp region between ND2 and tRNATrp present in NC_038142.1, the only mitogenome available on Genbank for T. erytreae. The reference-based mapping using NC_038142.1, after the excision of the unannotated region, resulted in the mapping of 281,007 reads for TE-ETH, 259,315 reads for TE-UG, and 3.6 million reads for TE-SA. The reference based assembly resulted in an average coverage of 3,062x for TE-ETH, 2,835x for TE-UG, and 39,887x for TE-SA. The de novo and the reference-based assembly methods resulted in identical sequences for each mitogenome, indicating that the sequence NC_038142.1 should be revised, as the 1,689 bp region between ND2 and tRNATrp most probably represents a sequencing or assembly artefact.
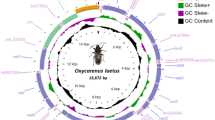
The main features were identical in the three mitogenomes, with the typical mitochondrial complement of 13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, two ribosomal RNA (rRNA) genes, and an AT-rich non-coding region generally assumed to contain the control for transcription and replication (Fig. 1). Gene order was conserved in all species included in the phylogenetic reconstruction (Table S1), and identical to the hypothetical ancestral mitogenome organisation in insects. The average size of the complete sequences (15,087 bp) was similar to the average size of the complete mitogenomes of other triozids (15,028 bp). Twenty-three genes were located on the majority (J) strand, and 14 genes were located on the minority (N) strand.

Mitogenome organization of Trioza erytreae. Circular map of the mitochondrial genome of Trioza erytreae. Protein-coding, transfer RNAs and ribosomal genes are shown with standard abbreviations. The arrows indicate the direction of the genes.
The three complete mitogenomes had the high A + T content characteristic of insects, with values higher than 79.9% (complete sequence) in all genes (Table S2). The average A + T content of the AT-rich region (88.6%) was higher than the average for the complete sequences (80.0%), the combined tRNAs (79.0%) and the two rRNAs (79.8%). All PCGs had negative GC-skews, except for COI. Five PCGS (ATP8, ND4, ND4L, ND1 and ND5) had positive AT-skews, while eight PCGs (COII, ATP6, COIII, ND3, ND6, CYTB and ND2) had negative AT-skews (Table S2). The three mitogenomes were equally compact, and had 13 short gene overlaps mostly involving tRNAs. Gene overlaps ranged between 1 and 13 bp, with the largest between tRNAGlu and tRNAPhe. Intergenic regions were found at 15 locations, and represented a total average of 78 bp, the longest of which was between tRNASer2 and ND1 (25 bp). The largest non-coding region (average = 1,054 bp) was located between the 12S rRNA and the I-Q-M tRNA cluster, and was annotated as the putative control region. The large ribosomal RNA gene (16s rRNA; 1,146 bp) was located between tRNALeu1 and tRNAVal, and the small ribosomal RNA gene (12s rRNA; 685 bp) was located between tRNAVal and the AT-rich region. The total complement of 22 tRNAs was identified with ARWEN software, and the sizes of the genes varied between 52 bp (tRNAHis) and 69 bp (tRNALys and tRNAGln). The average combined length of the 13 PCGs was 10,822 bp, representing 71.7% of the total mitogenomes, similarly to other triozids (average 10,801 bp) (Table 1). The largest PCG was ND5 (1,618 bp), and the smallest was ATP8 (153 bp), also in line with the other triozids. The PCGs had an average A + T content of 79.4%, ranging from 75.6% in COI to 85.7% in ATP8. The most frequent start codon was ATG, and all genes were terminated by TNN codons (TAA, TTA, TAG and T) (Fig. S1).
Nucleotide pairwise comparisons showed high similarity between the mitogenomes of the specimens from Ethiopia and Uganda, with only 16 single nucleotide polymorphisms (SNPs) found across the 13 PCGs. All SNPs were transitions, four of which resulted in amino acid changes (Table S3). In PCGs, the largest number of nucleotide differences between TE-ETH and TE-UG was found in COIII and ND1, with three SNPs in each of the genes. The sequences of ND6, ND3, ATP8 and ND4L were identical in TE-ETH and TE-UG. In contrast, an average of 294 SNPs was found between TE-SA/TE-ETH and TE-SA/TE-UG, considering only the PCGs. These SNPs represented an average of 261 transitions and 33 transversions, of which 67 resulted in amino acid changes. The percentage of nucleotide differences between TE-SA and the other two mitogenomes was highest in ATP8 (4.58%), and the percentage of amino acid substitutions was highest in ND4L (2.08%) (Fig. 2). Comparison of tRNA sequences showed 16 SNPs between TE-SA and the other two mitogenomes in 10 genes (tRNAAsp, tRNAArg, tRNACys, tRNAGlu, tRNAGly, tRNALeu2, tRNAPro, tRNASer1, tRNASer2, tRNAThr). In contrast, only one SNP was found between the tRNAs of TE-ETH and TE-UG (tRNATrp).

Mitogenome comparisons. Comparison of the mitochondrial sequences of three Trioza erytreae specimens collected in Ethiopia (TE-ETH), Uganda (TE-UG) and South Africa (TE-SA), based on the total complement of 13 protein-coding genes. The number of differences is given as (A) percentage of single nucleotide polymorphisms, and (B) percentage of non-synonymous substitutions, relatively to the size of each gene.
Phylogeny of the family Triozidae
The phylogenetic position of T. erytreae within Triozidae was recovered in a ML tree, using eight other complete mitogenomes publicly available, and two Aphididae as outgroups (Fig. 3). The new T. erytreae sequences formed a monophyletic cluster, in accordance with their low level of divergence. The topology of the mitochondrial tree was not fully congruent with that presented in a recent reconstruction of the superfamily Psylloidea, which employed a combination of mitochondrial and nuclear sequence data across a large number of species27. However, some common features were recovered: T. erytreae and Aacanthocnema dobsoni were closely related, in accordance with their classification in group D by Percy et al. 201827, as well Trioza urticae, Bactericera cockerelli and Paratrioza sinica in group M, and Pariaconus pele and Trioza remota in group A. The differences consisted in the position of Trioza anthrisci (group A), which was not recovered as closely related to P. pele and T. remota (group A), and the relative order of the species clusters representing groups. For example, the cluster formed by T. urticae, B. cockerelli and P. sinica (group M), was not recovered as basal to the other taxa. The recovery of deeper phylogenetic nodes is a well-known limitation of mitochondrial-based phylogenies, and the inclusion of nuclear sequence data is often necessary for the assessment of higher divergences28. Therefore, these results should be interpreted conservatively.
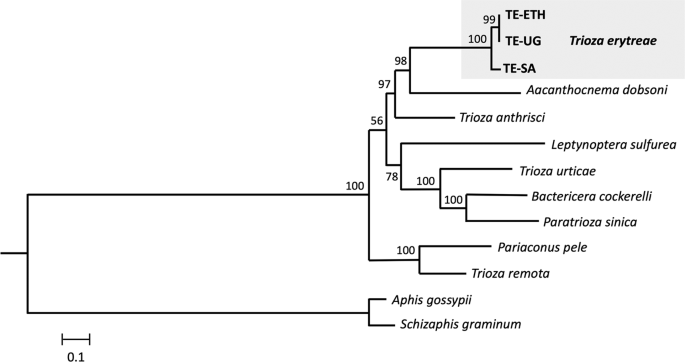
Mitochondrial phylogeny of Triozidae. Maximum likelihood tree representing the phylogenetic relationships within the family Triozidae, using the complete complement of the 13 mitochondrial protein-coding genes. Aphis gossypii and Schizaphis graminum (Aphididae) were used as outgroups. Values represent nodal support calculated from 1,000 bootstrap replicates. The length of the branches is proportional to the number of substitutions per site.
Intraspecific diversity of Trioza erytreae
The intraspecific genetic diversity of T. erytreae was assessed using a dataset of COI barcoding sequences identified as T. erytreae collected in Africa and Europe in two previous studies25,26 (Table S4). The final COI dataset (n = 89) included the three sequences generated in the present study, and was strongly biased towards Kenya, which represented 75.2% of all sequences (n = 66), followed by South Africa (14.6%; n = 13). Europe was represented by specimens collected in Portugal and Spain (n = 7), the only countries where T. erytreae has been found outside Africa. The COI dataset was analysed using a MJ network, a NJ tree, and estimates of genetic divergence (p-distances).
The MJ network revealed phylogeographic structuring of T. erytreae haplotypes, and their association with the different plant hosts (Fig. 4). Branch I was formed by two closely related haplotypes (Hap1 and Hap2), and included the majority of specimens found on citrus, some specimens found on Clausena anisata, Murraya koenigii and Stephania abyssinica in Kenya, and the single specimen from Tanzania found on citrus. Branch II (Hap3, Hap4 and Hap5) included all specimens collected from citrus plants in Portugal, Spain and South Africa, and one specimen collected in Kenya, also found on citrus. The specimens collected in Portugal and Spain had a single haplotype (Hap4) which was shared with South Africa, and closely related to Hap5 (South Africa) and Hap3 (shared between South Africa and Kenya). This haplotype similarity suggests that T. erytreae found in Europe most likely originated from South Africa, although the possibility of a Kenyan origin cannot be excluded. Branch III (Hap6) included a single specimen found in Kenya on C. anisata, and Branch IV represented the haplotype found in Ethiopia and Uganda on citrus. Branches V, VI and VII (Hap8, Hap9 and Hap10) represented the highly diverged haplotypes found in Kenya on C. anisata.

Network of Trioza erytreae haplotypes. Median-joining network of cytochrome c oxidase subunit 1 (COI) gene (607 bp) of Trioza erytreae (n = 89), showing the relationships between haplotypes according to geographic origin and plant host. The size of the circles is proportional to the number of individuals sharing the same haplotype. The length of the branches is proportional to the number of nucleotide substitutions between haplotypes. Numbers along branches indicate the number of substitutions between haplotypes.
The p-distances among all pairs of haplotypes represented in the network (Hap1 to Hap10) allowed for further inferences on the genetic diversity of T. erytreae (Table 2). The high p-distances between Hap8, Hap9 and Hap10 and other sequences, presented in the MJ network in the previous section, was confirmed. Hap10 was the most diverged haplotype, with a p-distance from all other haplotypes higher than 7.65%, followed by Hap9 which diverged from all other haplotypes by more than 5.24%. These values suggest that Hap9 and Hap10 represent two species distinct from all other haplotypes. Hap8 had a different pattern of p-distances relatively to the other haplotypes, as it diverged from Hap1, Hap2, Hap3 and Hap4 by more than 5.06%, suggesting that it represents a distinct species, but had a lower divergence from Hap6 (3.63%, Kenya) and Hap7 (2.82%, Ethiopia and Uganda). Overall, the high divergence among some of the COI sequences displayed in the MJ network and the p-distances among the haplotypes indicated that not all specimens morphologically identified as T. erytreae in the publicly available dataset belong to the same species. This pattern of high intra-specific divergence found in T. erytreae (maximum p-distance = 11.71%) was not observed in the majority of the other 30 Trioza species analysed, for which the average maximum intraspecific p-distance was 1.11%. The exceptions were Trioza urticae (4.18%), and Trioza anceps (14.27%), which may represent cases of accidental misidentification or cryptic diversity (Fig. 5; Table S5).

Genetic divergence within Trioza species. Intra-specific pairwise distances (K2P) in 31 species of the genus Trioza (Triozidae), based on cytochrome c oxidase 1 (COI) sequences.
The relationships among COI sequences of T. erytreae were also represented using an NJ tree (Fig. 6), which recovered the pattern of genetic divergence among haplotypes evidenced in the MJ network. The most basal sequences in the NJ tree were Hap8, Hap9 and Hap10 - all represented by specimens found in Kenya on C. anisata - in accordance with the high p-distances among these haplotypes and the other sequences. The bootstrap support for these basal nodes was low, as typically occurs in trees constructed using short COI sequences among distantly related taxa29.
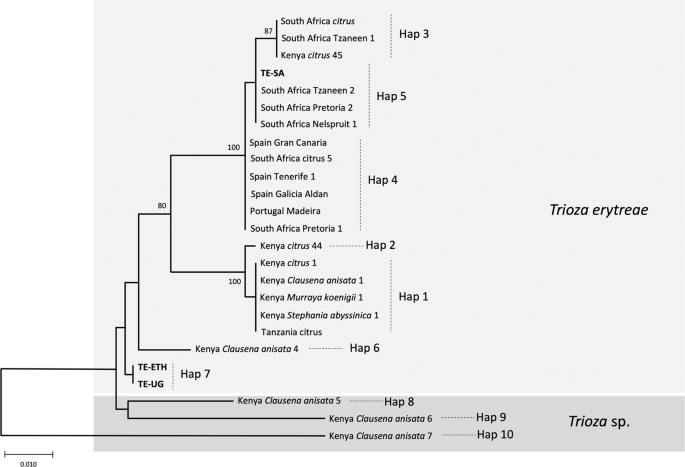
NJ tree of Trioza erytreae haplotypes. Neighbour-joining tree representing the relationships among haplotypes of Trioza erytreae collected from citrus and other plant hosts in Africa and Europe. The tree was constructed using a 571-bp alignment of cytochrome c oxidase subunit 1 (COI) sequences. Nodal support was calculated using 1,000 bootstrap replicates. The length of the branches is proportional to the number of substitutions per site.
African citrus triozids may represent a species complex
In face of the overall results, a hypothesis explaining the genetic diversity of T. erytreae is presented in the NJ tree, and proposes that Hap8, Hap9 and Hap10 constitute three cryptic Trioza sp. distinct from T. erytreae, which is represented by the remaining haplotypes. However, this hypothesis is based on the analyses of a low number of sequences representing a short region of COI, a genetic marker that should not be used for species delimitation30. Therefore, our proposal is probably an oversimplification of the complex phylogenetic relationships among African citrus triozids, and additional genetic, morphological and ecological data will be necessary to draw definite conclusions. Nevertheless, these results indicate cryptic diversity in T. erytreae, and suggest that the degree of association between specific insect genetic subgroups and specific plant hosts should be further investigated.
The genetic diversity of African citrus triozids collected from C. anisata was particularly striking, as this plant hosted the highest number of haplotypes, including the haplotype most frequently found in Kenya on citrus (Hap1), and the three haplotypes that potentially represent cryptic species (Hap8, Hap9 and Hap 10). This result indicates that C. anisata is an important reservoir for African citrus triozid diversity, and that future insights into the genetic diversity of the insect could be obtained from the survey of specimens collected from this particular plant host. In South Africa, where C. anisata is commonly infected with LafCl, the bacterium has not been detected on citrus, in spite of the two plant species being frequently found in proximity. In contrast, LafCl has been reported from citrus in Uganda and Tanzania18,19. This apparent contradiction requires further study, as it can be hypothesized that the T. erytreae haplotypes or cryptic species present on C. anisata in South Africa differ from those in Uganda and Tanzania, and do not necessarily feed on citrus.
It is also noteworthy that, among the diversity of genetic lineages found in T. erytreae, only one lineage (represented by the closely related Branches I and II) seems to be successful on citrus in Kenya, Tanzania, South Africa and Europe. Interestingly, the haplotype found in Uganda and Ethiopia (Hap7, citrus) represents a distinct branch not found in Kenya or South Africa. This suggests that triozids affecting citrus plants in Ethiopia and Uganda may represent local phylogeographic lineages. Another interesting aspect is the pattern of divergence of Hap8 (Kenya, C. anisata) relatively to the other haplotypes. Hap8 was genetically distinct from all other haplotypes with p-distances higher than 4.89%, except for Hap6 (3.63%), which was also found in Kenya on C. anisata, and Hap7 (2.82%), the haplotype found in Ethiopia and Uganda. Although the use of p-distances based on DNA barcodes for species delimitation is controversial, and universal threshold cannot be established30, this result warrants further investigation for elucidating the taxonomic status of African citrus triozids. For that purpose, phylogenetic, genetic diversity and morphological analyses should be performed using a larger sequence dataset across the geographical range and host diversity of T. erytreae. Due to the limitations of mitochondrial DNA for the recovery of deeper divergences, and detection of hybridization and introgression events, definite answers may require the analysis of nuclear sequence data. In addition, the evidence for cryptic diversity in African citrus triozids, currently classified as T. erytreae, opens questions regarding potential association between specific genetic lineages of the vector and the genetic diversity of Liberibacter.
Finally, elucidating the phylogeographic structure and genetic diversity of T. erytreae can contribute to understand the dynamics of transcontinental dispersal of the insect and associated citrus pathogens. This aspect is critical for the management of triozid-mediated citrus disease in the current scenario of global trade, and the recent evidence for the high potential for invasion of African citrus triozids. For example, China was excluded as the probable source of D. citri in California, based on the close relationship of the Californian mitochondrial haplotypes, where the psyllid was detected in 2008, with those found in Florida, where the psyllid was detected in 199831. The characterization of the phylogeography of T. erytreae will contribute to elucidate the origin of the recent invasion of Europe, and to model potential routes for global dissemination of citrus diseases transmitted by African citrus triozids.
Methods
Sample collection and DNA extraction
Adult triozids were collected during field surveys on citrus orchards and backyard gardens in Ethiopia, Uganda and South Africa (Table S6). Identification of the specimens followed standard of the European and Mediterranean Plant Protection Organization for the identification of T. erytreae32. Specimens were aspirated from the trees and stored in 96% ethanol until DNA extraction. Geographical location of the sampling sites were recorded using a Garmin eTrex20 instrument (Garmin, USA). One adult specimen per country was randomly chosen for next-generation sequencing (NGS). Total DNA was individually extracted from each adult specimen using the Isolate II Genomic DNA Kit (Bioline, UK), and extracts were stored at −20 °C until further analyses.
Mitogenome sequencing, assembly and annotation
Total DNA from each of the three specimens was sequenced separately using the Ion Torrent Proton platform (Thermo Fisher Scientific, USA) available at the Central Analytical Facilities of Stellenbosch University, South Africa. Sequence libraries were prepared using the NEXTflex DNA Sequencing Kit for Ion Platforms (PerkinElmer, USA), according to the BI00 Scientific v15.12 protocol. Libraries were pooled and sequenced using the Ion PI HiQ Sequencing Solutions Kit (Life Technologies, USA). The de novo assembly of each mitogenome was performed using SPAdes v.3.13.033, and the resulting contigs were identified by BLAST+34. Each mitogenome was also mapped and assembled using NC_038142.1 as reference sequence, after excision of a non-annotated 1,689 bp region between ND2 and tRNATrp, using Geneious Prime v2019.1 (https://www.geneious.com)35.
Open reading frames of PCGs in both sets of mitogenomes (de novo and reference-based assembly) were identified using Geneious Prime, with the invertebrate mitochondrial genetic code. Transfer RNAs (tRNAs) were predicted with ARWEN software (http://130.235.244.92/ARWEN/)36, using the default composite metazoan mitochondrial code. Ribosomal RNAs (rRNAs) were estimated by BLASTn search on NCBI (https://blast.ncbi.nlm.nih.gov). Overlapping regions and intergenic spacers were counted manually. Nucleotide composition and AT- and GC-skews were calculated using Geneious Prime, as AT-skew = (A − T)/(A + T) and GC-skew = (G − C)/(G + C).
Phylogeographic structure and genetic diversity
In order to obtain further insights into the phylogeographic structure of T. erytreae, all COI sequences publicly available on Genbank as of October 2019 were compiled (Table S4)25,26. Multiple sequence alignment was performed using the MAFFT algorithm available on Geneious Prime. The final sequence alignment (n = 89; 571 bp) was used to construct a median-joining network using Network 10 software (http://www.fluxus-engineering.com/sharenet.htm), under the default settings37. Genetic divergence among the haplogroups displayed in the network were calculated as pairwise distances (p-distances) under the Kimura 2-parameter model (K2P)38, using MEGA X39. A neighbour-joining (NJ) tree was also constructed as an alternative display of the relationships among the sequences, using one sequence representative for each geographical region and each host, when the information was available. The NJ tree was constructed using MEGA X based on p-distances under the K2P model, with 1,000 bootstrap replicates. COI sequences for 30 other Trioza species represented by at least two individuals were retrieved from Genbank for the assessment of intra-specific p-distances within the genus, as described above for the p-distances within T. erytreae.
Phylogenetic analyses
Phylogenetic relationships among Triozidae were reconstructed using the new mitogenomes generated in this study, along with the nine complete mitogenomes available on Genbank for the family as of October 2019, with Aphis gossypii and Schizaphis graminum (Aphididae) as outgroups (Table S1). The phylogenetic analysis was based on the complement of the 13 PCGs, excluding stop codons. PCG sequences were extracted based on annotations, and concatenated using Geneious Prime. The concatenated sequences were aligned using the MAFFT algorithm40 available on Geneious Prime. The best-fitting evolutionary model (GTR + I + G) for the construction of the maximum-likelihood (ML) tree was selected using jModelTest241, according to the AIC and BIC criteria. The ML tree was run using the PhyML 3.1/3.0 aLRT algorithm42 available at https://www.phylogeny.fr/. Nodal support was based on 1,000 bootstrap replicates.
Data availability
The sequences generated in this study were deposited in Genbank under the accession numbers MT416549, MT416550 and MT416551.
Change history
13 November 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Food and Agriculture Organization of the United Nations. FAOSTAT; http://www.fao.org/faostat/en/#home Accessed November 11, 2019 (2019).
Kilalo, D., Olubayo, F., Obukosia, S. & Shibairo, S. Farmer management practices of citrus insect pests in Kenya. Afr. J. Hort. Sci. 2, 168–176 (2009).
Nyambo, B. Integrated pest management plan. The Agricultural Sector Development Program - Republic of Tanzania, pp 57 (2009).
Ekesi, S. Arthropod pest composition and farmers perceptions of pest and disease problems on citrus in Kenya. XII International Citrus Conference. pp 283 (2012).
Jagoueix, S., Bove, J.-M. & Garnier, M. The phloem-limited bacterium of greening disease of citrus is a member of the a subdivision of the Proteobacteria. Int. J. Syst. Bacteriol. 44, 379–386 (1994).
European and Mediterranean Plant Protection Organization. EPPO Global Database; https://gd.eppo.int/ Accessed November 11, 2019 (2019).
Fernandes, A. & Franquinho Aguiar, A. Evoluçao das pragas de quarentena Toxoptera citricida (Kirkaldy) e Trioza erytreae (Del Guercio) no Arquipelago da Madeira. Bol. Sanid. Veg. Plagas 27, 51–58 (2001).
Cocuzza, G. E. M. et al. A review on Trioza erytreae (African citrus psyllid), now in mainland Europe, and its potential risk as vector of Huanglongbing (HLB) in citrus. J. Pest Sci. 90, 1–17 (2017).
Bové, J. M. Huanglongbing: a destructive, newly-emerging, century-old disease of citrus. J. Plant Pathol. 88, 7–37 (2006).
Pole, F. N., Ndung’u, J. M., Kimani, J. M. & Kagunu, E. Citrus farming in Kwale district: a case study of Lukore location. Proceedings of the 12th KARI Biennial conference, pp 629-635 (2010).
Swai, I. S. Occurrence of Citrus Greening Disease in Tanzania. Plant Dis. 76, 1185 (1992).
McClean, A. P. D. & Oberholzer, P. C. Greening disease of the sweet orange: evidence that it is caused by a transmissible virus. S. Afr. J. Agric. Sci. 8, 253–276 (1965).
Massonie, G., Garnier, M. & Bove, J. M. Transmission of Indian citrus decline by Trioza erytreae (Del Guercio), the vector of South African greening. Proceedings of the 7th Conference of the International Organization of Citrus Virologists, pp 18–20 (1976).
Halbert, S. E. & Manjunath, K. L. Asian citrus psyllids (Sternorrhyncha: Psyllidae) and greening disease of citrus: a literature review and assessment of risk in Florida. Florida Entomol. 87, 330–353 (2004).
Hall, D. G., Richardson, M. L., Ammar, E. D. & Halbert, S. E. Asian citrus psyllid, Diaphorina citri, vector of citrus Huanglongbing disease. Entomol. Exp. Appl. 146, 207–223 (2013).
Grafton-Cardwell, E. E., Stelinski, L. L. & Stansly, P. A. Biology and management of Asian citrus psyllid, vector of the Huanglongbing pathogens. Annu. Rev. Entomol. 58, 413–432 (2013).
Saponari, M. et al. First Report of ‘Candidatus Liberibacter asiaticus’ associated with Huanglongbing in sweet orange in Ethiopia. Plant Dis. 94, 482 (2010).
Kalyebi, A. et al. Detection and identification of etiological agents (Liberibacter spp.) associated with citrus greening disease in Uganda. Uganda J. Agric. Sci. 16, 43–54 (2015).
Shimwela, M. M. et al. First occurrence of Diaphorina citri in East Africa, characterization of the Ca. Liberibacter species causing Huanglongbing (HLB) in Tanzania, and potential further spread of D. citri and HLB in Africa and Europe. Eur. J. Plant Pathol. 146, 349–368 (2016).
Roberts, R. et al. Resolution of the identity of ‘ Candidatus Liberibacter’ species from Huanglongbing-affected citrus in East Africa. Plant Dis. 101, 1481–1488 (2017).
Roberts, R., Steenkamp, E. & Pietersen, G. Three novel lineages of ‘Candidatus liberibacter africanus’ associated with native rutaceous hosts of Trioza erytreae in South Africa. Int. J. Syst. Evol. Microbiol. 65, 723–731 (2015).
Roberts, R. Genetic and biological diversity of ‘Candidatus Liberibacters’ from South Africa. PhD thesis. University of Pretoria, South Africa (2019).
Moran, V. C. Preliminary observations on the choice of host plants by adults of the citrus psylla, Trioza erytreae (Del Guercio) (Hornoptera: Psyllidae). J. Ent. S. Afr. 31, 403–410 (1968).
Samways, M. J. & Manicom, B. Q. Immigration, frequency distributions and dispersion patterns of the psyllid Trioza erytreae (Del Guercio) in a citrus orchard. J. Appl. Ecol. 20, 463–472 (1983).
Khamis, F. M. et al. DNA barcode reference library for the African citrus triozid, Trioza erytreae (Hemiptera: Triozidae): vector of African Citrus Greening. J. Econ. Entomol. 110, 2637–2646 (2017).
Pérez-Rodríguez, J. et al. Classical biological control of the African citrus psyllid Trioza erytreae, a major threat to the European citrus industry. Sci. Rep. 9, 9440, https://doi.org/10.1038/s41598-019-45294-w (2019).
Percy, D. M. et al. Resolving the psyllid tree of life: phylogenomic analyses of the superfamily Psylloidea (Hemiptera). Syst. Entomol. 43, 762–776 (2018).
Rubinoff, D. & Holland, B. S. Between two extremes: mitochondrial DNA is neither the panacea nor the nemesis of phylogenetic and taxonomic inference. Syst. Biol. 54, 952–961 (2005).
Powell, C. et al. Barcoding of parasitoid wasps (Braconidae and Chalcidoidea) associated with wild and cultivated olives in the Western Cape of South Africa. Genome 62, 183–199 (2019).
Collins, R. A. & Cruickshank, R. H. The seven deadly sins of DNA barcoding. Mol. Ecol. Resour. 13, 969–75 (2013).
Wu, F. et al. Analyses of mitogenome sequences revealed that Asian citrus psyllids (Diaphorina citri) from California were related to those from Florida. Sci. Rep. 7, 10154, https://doi.org/10.1038/s41598-017-10713-3 (2017).
European and Mediterranean Plant Protection Organization. EPPO Standards PM/57. Diagnostics. Trioza erytreae. EPPO Bull. 35, 357–360 (2005).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics. 10, 421, https://doi.org/10.1186/1471-2105-10-421 (2009).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Laslett, D. & Canbäck, B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175 (2008).
Bandelt, H. J., Forster, P. & Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48 (1999).
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120 (1980).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and high-performance computing. Nat. Methods 9, 772 (2015).
Dereeper, A. et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–469, https://doi.org/10.1093/nar/gkn180 (2008).
Acknowledgements
Inusa Ajene was supported by a German Academic Exchange Service (DAAD) In-Region Postgraduate Scholarship and the German Ministry for Economic Cooperation and Development (BMZ) through GIZ to the project ‘Strengthening Citrus Production Systems through the Introduction of Integrated Pest Management (IPM) Measures for Pests and Diseases in Kenya and Tanzania (SCIPM)’. We would also like to acknowledge icipe core funding provided by the UK Aid from the UK Government, the Swedish International Development Cooperation Agency (Sida), the Swiss Agency for Development and Cooperation (SDC), the Federal Democratic Republic of Ethiopia, and the Kenyan Government.
Author information
Authors and Affiliations
Contributions
B.V.A. designed the study, and I.A., F.K. and G.P. performed the analyses. All authors discussed the results and contributed to the writing of the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ajene, I., Khamis, F.M., Pietersen, G. et al. Mitochondrial genetic variation reveals phylogeographic structure and cryptic diversity in Trioza erytreae. Sci Rep 10, 8893 (2020). https://doi.org/10.1038/s41598-020-65880-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-65880-7
This article is cited by
-
Addition of Selected Plant-Derived Semiochemicals to Yellow Sticky Traps Does Not Improve Citrus Psyllid Captures
Journal of Chemical Ecology (2024)
-
Documenting the biodiversity of edible grasshoppers in South Africa with traditional knowledge, classic taxonomy and genetic information
Biodiversity and Conservation (2023)
-
Insights into the origin of the invasive populations of Trioza erytreae in Europe using microsatellite markers and mtDNA barcoding approaches
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
