
Abstract
In order to identify candidate genes or loci associated with growth performance of the newly established common carp strain, Xinlong, we conducted a genome-wide association analysis using 2b-RAD technology on 123 individuals. We constructed two sets of libraries associated with growth-related parameters (weight, length, width and depth) measured at two different grow-out stages. Among the 413,059 SNPs identified using SOAP SNP calling, 147,131 were tested for GWAS after quality filtering. Finally, 39 overlapping SNPs, assigned to four genomic locations, were associated with growth traits in two stages. These loci were assigned to functional classes related to immune response, response to stress, neurogenesis, cholesterol metabolism and development, and proliferation and differentiation of cells. By overlapping results of Plink and EMMAX analyses, we identified three genes: TOX, PLK2 and CD163 (both methods P < 0.05). Our study results could be used for marker-assisted selection to further improve the growth of the Xinlong strain, and illustrate that largely different sets of genes drive the growth of carp in the early and late grow-out stages.
Similar content being viewed by others


Introduction
Genome-wide association studies facilitate identification of single-nucleotide polymorphisms (SNPs) and genes associated with important economic traits. In particular, growth is one of the most economically important traits for the aquaculture industry. At the genomic level, genes and loci controlling this quantitative trait received a lot of interest in fish; for example growth rate in Atlantic salmon and rainbow trout1,2,3, head size in catfish and common carp4,5, etc. As a powerful statistical tool for connecting traits to their corresponding genes, genome-wide association study (GWAS) offers the possibility to analyze a massive amount of data. This allows identification of single nucleotide polymorphisms (SNP) or genes that may be related to important economic traits, or other traits of interest6. However, GWAS employs methods that require genome-wide SNP data produced by genome re-sequencing, so it is relatively costly, which limits its applicability7. Although the genechip used to capture the genome wide molecular markers has been established for some farmed animals (e.g. cattle and pigs), the application of GWAS in aquatic animals still faces higher costs and practical problems. To account for this, a simplified and cheaper genotyping method, 2b-RAD, was developed8. This method is based on the sequencing of uniform DNA fragments produced by type IIB restriction endonuclease. Its effectiveness in associated analysis and genotyping has been established in several freshwater and marine fish species, including bighead carp (Hypophthalmichthys nobilis), Nile tilapia (Oreochromis niloticus), and yellowfin tuna (Thunnus albacares)9,10,11,12. The common carp (Cyprinus carpio L.) is one of the oldest and most important farmed fish species, and the most widely distributed freshwater fish in the world13,14. Ranking third among the most commercially important fish species in China, its production exceeded 349,800 tons in 201615. With such importance, common carp has been a subject of several breeding programs. A carp strain named “Huanghe” has a long tradition and historical importance in Chinese culture, and retains notable economic importance. As a result, despite the fact that its growth rate is not as fast as that of more modern strains16,17, it has received a lot of interest from researchers. Recently, a new derivative of the Huanghe strain, provisionally named “Xinlong”, has been established using a combination of the best linear unbiased prediction (BLUP) with marker-assisted technology. After six years of continuous selection, its growth rate improvement (compared to the Huanghe strain) was 20.84% after an 8 months-long growth trial16. In order to explore the genetic basis for this fast-growing phenotype, we previously conducted a genome-wide association study using 2b-RAD sequence assay for four growth-related traits at 3 and 8 months of age, and found that genes contributing to this improvement are related to sex, neural pathways and fatty acid metabolism18. However, for the aquaculture industry purposes, growth performance at the end of the culture period, i.e. when fish reach the marketable size (17 months after stocking for carp) is more important than growth rate in the middle of that period. Furthermore, it is also important to know whether growth rate is controlled by the same set of genes during the entire grow-out period, or whether different genes may be controlling growth rates in different growth stages.
In this study, we set out to identify genes driving the growth performance during the second half of the grow-out period (8–17 months). We also determined genes and loci that exhibited a strong correlation with improved growth parameters during both grow-out stages. To achieve this, we conducted a genome-wide comparative analysis of growth parameters-associated loci using the cost-effective 2b-RAD method. This allowed us to discover thousands of SNPs associated with growth performance in two different growth periods, as well as identify those overlapping between the two stages. These results shall help us better understand the genetic basis for the improved growth rate of the new Xinlong strain.
Results
Evaluation of growth-related traits
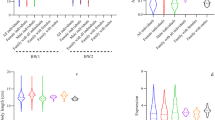
We analyzed four growth parameters in two different grow-out stages: five months after tagging, and at the end of the production cycle (17 months). Within-stage comparisons revealed a statistically significant linear relationship between the four growth traits. Notably, we observed particularly strong correlation between Blen5m/Bwid5m (r = 0.94) and Blen5m/Bdep5m (r = 0.91) in the first stage, and between hBwt and hBdep (r = 0.93) in the second stage (Table 1). At the end of the growth cycle, all four growth parameters were statistically significantly higher in the Selection group, whereas control group exhibited higher SD values (Fig. 1).

Comparison of mean growth rates of two carp populations at harvest. Con; Control group; Sel: selection group; hBwt: body weight (g); hBlen: body length (cm); hBwid: body width (cm); hBdep: body depth (cm). Data are expressed as the mean ± SD.
2b-RAD raw data processing
Raw 2b-RAD data were filtered via several quality control steps (see methods for details), clean data were then mapped to the genome of domesticated common carp19, and 413,059 SNPs detected after SNP calling by RADtyping program. After the SNP quality checking (QC), 147,131 SNPs were left and successfully assigned to 50 chromosomes and 3,377 scaffolds of the draft genome. The average sequencing depth was 44.56 and mapping successfulness rate ranged from 46.89% to 50.83%. We obtained 27,730,499 unique tags (225,451 tags per specimen) (Supplementary Table S1). Five regions accounted for 96.01% of all SNPs; with the highest proportion annotated in the intron (34.00%), followed by the intergenic (27.18%), upstream genes in the annotation file of reference genome (13.37%), downstream genes (13.18%), and finally exon regions (8.28%) (Supplementary Table S2).
Exploring genomic markers associated with growth traits in two grow-out stages
Analyses with Plink and EMMAX were conducted for candidate SNPs associated with the four growth performance traits in each stage: 44,306 SNPs were associated with growth traits in the first stage and 33,039 in the second stage (P < 0.05). After the Bonferroni correction, statistically significant values (P < 0.05) were found for only 109 SNPs in the first stage and only one in the second stage (Table 2).
Manhattan graphs plotted for parameters of all growth stages [−log10 (P value)> 5] allowed us to isolate 345 related SNPs. Using EMMAX software, which can account for full sib information, we identified 39 SNPs that were associated with the studied growth parameters via comparison of 45 (10 traits * 9/2 combination pairs) pairs of growth traits in both grow-out stages (Table 3). No overlapping loci were identified in pairwise comparisons of loci associated with growth parameters between first and second stages. These 39 SNPs were annotated using the reference genome (Table 4). Among these 39 SNPs, 36 were identified only by EMMAX, and three were further corroborated by plink analysis. These 39 loci were located in the 5' flank regions, 3' flank regions, CDS and intron regions of corresponding genes, and belonged to the following five functional classes:
-
1)
Immune response to pathogens. In response to the microbial infection, interferon alpha/beta receptor 2 (IFNA/betaR2) triggers a complex cascade of events20. Many genes associated with these downstream events are involved in cellular immune processes. For T-cell development and trafficking, thymocyte selection-associated high mobility protein (TOX) is involved in chromatin assembly, transcription, and modulation of T-cell development, and growth regulation21. DCs and T cells in semaphorin-4A (Sema4a)-knockout mice displayed poor allostimulatory activities and T helper cell (Th) differentiation, respectively22. Spinster homolog 2 (SPNS2) regulates the levels of Sphingosine-1-phosphate (S1P) gradient that controls lymphocyte trafficking23. In addition, dnaJ homolog subfamily C member 25 (DNAJC25) can significantly increase cell apoptosis, and its overexpression inhibits cell growth24. Deletion of inositol polyphosphate multikinase (IPMK) in cell lines and mice virtually abolished lipophagy, caused liver damage and inflammation, and impaired hepatocyte regeneration25. Among the regulation factors, laccase domain-containing protein 1 (LACC1) regulates TNF and IL-17 in mouse models of arthritis and inflammation26. A knockout of scavenger receptor cysteine-rich type 1 protein M130 (CD163) improved the resistance to viral infection in pigs27. A knockout of apolipoprotein A-I (APOA1) decreased parenchymal and vascular β-amyloid pathology in the Tg2576 mouse model of Alzheimer’s disease28.
-
2)
Development, proliferation and differentiation of cells. In the embryonic development of mice, tyrosine-protein kinase JAK1 deficiency causes arrested development, and apnea and death in newborn pups29. In the cell proliferation and differentiation category, nudC domain-containing protein 1 (NUDCd1) expression was positively correlated with cell proliferation, migration, and invasion in A498 cells30. Compared to smarcb1a NUDCd1 function, SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily B, member 1-A (smarcb1a) is a critical component of the mammalian SWItch/Sucrose Non-Fermentable (mSWI/SNF) protein, which promotes MyoD-mediated muscle differentiation by altering the chromatin structure in promoter regions of endogenous loci31. APC membrane recruitment protein 1 (Amer1) acts as a scaffold protein for the β-catenin destruction complex and promotes stabilization of Axin at the plasma membrane, thereby exerting negative regulatory role in Wnt signaling (key role in embryonic development)32. Complete loss of DNA damage checkpoint protein RAD9A in male mice caused a radical loss of spermatogenic cells (infertility or sub-fertility)33.
-
3)
Cholesterol metabolism. Phosphatidylinositol 5-phosphate 4-kinase type-2 alpha (PIP4K2A) regulates intracellular cholesterol transport34. As a result, hormone-sensitive lipase (HSL) has negative correlation with cholesterol content in the testes35, and dela(24)-sterol reductase (DHCR24) knockout in brain caused cholesterol deficiency in mice36.
-
4)
Neurogenesis. The genes involved in this category can be divided into two groups, one of which is related to the neurons and neuritogenesis. Rotein kinase C-binding protein (NELL2) promotes survival of neurons37. Homeobox protein (DBX1-B) is crucial for production of dorsal habenular neurons38. Synapse differentiation-inducing gene protein 1 (SYNDIG1) regulates the excitatory synapse maturation in rats39. Mice lacking Nrxn2α (α-variant of NRXN2 (neurexin 2)) exhibit behavioral abnormalities (social interaction deficits and increased anxiety)40. The other group is associated with signal transmission and regulation factors. Septin 8 B (sept8b) expression pattern in glial cells of zebrafish indicated that it was involved in signal transmission in nervous systems41. Mtss1 impinges on directional persistence and neuritogenesis42. Rap guanine nucleotide exchange factor 6(Rapgef6)-knockout mice exhibited mild behavioral abnormalities (hyperlocomotion and working-memory defects)43.
-
5)
Response to stress, comprising genes included in the interaction between environment and genetics. Catechol-O-methyltransferase (COMT) was identified as gene × environment interaction candidate gene (probably a regulatory factor), associated with childhood adversity, posttraumatic stress disorder, and major depressive disorder44. Mixed lineage kinase dual leucine zipper kinase (DLK) regulates the JNK-based stress response pathway45. Overexpression of IMPACT in yeast cells inhibited growth under all stress conditions that require GCN2 (eIF-2-alpha kinase) and GCN1 for cell survival, probably by the IMPACT promoting the dissolution of the GCN2–GCN1 complex46. Serine/threonine-protein kinase PLK2-like (PLK2-like) is involved in catalysis of the following reaction in response to stress: ATP + protein serine = ADP + protein serine phosphate, and ATP + protein threonine = ADP + protein threonine phosphate47.
We calculated pairwise R2 values between each of the three common SNPs associated with growth traits and other SNPs identified on the same chromosome or scaffold, and selected all pairs with R2 > 0.1. Manhattan plots for Blen5m (NW_017538109.1–569817 and NW_017541427.1–19670) and Bwid5m (W_017538165.1–106199 and NW_017541427.1–19670) are shown in Figures S1 and S2, and linkage disequilibrium for the three SNPs in Figure S3. Selected pairs were located on the position 521711 (annotation: exon of mitochondrial ribosomal protein L57 (MRPL57), transcript variant X1) in scaffold NW_017538109.1 (R2 = 0.11), position 396390 (annotation: intron of 1-acyl-sn-glycerol-3-phosphate acyltransferase epsilon-like (AGPAT5)) in scaffold NW_017538165.1 (R2 = 0.12), and position 65778 (annotation: uncharacterized LOC109077101) in scaffold NW_017541427.1 (R2 = 0.44).
Validation of growth-related markers
To verify the authenticity of the three genes (TOX, PLK2 and CD163) identified by overlapping the results of Plink and EMMAX analyses in 188 specimens, three primer pairs were designed to amplify and sequence the PCR products. Among the sequenced products, two PLK2 genotypes, AG and GG, were identified. We found that two genotypes exhibited significant differences in the BWid5m growth parameter (Fig. 2).

Effect of PLK2 genotype observed by GWAS on four growth parameters of the Xinlong carp strain after 8 months of grow-out culture. (a) Bwt5m (body weight, g), Blen5m (body length, mm), Bdep5m (body depth, mm) and BWid5m (body width, mm). Different characters represent significant differences between two genotypes. (b) DNA sequencing electropherograms of the AG and GG genotypes. The targeted SNP sites are indicated by black arrows.
Discussion
Genome-wide association studies are a very efficient approach for associating SNPs with important economic traits, as they allow analysis of large amounts of SNP data and identification of specific markers6,48. Genomic selection based on specific genes can be a cost-effective and fast (regarding the number of genome-wide markers used) method to considerably facilitate the selection for complex quantitative traits by precisely selecting favourable alleles. Obtaining thousands of SNP markers is becoming easier with the development of new technologies for population genotyping, thus allowing construction of high-density genetic maps for domesticated animals, as well as identification of QTL and specific markers for economically important traits49. High and low-density genome-wide association assays have been constructed both for terrestrial and aquatic farmed animals. Examples include the bovine SNP50 chip with 54,001 SNPs50, the SNP linkage map developed for Atlantic salmon, with around 5500 markers resulting from 16.5 K SNPs, and the SNP linkage map of common carp with 28,194 SNPs from a 250 K genechip51,52. These SNP arrays were used to explore QTL related to important economic traits, such as body weight and age at sexual maturation4. Xu et al. relied on QTL mapping to identify a number of growth performance regulators, including those associated with cell-proliferation (IGF1) and growth and energy metabolism (ERBB4, BMPR1B, SMTLB, GS, KISS2 and NPFFR1)52. In our previous genome-wide analysis of the Xinlong strain, we found a number of genes associated with growth parameters at the early grow-out stage, including those related to neural pathways (RHEB, MDGA2, NRXN1a, BDNF, SHANK2 and REEP2), sex dimorphism (NRXN1A, SMOC2, RAB10 and HSD17B), and fatty acid metabolism (RAB10, TNMD, VPAC2 and ACSF2)18. However, it remained unknown which candidate genes or loci contribute to the growth performance during later growth stages.
In this study, we were interested in genes controlling the growth parameters in later grow-out stages, up until the marketable fish size. By studying ten growth traits in two different grow-out stages, and then merging the results of Plink and EMMAX analyses, we identified 39 growth-related SNPs. SNPs associated with five functional classes were associated with four growth parameters in the first grow-out stage. No overlapping SNPs were found between growth performance parameters in the first and second grow-out stages. This may be a genetic molecular marker contribution to the comparatively high correlation coefficients among the former four parameters and lower coefficients between the growth parameters in the two grow-out stages (Table 1). These overlapping genes may be interacting in a way that produces the observed increased growth performance. Firstly, both HSL (regulates the cholesterol content35) and PIP4K2A (regulates the intracellular cholesterol transport34) can influence the availability of cholesterol. The cholesterol level appears to limit development of the central nervous system (CNS)36,53. The LENSSPQAPARRLLPP (BigLEN) neuropeptide delivered to synapse is believed to act through the G protein-coupled receptor 171 (GPR171) to regulate body weight by food intake and metabolism54. Beside the body weight, hypothalamic neurons may be involved in the regulation of metabolism, energy balance, and social behavior55. Within the brain, endogenous leptin plays a physiologically important role in the control of food intake56. In the neural cell lines, leptin-induced transactivation of NPY gene (involved in the central and sympathetic regulation of food intake) promoter can be mediated by JAK157. This gene was singled out in our pairwise comparisons of growth parameters, and six other genes were involved in pathways associated with neurons and neurogenesis (NELL2, dbx1b, Mtss1, SYNDIG1, NRXN2 and BDNF). Thus, cholesterol content and transportation can affect the central nervous system and the brain, both of which play a role in the control of food intake via leptin, JAK1 and GPR171 genes.
By overlapping Plink and EMMAX results, we identified three genes. TOX is required for a critical transitional step in selection and maturation of thymocytes, so this gene may have a direct effect on the cell-mediated immunity58. Therefore, we speculate that this gene may play a role in immune responses and growth of the common carp. PLK2-like is associated with metabolic responses to stress, involved in cell proliferation by contributing to the mitosis and centrosome cycle, and plays an important role in embryonic and skeletal development, as evidenced by experiments on cultured PLK2 embryonic fibroblasts, where the proliferation was stronger in cells expressing PLK259,60. And finally, CD163-knockout pigs were completely resistant to viral infection during the PRRSV challenge due to the fact that this gene acts as a cellular receptor for PRRSV27.
By LD analysis of three common loci, two candidate genes were found: AGPAT5 and MRPL57. Polymorphism of AGPAT5 gene was associated with the pig and cattle meat quality61,62. Liver-specific Agpat5 knockout mice had significantly reduced fasting plasma insulin and hepatic triglycerides after 12 weeks of high-fat diet63. AGPAT2 enzyme catalyzes the acylation of lysophosphatidic acid to form phosphatidic acid, a key intermediate in the biosynthesis of triacylglycerol and glycerophospholipids64. Therefore, regulation of biosynthesis of triacylglycerol and glycerophospholipids may be the genetic explanaiton behind higher content of polyunsaturated fatty acids in the muscle of the Jinlong carp18.
The candidate genes explored in this paper indicate that growth of common carp is a complex trait, associated with immunity, metabolism, stress and development, and contingent upon the interaction between environment and genetic backgroud65. Therefore, genome-based selective breeding should not only focus on traditional growth-related genes, but also use reliable breeding population to explore additional genes that may affect growth. As many of these genes remain unknown for the time being, it may be a promising direction for future studies.
Conclusions
The objective of this study was to use the cost-effective 2b-RAD technology to investigate the genomic regions related to the growth performance of a new common carp strain. Growth-related parameters, measured at 8 months of age and at the end of the grow-out cycle, were evaluated for 123 specimens. According to these results, we analyzed 2b-RAD data and identified a total of 147,131 SNPs. A comparison of loci associated with all growth parameters in the first and second grow-out stages identified 39 SNPs. None of these SNP overlapped between the first grow-out stage and second grow-out stage. Genes associated with these SNPs belonged to the following five functional classes: immune response to pathogens, development, proliferation and differentiation of cells, cholesterol metabolism, neurogenesis and response to stress. Plink and EMMAX analyzes singled-out three genes: TOX, PLK2 and CD163.
Methods
Experimental material and sampling
Focusing on growth performance as the target trait, a genetically distinct strain was developed from the traditional Huanghe carp strain at the Freshwater Fisheries Research Center, Chinese Academy of Fishery Sciences using BLUP66 method according to the best-predicted breeding values. A population of the new strain was obtained using artificial breeding (spawning in females was stimulated with hormonal injections, and then roe and milt mixed manually), fertilized eggs were incubated in separate cage settle nets for one week, and larvae then transferred to labelled (ID + spawning time) nursery happas. After 3 months in the nursery, parents and 8 of their full-sib F1 progenies randomly selected from each family were anesthetized with clove oil (75 mg/L)67 and tagged with passive integrated transponder tags (PIT) produced by Biomark. In total, 123 tagged full-sib common carp progeny specimens belonging to 25 families were selected for the experiment. Xinlong strain was represented by 95 specimens belonging to 19 families, and the control population (Huanghe strain) by 28 specimens belonging to 6 families. These two populations were cultivated in concrete tanks supplied with aerated and filtered water. During 17 months of the culture period, counting from the date of tagging (3 months of age) to the harvesting time (20 months of age), the fish were fed with a commercial feed (30% protein content) at a daily dose of 5% of their approximate body weight. Physicochemical parameters of the water were regularly monitored: dissolved oxygen, pH and temperature.
Growth parameters were measured after 5 (5 m) and 17 (h) months of the growth trial: body weight (Bwt5m and hBwt), body length (Blen5m and hBlen), body depth (Bdep5m and hBdep) and finally body width (BWid5m and hBWid), respectively. Fish were anesthetized with clove oil (75 mg·l−1) during the measurement procedure. Between 50 and 100 mg of caudal fin tissue was collected from each individual at the harvesting stage and preserved in 95% alcohol at 4 °C until DNA extraction.
Experimental procedures and animal handling were carried out in accordance with guidelines for the care and use of animals for scientific purposes set by the Institutional Animal Care and Use Committee of the Freshwater Fisheries Research Center and approved by the animal ethics committee of Chinese Academy of Fishery Sciences.
DNA extraction and 2b-RAD sequencing
DNA was isolated using universal genomic DNA kit (CWBIO, China) following the manual. Briefly, around 25 mg of fin tissue was digested for 1 hour at 56 °C with 20 µl Proteinase K to remove the proteins, and then incubated for 10 minutes at 70 °C after adding Buffer GL and 100% ethanol. Once the solutions were transferred to a column with a collection tube (Spin Columns DM), they were processed with Buffer GW1 and Buffer GW2 in order to improve the DNA purity. Finally, Buffer GE was added and DNA precipitated, collected and stored at −20 °C. Quality and concentration of the extracted DNA were checked by spectrophotometry (optical density reading at 260 and 280 nm) and electrophoresis on 1.0% agarose gel.
In order to construct the 2b-RAD libraries for each of the 123 individuals, we followed a simplified RAD (restriction site–associated DNA) genotyping method, as described by Su et al.18, with minor modifications. This method is based on sequencing of uniform fragments produced by type IIB restriction endonucleases8, as described in the 2b-RAD protocol3. PCR products were purified and quantified using SPRI select purification kit (Beckman Coulter, Pasadena, CA, USA) and Qubit 2.0 fluorometer (Invitrogen). The quality of all amplicon libraries was checked on 1.8% agarose gels and verified on Agilent 2100 Bioanalyzer.
2b-RAD raw data processing
In order to filter the sequenced reads and get high-quality reads, quality check (QC) (conducted on the basis of two criteria: max-missing (integrity parameter) <0.8 and Minor Allele Frequency (maf)> 0.01) and adapter trimming9 were performed. SNPs were discovered by aligning reads against a reference genome68 using STACKS v1.23 (parameters m3, M2 and N4). Genotyping was then done following steps reported in Jiao et al.69, excluding 3-bp positions, ambiguous reads, long homopolymer regions, and excessive numbers of low-quality positions (between 5 to 10).
The paired-end reads were merged by Pear software (Version 0.9.6)70. The merged reads were processed using a custom Perl script to trim adaptor sequences. The terminal 3-bp positions were also excluded from each read to eliminate artifacts that might have arisen from ligation sites. Reads with ambiguous bases (N) exceeding 8%, poor quality (15% nucleotide positions with a Phred quality <30), or without restriction sites were removed. The BsaXI tags in the genome of common carp were extracted based on the enzyme’s recognition site, which served as a reference for SNP discovery. High quality reads of each individual were aligned to the reference genome using SOAP2 (version 2.21)71 with the following parameters: r = 0, M = 4, v = 2. The aligned data for each individual were then used for SNP detection by RADtyping72 program with default parameters. For co-dominant markers, we used an ML algorithm to estimate for homozygotes or heterozygotes. In order to obtain robust results in the subsequent analyses, the following criteria were applied for SNP filtering. (1) Segregating markers that could be genotyped in at least 80% of the individuals were kept for analyses. (2) SNPs with a minor allele frequency (MAF) < 0.01 were discarded. (3) Polymorphic loci with more than two alleles possibly derived from sequencing or clustering errors were excluded. (4) Tags with more than two SNPs were excluded
Genome-wide analysis and candidate SNP identification
Two programs were used to identify the candidate SNPs. Plink applies a general linear model and EMMAX applies a mixed linear model, which can control for the false positive rate. The BH method in p.adjust function in R was used to calculate the false discovery rate. Using Bonferroni correction (P < 0.05 and P < 0.01), −log10 (P value) was calculated and plotted on Manhattan graphs with 100,000 window size using Plink73, EMMAX74 and qqman package75 in R. In order to identify the candidate SNPs, two models (linear and mixed linear model) were considered. Plink applies the general linear model, and EMMAX applies the mixed linear model, which can control for the false positive rate. The BH method in p.adjust function in R language was used to calculate the false discovery rate. For each stage, we identified significant SNPs, and their chromosomal locations. After we selected the SNPs for these two stages, we filtered the common SNPs according to their positions on chromosomes in the reference genome.
EMMAX conducts GWAS analysis on the basis of variance component approach. The algorithm conducts association analysis of quantitative traits as described below:
Let n be the sample size, p the total number of genotyped SNPs and Y the vector of observed phenotypes. We used the genotype data to calculate the n x n matrix \(\mathop{S}\limits^{\wedge }\) pairwise genetic relatedness between individuals, such as IBS or Balding-Nichols matrix, and normalized \(\mathop{S}\limits^{\wedge }\) to have sample variance 1 using a Gower’s centered matrix. We also used BN matrix as recommended by the authors.
P = I-11'/n and 1 is vector of number one. \(\mathop{S}\limits^{\wedge }\) is a positive-semidefinite matrix and can be replaced by other pairwise relatedness matrices estimated from the genotypes if they are also positive-semidefinite. Then we used a variance component model to estimate the restricted maximum likelihood parameters (or alternatively, maximum likelihood parameters) of \({\sigma }_{{\rm{a}}}^{2}\) and \({\sigma }_{{\rm{e}}}^{2}\) in
This tests the hypothesis H0: \({\sigma }_{{\rm{a}}}^{2}=0\). If the null hypothesis is rejected, it proceeds to step 3; otherwise, use the ordinary least squares to estimate the coefficients of each of the SNPs genotyped. For each marker, we used GLS F-test, or alternatively a score test, to estimate the effects \({\beta }_{k}\) and test the hypothesis \({\beta }_{k}\ne 0\) in the following model:
We substituted β0 by a multicolumn matrix containing the group information, as we thought that group information may be a confounding variable. We suspected that groups may cause population stratification (e.g. selection line and control line), which in turn may cause false positive signals in GWAS. β0 was accepted by –c parameter of EMMAX, which was a multicolumn file, such as:
100211 100211 1 1
100611 100611 1 1
100711 100711 1 0
100811 100811 1 0
101611 101611 1 1
101711 101711 1 1
First and second columns are sample names, third column is always 1, and fourth column is group information, with selected group coded as 1 and control group coded as 0.
In order to identify the candidate growth-associated genes, loci containing the selected SNP were annotated by SnpEff (version 4.1 g)76 and designated according to the published reference genome of the common carp (Songpu strain)19. Finally, plink and EMMAX results were merged by seeking a mathematic Union set. For each stage, we found significant SNPs, and identified their chromosome positions. After we selected the SNPs for these two stages, we filtered the common SNPs, i.e. the ones with corresponding relevant information (positions on chromosomes) and significant P values between the two stages. For these significant SNPs, we conducted LD analysis using Plink. The overlapping loci identified by these two methods were used to conduct the validation test. The common SNPs were used to conduct the linkage disequilibrium (LD) analysis, with ±0.5 M window along the chromosome or scaffold, using Plink and LDheatmap package77. Position name was constructed using chromosome or scaffold name plus position.
Validation of growth performance markers
On the basis of the genomic region of DNA designated from the reference genome of common carp, specific primers for the genome-wide loci significantly associated with growth-related traits were designed using Primer3web 4.1.0 (for picking primers from DNA sequences) and Primer-BLAST (NCBI; for finding specific primers). PCR was conducted using 2×Es Taq MasterMix (CWBio, China), and results were checked on 1.8% agarose gels and verified on Agilent 2100 Bioanalyzer.
In total 180 individuals from the F3 generation of Xinlong carp strain (no relationship with the 2b-RAD sequence data) were randomly selected and used to validate the efficiency of the identified markers. After amplification of the SNPs of the three genes in these 188 individuals, three primer pairs were designed on the basis of corresponding sequences in the reference common carp genome (Table 5). PCR products were sequenced (one–way) and aligned using DNAMAN6.0 software.
Statistical data analysis
All data were represented as mean ± SE and subjected to analysis by Chi-square tests and Student Test using Statistical Package for Social Sciences (SPSS) for Windows (SPSS Inc., Chicago, IL, USA). Differences were considered statistically significant when P values were <0.05.
Data availability
All of the 2b-RAD sequences used during this study are available from the NCBI Sequence Reads Archive (SRA) database under the accession numbers SRR6241620 and SRR6262716. The other datasets supporting the conclusions are included within the article and its additional files..
References
Tsai, H. Y. et al. Genome wide association and genomic prediction for growth traits in juvenile farmed Atlantic salmon using a high density SNP array. BMC Genomics. 16, (2015).
Gutierrez, A. P., Yáñez, J. M., Fukui, S., Swift, B. & Davidson, W. S. Genome-wide association study (GWAS) for growth rate and age at sexual maturation in Atlantic salmon (Salmo salar). PLoS One. 10, e0119730–e0119730 (2015).
Gonzalez-Pena, D. et al. Genome-wide association study for identifying loci that affect fillet yield, carcass, and body weight traits in Rainbow Trout (Oncorhynchus mykiss). Front. Genet. 7, 203 (2016).
Geng, X. et al. A Genome-wide association study identifies multiple regions associated with head size in Catfish. G3 (Bethesda). 6, 3389–3398 (2016).
Chen, L. et al. Genetic mapping of head size related traits in common carp (Cyprinus carpio). Front. Genet. 9, 448 (2018).
Korte, A. & Farlow, A. The advantages and limitations of trait analysis with GWAS: a review. Plant Methods. 9, 29 (2013).
Xu, J. et al. Development and evaluation of the first high-throughput SNP array for common carp (Cyprinus carpio). BMC Genomics. 15, 307 (2014).
Wang, S., Meyer, E., McKay, J. K. & Matz, M. V. 2b-RAD: a simple and flexible method for genome-wide genotyping. Nat. Methods. 9, 808–810 (2012).
Pecoraro, C. et al. Methodological assessment of 2b-RAD genotyping technique for population structure inferences in yellowfin tuna (Thunnus albacares). Mar. Genomics. 25, 43–48 (2016).
Seetharam, A. S. & Stuart, G. W. Whole genome phylogeny for 21 Drosophila species using predicted 2b-RAD fragments. PeerJ. 1, e226 (2013).
Fu, B., Liu, H., Yu, X. & Tong, J. A high-density genetic map and growth related QTL mapping in bighead carp (Hypophthalmichthys nobilis). Sci. Rep. 6, 28679 (2016).
Palaiokostas, C. et al. Mapping and validation of the major sex-determining region in Nile tilapia (Oreochromis niloticus L.) Using RAD sequencing. PLoS One. 8, e68389 (2013).
Casal, C. M. V. Global documentation of fish introductions: the growing crisis and recommendations for action. Biol. Invasions. 8, 3–11 (2006).
FAO. FAO Yearbook: Fisheries and Aquaculture Statistics. (Food and Agriculture Organization of the United Nations, 2015).
AFFAB. China Fishery Statistics Yearbook. Fisheries and Fishery Administration Bureau of the Ministry of Agriculture. (China Agriculture Press Chinese Agricultural Press, 2017).
Su, S. Y., Zhang, C. F., Dong, Z. J., Xu, P. & Yuan, X. H. The breeding gain of Huanghe carp (Cyprinus carpio hacmalopterus Temminck et Schlegel) new strain G3- The effect of higher carbohydrate diet on growth and fatty acid profile of huanghe carp new strain. J. Yangzhou Univ. 39, 63–66 (2018).
Chengfeng, Z., Shengyan, S., Jian, Z., Wenbin, Z. & Zaijie, D. Growth analysis of Huanghe carp during two grow-out stages. Acta Hydrobiol. Sin. 722–727 (2013).
Su, S. et al. Combined QTL and genome scan analyses with the help of 2b-RAD identify growth-associated genetic markers in a new fast-growing carp strain. Frontiers in Genetics. 9, 592 (2018).
Xu, P. et al. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat. Genet. 46, 1212–1219 (2014).
Ng, C. T., Mendoza, J. L., Garcia, K. C. & Oldstone, M. B. Alpha and beta type 1 interferon signaling: passage for diverse biologic outcomes. Cell. 164(3), 349–352 (2016).
Liu, T., Jiang, W., Han, D. & Yu, L. DNAJC25 is downregulated in hepatocellular carcinoma and is a novel tumor suppressor gene. Oncol. Lett. 4(6), 1274–1280 (2012).
Guha, P. et al. IPMK mediates activation of ULK signaling and transcriptional regulation of autophagy linked to liver inflammation and regeneration. Cell. Rep. 26(10), 2692–2703.e7 (2019).
Skon-Hegg, C. et al. LACC1 regulates TNF and IL-17 in mouse models of arthritis and inflammation. J. Immunol. 202(1), 183–193 (2019).
Contu, L., Carare, R.O. & Hawkes, C. A. Knockout of apolipoprotein A-I decreases parenchymal and vascular β-amyloid pathology in the Tg2576 mouse model of Alzheimer’s disease. Neuropathol. Appl. Neurobiol. (2019).
Lobbardi, R. et al. TOX regulates growth, DNA repair, and genomic instability in T-cell acute lymphoblastic leukemia. Cancer. Discov. 7, 1336–1353 (2017).
Yang, H. et al. CD163 knockout pigs are fully resistant to highly pathogenic porcine reproductive and respiratory syndrome virus. Antiviral Res. 151, 63–70 (2018).
Kumanogoh, A. et al. Nonredundant roles of Sema4Ain the immune system: defective T cell priming and Th1/Th2 regulation in Sema4A-deficient mice. Immunity. 22(3), 305–316 (2005).
Spiegel, S., Maczis, M. A., Maceyka, M. & Milstien, S. New insights into functions of the sphingosine-1-phosphate transporter SPNS2. J. Lipid. Res. 60(3), 484–489 (2019).
De la Serna, I. L., Carlson, K. A. & Imbalzano, A. N. Mammalian SWI/SNF complexes promote MyoD-mediated muscle differentiation. Nat.Genet. 27, 187–190 (2001).
Tanneberger, K. et al. Structural and functional characterization of the Wnt inhibitor APC membrane recruitment 1(Amer1). J Biol Chem. 286(22), 19204–19214 (2011).
Sakamoto, K., Wehde, B. L., Rädler, P. D., Triplett, A. A. & Wagner, K. U. Generation of Janus kinase 1 (JAK1) conditional knockout mice. Genesis. 54(11), 582–588 (2016).
Wang, R. J. et al. The impact of NudCD1 on renal carcinoma cell proliferation, migration, and invasion. Eur. Rev. Med. Pharmacol. Sci. 22(3), 671–677 (2018).
Vasileva, A. et al. The DNA damage checkpoint protein RAD9A is essential for male meiosis in the mouse. J. Cell. Sci. 126(Pt 17), 3927–3938 (2013).
Wang, F. et al. Hormone-sensitive lipase deficiency alters gene expression and cholesterol content of mouse testis. Reproduction. 153(2), 175–185 (2017).
Hu, A. et al. PIP4K2A regulates intracellular cholesterol transport through modulating PI(4,5)P2 homeostasis. J. Lipid. Res. 59(3), 507–514 (2018).
Kuehnle, K. et al. Age-dependent increase in desmosterol restores DRM formation and membrane-related functions in cholesterol-free DHCR24-/- mice. Neurochem. Res. 34(6), 1167–1182 (2009).
Saharan, M. et al. Enhanced long-term potentiation in vivo in dentate gyrus of NELL2-deficient mice. Neuroreport 15(3), 417–420 (2004).
Dean, B. J., Erdogan, B., Gamse, J. T. & Wu, S. Y. Dbx1b defines the dorsal habenular progenitor domain in the zebrafish epithalamus. Neural. Dev. 9, 20 (2014).
Díaz, E. SynDIG1 regulation of excitatory synapse maturation. J. Physiol. 590(1), 33–38 (2012).
Sistig, T. et al. Mtss1 promotes maturation and maintenance of cerebellar neurons via splice variant-specific effects. Brain. Struct. Funct. 222(6), 2787–2805 (2017).
Berger, C., Helmprobst, F., Chapouton, P., Lillesaar, C. & Stigloher, C. Sept8a and sept8b mRNA expression in the developing and adult zebrafish. Gene Expr Patterns. 25-26, 8–21 (2017).
Born, G. et al. Genetic targeting of NRXN2 in mice unveils role in excitatory cortical synapse function and social behaviors. Front. Synaptic. Neurosci. 7, 3 (2015).
Maeta, K. et al. Comprehensive behavioral analysis of mice deficient in Rapgef2 and Rapgef6, a subfamily of guanine nucleotide exchange factors for Rap small GTPases possessing the Ras/Rap-associating domain. Mol. Brain. 11(1), 27 (2018).
Ghosh, A. S. et al. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J. Cell. Biol. 194(5), 751–64 (2011).
Cambiaghi, T. D. et al. Evolutionarily conserved IMPACT impairs various stress responses that require GCN1 for activating the eIF2 kinase GCN2. Biochem Biophys Res Commun. 443(2), 592–597 (2014).
Maul, S. et al. Genetics of resilience: Implications from genome-wide association studies and candidate genes of the stress response system in posttraumatic stress disorder and depression. Am. J. Med. Genet. B. Neuropsychiatr. Genet. (2019).
Cizmecioglu, O., Warnke, S., Arnold, M., Duensing, S. & Hoffmann, I. Plk2 regulated centriole duplication is dependent on its localization to the centrioles and a functional polo-box domain. Cell. Cycle. 7, 3548–3555 (2008).
Visscher, P. M., Brown, M. A., McCarthy, M. I. & Yang, J. Five years of GWAS discovery. Am. J. Hum. Genet. 90, 7–24 (2012).
Goddard, M. E. & Hayes, B. J. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 10, 381–391 (2009).
Matukumalli, L. K. et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS One. 4, e5350 (2009).
Gidskehaug, L., Kent, M., Hayes, B. J. & Lien, S. Genotype calling and mapping of multisite variants using an Atlantic salmon iSelect SNP array. Bioinformatics. 27, 303–310 (2011).
Peng, W. et al. An ultra-high density linkage map and QTL mapping for sex and growth-related traits of common carp (Cyprinus carpio). Sci Rep. 6, (2016).
Mauch, D. H. et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 294, 1354–1357 (2001).
Gomes, I. et al. GPR171 is a hypothalamic G protein-coupled receptor for BigLEN, a neuropeptide involved in feeding. Proc. Natl. Acad. Sci. USA 110, 16211–16216 (2013).
Bakos, J., Zatkova, M., Bacova, Z. & Ostatnikova, D. The role of hypothalamic neuropeptides in neurogeneis and neuritogenesis. Neual. plasticity. 2016, 3276383 (2016).
Brunner, L. et al. Leptin is a physiologically important regulator of food intake. Int. J. Obes. Relat. Metab. Disord. 21, 1152 (1997).
Muraoka, O. et al. Leptin-induced transactivation of NPY gene promoter mediated by JAK1, JAK2 and STAT3 in the neural cell lines. Neurochem. Int. 42(7), 591–601 (2003).
Chaffey, N. et al. Molecular Biology of the Cell. 4th edn. Ann. Bot. 91, 401 (2003).
Archambault, V. & Glover, D. M. Polo-like kinases: conservation and divergence in their functions and regulation. Nat. Rev. Mol. Cell Biol. 10, 265–275 (2009).
Ma, S., Charron, J. & Erikson, R. L. Role of Plk2 (Snk) in mouse development and cell proliferation. Mol. Cell. Biol. 23, 6936–6943 (2003).
Park, W. B. et al. The rs196952262 Polymorphism of the AGPAT5 gene is associated with meat quality in berkshire pigs. Korean. J. Food. Sci. An. 37, 926 (2017).
Wei, X., Zhang, J., Sun, X., Zhao, F. & Zhang, J. Correlation analysis between the polymorphism of AGPAT5 gene and meat quality traits in German Simmental cattle. Hei. Anim. Sci. Vet. Med. 9, 12 (2013).
Clair, S. L. S., Belisle, S. L., Jaimes, F. B. L., Li, Z. & Parks, B. 1892-P: Hepatic Agpat5 Regulates Plasma Insulin in Obesity. Diabetes. 68, 1892-p (2019).
Agarwal, A. K. et al. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat. Genet. 31, 21–23 (2002).
Wang, C. H. & Li, S. F. Genetic effects and genotype x environment interactions for growth-related traits in common carp, Cyprinus carpio L. Aquaculture. 272, 267–272 (2007).
Ponzoni, R. W., Hamzah, A., Tan, S. & Kamaruzzaman, N. Genetic parameters and response to selection for live weight in the GIFT strain of Nile tilapia (Oreochromis niloticus). Aquaculture. 247, 203–210 (2005).
Velisek, J., Svobodova, Z., Piackova, V., Groch, L. & Nepejchalova, L. Effects of clove oil anaesthesia on common carp (Cyprinus carpio L.). Vet Med. 50, 269–275 (2005).
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A. & Cresko, W. A. Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140 (2013).
Jiao, W. et al. High-resolution linkage and quantitative trait locus mapping aided by genome survey sequencing: building up an integrative genomic framework for a bivalve mollusc. DNA Res. 21, 85–101 (2014).
Zhang, J., Kobert, K., Flouri, T. & Stamatakis, A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 30, 614–620 (2014).
Li, R. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 25, 1966–1967 (2009).
Fu, X. et al. RADtyping: An Integrated Package for Accurate De Novo Codominant and Dominant RAD Genotyping in Mapping Populations. PloS. One. 8, e79960 (2013).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Kang, H. M. et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010).
Turner, S.D. Qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. BioRxiv.005165
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly. 6, 80–92 (2012).
Shin, J. H. et al. The International HapMap Consortium. A haplotype map of the human genome. Nature. 437, 1299–1320 (2005).
Acknowledgements
This work was supported by grants from the Special Scientific Research Funds for Central Non-profit Institutes, Chinese Academy of Fishery Sciences (2020JBFJ02, 2016RC-LX03), and the Key Projects in the National Science & Technology Pillar Program during the twelfth Five-Year Plan Period (2012BAD26B02) and the National Nonprofit Institute Research Grant of CATAS-TCGRI (2013JBFM14, 2020TD37). We thank the students and staff of the Aquatic Genetic Laboratory, FFRC for their kind assistance in the study.
Author information
Authors and Affiliations
Contributions
SS designed this study and developed the statistical models. BR and SS carried out the DNA extraction, 2b-RAD library preparation, sequence data processing and drafted the manuscript. X.H., N.C., and X.L. analyzed the data and assisted in drafting the manuscript. J.Y., J.L., F.Y., M.W. and Y.T. revised the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Su, S., Raouf, B., He, X. et al. Genome Wide Analysis for Growth at Two Growth Stages in A New Fast-Growing Common Carp Strain (Cyprinus carpio L.). Sci Rep 10, 7259 (2020). https://doi.org/10.1038/s41598-020-64037-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-64037-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
