
Abstract
Bats provide key ecosystem services such as crop pest regulation, pollination, seed dispersal, and soil fertilization. Bats are also major hosts for biological agents responsible for zoonoses, such as coronaviruses (CoVs). The islands of the Western Indian Ocean are identified as a major biodiversity hotspot, with more than 50 bat species. In this study, we tested 1,013 bats belonging to 36 species from Mozambique, Madagascar, Mauritius, Mayotte, Reunion Island and Seychelles, based on molecular screening and partial sequencing of the RNA-dependent RNA polymerase gene. In total, 88 bats (8.7%) tested positive for coronaviruses, with higher prevalence in Mozambican bats (20.5% ± 4.9%) as compared to those sampled on islands (4.5% ± 1.5%). Phylogenetic analyses revealed a large diversity of α- and β-CoVs and a strong signal of co-evolution between CoVs and their bat host species, with limited evidence for host-switching, except for bat species sharing day roost sites. These results highlight that strong variation between islands does exist and is associated with the composition of the bat species community on each island. Future studies should investigate whether CoVs detected in these bats have a potential for spillover in other hosts.
Similar content being viewed by others


Introduction
The burden of emerging infectious diseases has significantly increased over the last decades and is recognized as a major global health concern. In 2018, the World Health Organization (WHO) established the “Blueprint priority disease list”, identifying viruses such as Ebola, Lassa fever, Middle East Respiratory Syndrome (MERS), and Nipah fever as significant threats to international biosecurity1. This list also highlights the potential pandemic risk from the emergence of currently unknown zoonotic pathogens, collectively referring to these unknown threats as “disease X″1. Investigation of the potential zoonotic pathogens in wild animals, particularly vertebrates, is thus critical for emerging infectious disease preparedness and responses.
Bats represent nearly 1,400 species and live on all continents except Antarctica2. They provide key ecosystem services such as crop pest regulation, pollination, seed dispersal, and soil fertilization3,4,5,6,7,8,9,10. Bats are also recognized as reservoirs of many zoonotic pathogens, including coronaviruses (CoVs)11,12,13. Indeed, several CoVs originating from bats have emerged in humans and livestock with sometimes major impacts to public health. For instance, in 2003, the Severe Acute Respiratory Syndrome (SARS) CoV emerged in humans, after spillover from bats to civets14,15,16,17,18, and led to the infection of 8,096 people and 774 deaths in less than a year19.
Our study area spans geographic locations across the islands of the Western Indian Ocean and southeastern continental Africa (SECA) (Fig. 1). These islands have diverse geological origins that have influenced the process of bat colonization and species distributions20. The ecological settings and species diversity on these islands for bats are notably different. On Madagascar, more than 45 bat species are known to occur, of which more than 80% are endemic to the island21,22,23. The smaller studied islands of the Western Indian Ocean, Mauritius, Mayotte, Reunion Island, and Mahé (Seychelles), host reduced bat species diversity (e.g. three species on Reunion Island), whereas SECA supports a wide range of bat species. To date, several studies have identified bat-infecting CoVs in countries of continental Africa, including Zimbabwe24, South Africa25,26, Namibia27, and Kenya28,29. CoVs have also been reported in fruit bats (Pteropodidae) in Madagascar, where β-coronaviruses belonging to the D-subgroup were identified in Eidolon dupreanum and Pteropus rufus30.

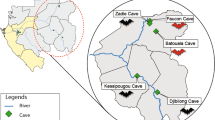
Geographic distribution of the tested samples. N: number of bats sampled for each location. The open-source GIS software, QGIS v.3.6.1, was used to generate the map. http://qgis.osgeo.org (2019).
In this study, we investigated the presence of CoVs in over 1,000 individual bats belonging to 36 species, sampled on five islands (Madagascar, Mauritius, Mayotte, Reunion Island, and Mahé) and one continental area (Mozambique). Based on molecular screening and partial sequencing of the RNA-dependent RNA polymerase gene, we (i) estimated CoV prevalence in the regional bat populations, (ii) assessed CoV genetic diversity, and (iii) identified associations between bat families and CoVs, as well as potential evolutionary drivers leading to these associations.
Results
Prevalence of CoV
A total of 1,013 bats were tested from Mozambique, Mayotte, Reunion Island, Seychelles, Mauritius and Madagascar (Fig. 1). In total, 88 of the 1,013 bat samples tested positive for CoV by Real-Time PCR (mean detection rate: 8.7%). The prevalence of positive bats was different according to the sampling locations (χ² = 77.0, df = 5; p < 0.001), with a higher prevalence in Mozambique (±95% confidence interval: 20.5% ± 4.9%) than on all Western Indian Ocean islands (4.5% ± 1.5%) (Fig. 2). A significant difference in the prevalence of positive bats was also detected between families (χ² = 44.8, df = 8; p < 0.001; Supplementary Figure S1). The highest prevalence was observed in the families Nycteridae (28.6% ± 23.6%) and Rhinolophidae (26.2% ± 11.0%). Bat species had a significant effect on the probability of CoVs detection (χ² = 147.9, df = 39; p < 0.001; Supplementary Figure S2). Finally, the prevalence of CoV positive bats in Mozambique was significantly different (N = 264, χ²= 22.8, df = 1; p < 0.001; Supplementary Figure S3) between February (37.4% ± 9.9%) and May (11.6% ± 4.8).

Mean CoV prevalence (±95% confidence interval) in bats in the Western Indian Ocean. Pairwise test; ***p < 0.001; NS: p > 0.05, not significant.
RdRp sequence diversity
Of the 88 positive samples, we obtained 77 partial RdRp sequences using the Real-Time PCR detection system (179 bp) and 51 longer partial RdRp sequences using a second PCR system (440 bp). Sequences generated with the second system were subsequently used for phylogenetic analyses. Details of the sequenced CoV-positive samples are provided in Supplementary Table S1. Pairwise comparison of these 51 sequences revealed 28 unique sequences, and sequences similarities ranging from 60.2% to 99.8%. The lowest sequence similarity was found in Mozambique (60.2% to 99.8%), then in Madagascar (64.0% to 99.8%). No genetic variation was observed for samples from Mayotte and Reunion Island.
Phylogenetic structure of CoVs
Sequence comparison indicated that Western Indian Ocean bats harbor a high diversity of both α and β-CoVs, with conserved groups clustering mostly by bat family (Fig. 3). Specifically, 25 sequences were identified as α-CoVs, and three sequences were genetically related to the β-CoVs. For α-CoVs, all sequences detected in our tested Molossidae formed a highly supported monophyletic group, including CoV sequences from Molossidae bats previously detected in continental Africa (Fig. 4). CoVs detected in Mops condylurus (Mozambique), Mormopterus francoismoutoui (Reunion Island), Chaerephon pusillus and Chaerephon sp. (Mayotte), and Mormopterus jugularis (Madagascar) shared 90%–98% nucleotide similarity with a CoV detected in Chaerephon sp. in Kenya (Supplementary Table S2). All CoVs found in Miniopteridae clustered in a monophyletic group, including Miniopteridae CoVs sequences from Africa, Asia, and Oceania (Supplementary Table S2). The great majority of α-CoVs detected in Rhinolophidae bats clustered in two monophyletic groups (Fig. 3); one with African Rhinolophidae CoVs and one with Asian Rhinolophidae CoVs. We also detected one CoV from Rhinolophus rhodesiae, which was 100% similar to a Miniopteridae CoV from this study. Rhinonycteridae CoVs formed a single monophyletic group with NL63 Human CoVs. The Rhinonycteridae CoVs detected clustered with NL63-related bat sequences found in Triaenops afer in Kenya (Fig. 5) and showed 85% similarity to NL63 Human CoVs (Supplementary Table S2). Hipposideridae α-CoVs mainly clustered into a single monophyletic group, including 229E Human CoV-related bat sequence found in Hipposideros vittatus from Kenya (Fig. 6; Supplementary Table S2).

Maximum Likelihood (ML) consensus tree derived from 202 coronavirus (CoV) RNA-dependent RNA-polymerase partial nucleotide sequences (393 bp). Colored circles at the end of branches indicate bat family origin. Sequences in bold refer to bat CoVs detected in this study. Bootstrap values >0.7 are indicated on the tree. Scale bar indicates mean number of nucleotide substitutions per site. The tree was generated with the General Time Reversible evolutionary model (GTR + I + Г, I = 0.18, α = 0.64) and 1,000 bootstrap replicates.

Detail of the α-CoV clade. Molossidae CoVs generated in the study are indicated in bold. This sub-tree is a zoom on Molossidae CoV clade from the tree depicted in Fig. 3. Bootstrap values >0.7 are indicated on the tree. Scale bar indicates mean number of nucleotide substitutions per site.

Detail of the α-CoV clade. NL63-like CoVs generated in the study are indicated in bold. This sub-tree is a zoom on NL63 CoV clade from the tree depicted in Fig. 3. Only bootstrap values >0.7 are indicated on the tree. Scale bar indicates mean number of nucleotide substitutions per site.

Detail of the α-CoV clade. 229E-like CoVs generated in the study are indicated in bold. This sub-tree is a zoom on NL63 CoV clade from the tree depicted in Fig. 3. Bootstrap values >0.7 are indicated on the tree. Scale bar indicates mean number of nucleotide substitutions per site.
For β-CoVs, two sequences obtained from Nycteris thebaica clustered in the C-subgroup together with other CoVs previously reported in African Nycteris sp. bats (Fig. 7). The sequences showed 88% nucleotide identity to a β-C CoV found in Nycteris gambiensis in Ghana (Supplementary Table S2). Rousettus madagascariensis CoVs clustered with Pteropodidae CoVs belonging to the D-subgroup of β-CoVs (Fig. 8). BLAST queries against the NCBI database showed 98% nucleotide identity between CoV sequences from Rousettus madagascariensis and a β-D CoV sequence detected in Eidolon helvum from Kenya (Supplementary Table S2).

Detail of the β-C CoV clade. CoVs generated in the study are indicated in bold. This sub-tree is a zoom on β-C CoV clade from the tree depicted in Fig. 3. Bootstrap values >0.7 are indicated on the tree. Scale bar indicates mean number of nucleotide substitutions per site.

Detail of the β-D CoV. CoVs generated in the study are indicated in bold. This sub-tree is a zoom on β-D CoV clade from the tree depicted in Fig. 3. Bootstrap values >0.7 are indicated on the tree. Scale bar indicates mean number of nucleotide substitutions per site.
Co-phylogeny between bats and CoVs
Co-phylogeny tests were conducted using 11 Cyt b sequences obtained from the 11 CoVs positive bat species and 27 partial CoV RdRp sequences (440 bp). Results supported co-evolution between the Western Indian Ocean bats and their CoVs (ParaFitGlobal = 0.04; p = 0.001) and a high level of phylogenetic congruence (Fig. 9).

Tanglegram representing host-virus co-evolution between bats of the Western Indian Ocean and their associated CoVs. Phylogeny of bats (left) was constructed with an alignment of 11 Cyt b sequences of 1,030 bp by Neighbor-Joining with 1,000 bootstrap iterations. Pruned phylogeny of Western Indian Ocean bats CoVs (right) was constructed with an alignment of 27 unique sequences of 393 bp from Western Indian Ocean bats CoVs, by Neighbor-Joining with 1,000 bootstrap iterations.
Discussion
We provide evidence for a high diversity of CoVs in bats on Western Indian Ocean islands. The overall prevalence of CoV positive bats was consistent with studies from continental Africa25 and from islands in the Australasian region31, although we detected significant variation in the prevalence of infected bats, according to their family, species, sampling location and season. Our study is nevertheless affected by the strong heterogeneity of bat communities in the island of the Western Indian Ocean, in particular in term of species richness. The high CoV genetic diversity detected in bats from Mozambique and Madagascar is likely to be associated with the higher bat species diversity in the African mainland and in Madagascar, has compared to small oceanic islands20. In addition, CoV prevalence in bat populations may significantly vary across seasons, as found in Mozambique with higher prevalence during the wet season than in the dry season. Several studies on bat CoV have indeed shown significant variations in the temporal infection dynamic of CoV in bats, potentially associated with bat parturition32,33,34.
Host specificity is well known for some bat CoVs subgenera35,36,37. For example, β-C CoVs are largely associated with Vespertilionidae, whereas β-D CoVs are found mostly in Pteropodidae36,38. In our study, we showed that Western Indian Ocean bats harbor phylogenetically structured CoVs, of both α-CoV and β-CoV subclades, clustering mostly by bat family. In the new CoV taxonomy based on full genomes proposed by the International Committee of Taxonomy of Viruses (ICTV), α-CoVs and β-CoVs are split in subgenera mostly based on host families39, reflected in the subgenera names (e.g. Rhinacovirus for a Rhinolophidae α-CoV cluster, Minuacovirus for a Miniopteridae α-CoV cluster, Hibecovirus for an Hipposideridae β-CoV cluster). Although our classification was based on a partial sequence of the RdRp region, we identified sequences from samples belonging to four of these subgenera (Minuacovirus, Duvinacovirus, Rhinacovirus, and Nobecovirus) and three that could not be classified according to this taxonomic scheme hence representing unclassified subgenera (we propose “Molacovirus”, “Nycbecovirus”, and “Rhinacovirus2”).
A strong geographical influence on CoVs diversity, with independent evolution of CoVs on each island, was expected in our study, because of spatial isolation and endemism of the tested bat species. Anthony et al.38 found that the dominant evolutionary mechanism for African CoVs was host switching. Congruence between host and viral phylogenies however suggests a strong signal for co-evolution between Western Indian Ocean bats and their associated CoVs. Geographical influence seems to occur within bat families, as for Molossidae. Endemism resulting from geographic isolation may thus have played a role in viral diversification within bat families.
Although co-evolution could be the dominant mechanism in the Western Indian Ocean, host-switching may take place in certain situations. For example, in Mozambique, we found a potential Miniopteridae α-CoV in a Rhinolophidae bat co-roosting with Miniopteridae in the same cave. These host-switching events could be favored when several bat species roost in syntopy40. A similar scenario was described in Australia where Miniopteridae α-CoV was detected in Rhinolophidae bats31. These infrequent host-switching events show that spillovers can happen but suggest that viral transmission is not maintained in the receiver host species. The host-virus co-evolution might thus have resulted in strong adaptation of CoVs to each bat host species. In addition, viral factors (mutation rate, recombination propensity, replication ability in the cytoplasm, changes in the ability to bind host cells), environmental factors (climate variation, habitat degradation, decrease of bat preys), and phylogenetic relatedness of host species are also critical for the viral establishment in a novel host41,42,43,44. Nevertheless, apparent evidence of host switching as a dominant mechanism of CoV evolution could be an artifact of a lack of data for some potential bat hosts, leading to incomplete phylogenetic reconstructions38.
Several bat CoVs we identified in Rhinonycteridae and Hipposideridae from Mozambique had between 85% and 93% nucleotide sequence similarity with NL63 Human CoVs and 229E Human CoVs, respectively. These two human viruses are widely distributed in the world and associated with mild to moderate respiratory infection in humans45. Tao et al. established that the NL63 Human CoVs and 229E Human CoVs have a zoonotic recombinant origin from their most recent common ancestor, estimated to be about 1,000 years ago46. During the past decade, they were both detected in bats in Kenya, and in Ghana, Gabon, Kenya, and Zimbabwe, respectively24,28,47,48. Intermediate hosts are important in the spillover of CoVs, despite major knowledge gaps on the transmission routes of bat infectious agents to secondary hosts49. This hypothesis has been formulated for the 229E Human CoV, with an evolutionary origin in Hipposideridae bats and with camelids as intermediate hosts48. The ancient spillover of NL63 from Rhinonycteridae bats to humans might have occurred through a currently unidentified intermediate host28,50,51. Because receptor recognition by viruses is the first essential cellular step to infect host cells, CoVs may have spilt over into humans from bats through an intermediate host possibly due to mutations on spike genes13,28. Further investigations of CoVs in Kenyan and Mozambican livestock and hunted animals could potentially provide information on the complete evolutionary and emergence history of these two viruses before their establishment in humans.
MERS-like CoV, with high sequence similarity (>85%) to human and camel strains of MERS-CoV, have been detected in Neoromicia capensis in South Africa and Pipistrellus cf. hesperidus in Uganda, suggesting a possible origin of camel MERS-CoV in vespertilionid bats25,38,52. This family has been widely studied, with 30% of all reported bat CoVs sequences from the past 20 years coming from vespertilionids53. No members of this family were positive for CoV in our study, which may be associated with the low number of individuals tested; additional material is needed to explore potential MERS-like CoV in the Western Indian Ocean, in particular on Madagascar.
Knowledge on bat CoV ecology and epidemiology has significantly increased during the past decade. Anthony et al. estimated that there might be at least 3,204 bat CoVs worldwide38; however, direct bat-to-human transmission has not been demonstrated so far. As for most emerging zoonoses, CoV spillover and emergence may be associated to human activities and ecosystem changes such as habitat fragmentation, agricultural intensification and bushmeat consumption. The role of bats as epidemiological reservoir of infectious agents needs to be balanced with such human driven modifications on ecosystem functioning, in order to properly assess bat-borne CoV emergence risks.
Materials and Methods
Origin of the tested samples
Samples obtained from vouchered bat specimens during previous studies in Mozambique (February and May 2015), Mayotte (November to December 2014), Reunion Island (February 2015), Seychelles (February to March 2014), Mauritius (November 2012) and Madagascar (October to November 2014) were tested54,55,56,57 (Supplementary Information). We also collected additional swab samples from several synanthropic bat species on Madagascar, in January 2018 (Supplementary Information). Details on sample types, bat families, species, and locations are provided in Supplementary Table S3.
Ethical statement
The ethical terms of these research protocols were approved by the CYROI Institutional Animal Care and Use Committee (Comité d’Ethique du CYROI no.114, IACUC certified by the French Ministry of Higher Education, of Research and Innovation). All protocols strictly followed the terms of research permits and regulations for the handling of wild mammals and were approved by licencing authorities (Supplementary Information).
Molecular detection
RNA was extracted from 140 μL of each sample using the QIAamp Viral RNA mini kit (QIAGEN, Valencia, California, USA), and eluted in 60 μL of Qiagen AVE elution buffer. For bat organs, approximately 1 mm3 of tissue (either lungs or intestines) was placed in 750 µL of DMEM medium and homogenized in a TissueLyser II (Qiagen, Hilden, Germany) for 2 min at 25 Hz using 3 mm tungsten beads, prior to the RNA extraction. Reverse transcription was performed on 10 μL of RNA using the ProtoScript II Reverse Transcriptase and Random Primer 6 (New England BioLabs, Ipswich, MA, USA) under the following thermal conditions: 70 °C for 5 min, 25 °C for 10 min, 42 °C for 50 min, and 65 °C for 20 min58. cDNAs were tested for the presence of the RNA-dependent RNA-polymerase (RdRp) gene using a multi-probe Real-Time PCR59. The primer set with Locked Nucleic Acids (LNA; underlined position in probe sequences) was purchased from Eurogentec (Seraing, Belgium): 11-FW: 5′-TGA-TGA-TGS-NGT-TGT-NTG-YTA-YAA-3′ and 13-RV: 5′-GCA-TWG-TRT-GYT-GNG-ARC-ARA-ATT-C-3′. Three probes were used: probe I (ROX): 5′-TTG-TAT-TAT-CAG-AAT-GGY-GTS-TTY-AT-3′, probe II (FAM): 5′-TGT-GTT-CAT-GTC-WGA-RGC-WAA-ATG-TT-3′, and probe III (HEX): 5′-TCT-AAR-TGT-TGG-GTD-GA-3′. Real-Time PCR was performed with ABsolute Blue QPCR Mix low ROX 1X(Thermo Fisher Scientific, Waltham, MA, USA) and 2.5 µL of cDNA under the following thermal conditions: 95 °C for 15 min, 95 °C for 30 s, touchdowns from 56 °C to 50 °C for 1 min and 50 cycles with 95 °C for 30 s and 50 °C for 1 min in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA).
Because of the limited size of sequences generated from the Real-Time PCR, a second PCR targeting 440 bp of the RdRp gene was performed with 5 µL of cDNA of each positive sample, with the following primer set: IN-6: 5′-GGT-TGG-GAC-TAT-CCT-AAG-TGT-GA-3′ and IN-7: 5′-CCA-TCA-TCA-GAT-AGA-ATC-ATC-ATA-3′60. PCRs were performed with the GoTaq G2 Hot Start Green Master Mix (Promega, Madison, WI, USA) in an Applied Biosystems 2720 Thermal Cycler (Thermo Fisher Scientific, Waltham, MA, USA), under the following thermal conditions: 95 °C for 2 min, 45 cycles with 95 °C for 1 min, 54 °C for 1 min, 72 °C for 1 min, and a final elongation step at 72 °C for 10 min. After electrophoresis in a 1.5% agarose gel stained with 2% GelRed (Biotium, Hayward, CA, USA), amplicons of the expected size were sequenced on both strands by Genoscreen (Lille, France).
Statistical analysis
We have performed Pearson χ² tests on all samples (1,013 bats) to explore the effect of (i) location, (ii) bat family, and (iii) bat species on the detection of coronavirus RNA. Two sampling campaigns, at two different season, in the same location, were available for Mozambique. We thus investigated the effect of the sampling season, between the wet (February) and dry (May) season, on CoV detection in Mozambique in 2015 (264 bats). Analyses were conducted with R v3.5.1 software61.
Phylogenetic analyses
Sequences obtained with the second PCR system60 were edited with the Chromas Lite Software package version 2.6.462. We explored CoV diversity of the sequences with pairwise identity values obtained from seqidentity function in R bio3d package v2.3-463 and identified the most similar CoV RdRp sequences referenced in GenBank using BLASTN 2.2.29 + . An alignment was then generated using the 51 nucleotide sequences obtained in this study and 151 reference CoV sequences representing a large diversity of hosts and geographic origins (Europe, Asia, Oceania, America and Africa), with CLC Sequence viewer 8.0 Software (CLC Bio, Aarhus, Denmark). A phylogenetic tree was obtained by maximum likelihood using MEGA Software v10.0.464, with 1,000 bootstrap iterations, and with the best evolutionary model for our dataset as selected by modelgenerator v0.8565.
Host-virus associations were investigated using the phylogeny of Western Indian Ocean bats and their associated CoVs. Bat phylogeny was generated from an alignment of 1,030 bp of mitochondrial Cytochrome b (Cyt b) gene sequences (Supplementary Table S4), for each CoV positive bat species. Finally, bat and pruned CoV phylogenies based on each 393 bp RdRp unique sequence fragment were generated by Neighbor-Joining with 1,000 bootstrap iterations, using CLC Sequence viewer 8.0 Software (CLC Bio, Aarhus, Denmark)66. Phylogenetic congruence was tested to assess the significance of the coevolutionary signal between bat host species and CoVs sequences, using ParaFit with 999 permutations in the ape package v5.0 in R 3.5.167,68. Tanglegram representations of the co-phylogeny were visualized using the Jane software v4.0169.
Data availability
DNA sequences: Genbank accessions MN183146 to MN183273.
References
Reusken, C. et al. WHO Research and Development Blueprint 2018 Annual review of diseases prioritized under the Research and Development Blueprint Informal consultation. http://www.who.int/csr/research-and-development/documents/prioritizing_diseases_progress/en/ (2018).
American Society of Mammalogist. Chiroptera (ASM Mammal Diversity Database). https://mammaldiversity.org/#Y2hpcm9wdGVyYSZnbG9iYWxfc2VhcmNoPXRydWUmbG9vc2U9dHJ1ZQ (2019).
Muscarella, R. & Fleming, T. H. The role of frugivorous bats in tropical forest succession. Biol. Rev. 82, 573–590 (2007).
Kunz, T. H., Braun de Torrez, E., Bauer, D., Lobova, T. & Fleming, T. H. Ecosystem services provided by bats. Ann. N. Y. Acad. Sci. 1223, 1–38 (2011).
Kasso, M. & Balakrishnan, M. Ecological and economic importance of bats (Order Chiroptera). ISRN Biodivers. 2013, Article ID 187415 (2013).
Boyles, J. G., Cryan, P. M., McCracken, G. F. & Kunz, T. K. Economic importance of bats in agriculture. Science 332, 41–42 (2011).
Kemp, J. et al. Bats as potential suppressors of multiple agricultural pests: A case study from Madagascar. Agric. Ecosyst. Environ. 269, 88–96 (2019).
Jones, G., Jacobs, D. S., Kunz, T. H., Wilig, M. R. & Racey, P. A. Carpe noctem: The importance of bats as bioindicators. Endangered Species Research 8, 93–115 (2009).
Russo, D., Bosso, L. & Ancillotto, L. Novel perspectives on bat insectivory highlight the value of this ecosystem service in farmland: Research frontiers and management implications. Agric. Ecosyst. Environ. 266, 31–38 (2018).
Taylor, P. J., Grass, I., Alberts, A. J., Joubert, E. & Tscharntke, T. Economic value of bat predation services – A review and new estimates from macadamia orchards. Ecosyst. Serv. 30, 372–381 (2018).
Sulkin, S. E. & Allen, R. Virus Infections in Bats. in Monographs in Virology (ed. Karger, S.) 3–7 (Basel, 1974).
Calisher, C. H., Childs, J. E., Field, H. E., Holmes, K. V. & Schountz, T. Bats: Important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 19, 531–545 (2006).
Cui, J., Li, F. & Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192 (2019).
Guan, Y. et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science 302, 276–279 (2003).
Kan, B. et al. Molecular evolution analysis and geographic investigation of Severe Acute Respiratory Syndrome coronavirus-like virus in palm civets at an animal market and on farms. J. Virol. 79, 11892–11900 (2005).
Tu, C. et al. Antibodies to SARS coronavirus in civets. Emerg. Infect. Dis. 10, 2244–2248 (2004).
Lau, S. K. P. et al. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. 102, 14040–14045 (2005).
Li, W. et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 310, 676–679 (2005).
World Health Organization. Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003 Based on data as of the 31 December 2003. World Health Organization https://www.who.int/csr/sars/country/table2004_04_21/en/ (2003).
O’Brien, J. Bats of the Western Indian Ocean islands. Animals 1, 259–290 (2011).
Goodman, S. M. Les chauves-souris de Madagascar. (Association Vahatra, 2011).
Goodman, S. M. & Ramasindrazana, B. Description of a new species of the Miniopterus aelleni group (Chiroptera: Miniopteridae) from upland areas of central and northern Madagascar. Zootaxa 3936, 538–558 (2015).
Goodman, S. M. et al. An integrative approach to characterize Malagasy bats of the subfamily Vespertilioninae Gray, 1821, with the description of a new species of Hypsugo. Zool. J. Linn. Soc. 173, 988–1018 (2015).
Bourgarel, M. et al. Circulation of alphacoronavirus, betacoronavirus and paramyxovirus in Hipposideros bat species in Zimbabwe. Infect. Genet. Evol. 58, 253–257 (2018).
Ithete, N. L. et al. Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg. Infect. Dis. 19, 1697–1699 (2013).
Geldenhuys, M., Weyer, J., Nel, L. H. & Markotter, W. Coronaviruses in south african bats. Vector-Borne Zoonotic Dis. 13, 516–519 (2013).
Müller, M. A. et al. Coronavirus antibodies in African bat species. Emerg. Infect. Dis. 13, 1367–1370 (2007).
Tao, Y. et al. Surveillance of bat coronaviruses in Kenya identifies relatives of human coronaviruses NL63 and 229E and their recombination history. J. Virol. 91, e01953–16 (2017).
Waruhiu, C. et al. Molecular detection of viruses in Kenyan bats and discovery of novel astroviruses, caliciviruses and rotaviruses. Virol. Sin. 32, 101–114 (2017).
Razanajatovo, N. H. et al. Detection of new genetic variants of betacoronaviruses in endemic frugivorous bats of Madagascar. Virol. J. 12, 42 (2015).
Smith, C. S. et al. Coronavirus infection and diversity in bats in the Australasian region. Ecohealth 13, 72–82 (2016).
Annan, A. et al. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 19, 456–459 (2013).
Drexler, J. F. et al. Amplification of emerging viruses in a bat colony. Emerg. Infect. Dis. 17, 449–456 (2011).
Wacharapluesadee, S. et al. Longitudinal study of age-specific pattern of coronavirus infection in Lyle’s flying fox (Pteropus lylei) in Thailand. Virol. J. 15, 38 (2018).
Leopardi, S. et al. Interplay between co-divergence and cross-species transmission in the evolutionary history of bat coronaviruses. Infect. Genet. Evol. 58, 279–289 (2018).
Cui, J. et al. Evolutionary relationships between bat coronaviruses and their hosts. Emerg. Infect. Dis. 13, 1526–1532 (2007).
Drexler, J. F., Corman, V. M. & Drosten, C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res. 101, 45–56 (2014).
Anthony, S. J. et al. Global patterns in coronavirus diversity. Virus Evol. 3, vex012 (2017).
Ziebuhr, J. et al. ICTV Report 2017.013S. https://talk.ictvonline.org//taxonomy/p/taxonomy-history?taxnode_id=201851846 (2017).
Luis, A. D. et al. A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc. R. Soc. B Biol. Sci. 280, 20122753 (2013).
Willoughby, A. R., Phelps, K. L. & Olival, K. J. A comparative analysis of viral richness and viral sharing in cave-roosting bats. Diversity 9, 35 (2017).
Olival, K. J. et al. Host and viral traits predict zoonotic spillover from mammals. Nature 546, 646–650 (2017).
Streicker, D. G. et al. Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science 329, 676–9 (2010).
Parrish, C. R. et al. Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol. Mol. Biol. Rev. 72, 457–70 (2008).
Dijkman, R. & van der Hoek, L. Human coronaviruses 229E and NL63: close yet still so far. J. Formos. Med. Assoc. 108, 270–279 (2009).
Pyrc, K. et al. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J. Mol. Biol. 364, 964–973 (2006).
Pfefferle, S. et al. Distant relatives of Severe Acute Respiratory Syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg. Infect. Dis. 15, 1377–1384 (2009).
Corman, V. M. et al. Evidence for an ancestral association of human coronavirus 229E with bats. J. Virol. 89, 11858–11870 (2015).
Joffrin, L., Dietrich, M., Mavingui, P. & Lebarbenchon, C. Bat pathogens hit the road: But which one? PLoS Pathog. 14, e1007134 (2018).
El-Duah, P. et al. Development of a whole-virus ELISA for serological evaluation of domestic livestock as possible hosts of human coronavirus NL63. Viruses 11, v11010043 (2019).
Huynh, J. et al. Evidence supporting a zoonotic origin of human coronavirus strain NL63. J. Virol. 86, 12816–25 (2012).
Geldenhuys, M. et al. A metagenomic viral discovery approach identifies potential zoonotic and novel mammalian viruses in Neoromicia bats within South Africa. PLoS One 13, e0194527 (2018).
Chen, L., Liu, B., Yang, J. & Jin, Q. DBatVir: The database of bat-associated viruses. Database 2014, bau021 (2014).
Wilkinson, D. A. et al. Identification of novel paramyxoviruses in insectivorous bats of the Southwest Indian Ocean. Virus Res. 170, 159–163 (2012).
Wilkinson, D. A. et al. Highly diverse morbillivirus-related paramyxoviruses in wild fauna of the Southwestern Indian Ocean islands: evidence of exchange between introduced and endemic small mammals. J. Virol. 88, 8268–77 (2014).
Gomard, Y. et al. Malagasy bats shelter a considerable genetic diversity of pathogenic Leptospira suggesting notable host-specificity patterns. FEMS Microbiol. Ecol. 92, fiw037 (2016).
Mélade, J. et al. An eco-epidemiological study of Morbilli-related paramyxovirus infection in Madagascar bats reveals host-switching as the dominant macro-evolutionary mechanism. Sci. Rep. 6, (2016).
Lebarbenchon, C. et al. Astroviruses in bats, Madagascar. Emerg. Microbes Infect. 6, e58 (2017).
Muradrasoli, S. et al. Broadly targeted multiprobe QPCR for detection of coronaviruses: coronavirus is common among mallard ducks (Anas platyrhynchos). J. Virol. Methods 159, 277–287 (2009).
Poon, L. L. M. et al. Identification of a novel coronavirus in bats. J. Virol. 79, 2001–9 (2005).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.r-project.org/ (2018).
Technelysium Pty. Ltd. Chromas Lite Software. http://technelysium.com.au/wp/chromas/ (2018).
Grant, B. J., Rodrigues, A. P. C., ElSawy, K. M., McCammon, J. A. & Caves, L. S. D. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 22, 2695–2696 (2006).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Keane, T. M., Creevey, C. J., Pentony, M. M., Naughton, T. J. & Mclnerney, J. O. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol. 6, 29 (2006).
QIAGEN. CLC Sequence Viewer 8. https://clc-sequence-viewer.software.informer.com/8.0/ (2018).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2018).
Legendre, P., Desdevises, Y. & Bazin, E. A Statistical test for host-parasite coevolution. Syst. Biol 51, 217–234 (2002).
Conow, C., Fielder, D., Ovadia, Y. & Libeskind-Hadas, R. Jane: a new tool for the cophylogeny reconstruction problem. Algorithms Mol. Biol. 5, 16 (2010).
Acknowledgements
We are very grateful to S. Muradrasoli for recommendations on the optimization of the multiprobe PCR protocol as well as for providing PCR controls and to C. Cordonin for providing bat Cyt b sequences. L. Biscornet, C. Dionisio, L. Domergue, M. Dietrich, T. Mbohoahy, T. Nekena, J. Rakotoarivelo, M. Rakotomanga, C. F. Rakotondramanana, and A. Randrenjarison are thanked for their assistance in the field. We also thank S. Bos and A. Hoarau for their help in the laboratory, and K. Dellagi and H. Pascalis for the development and the management of the ‘partenariat Mozambique-Réunion dans la recherche en santé: pour une approche intégrée d’étude des maladies infectieuses à risque épidémique (MoZaR)’ research program. Field research was funded by the ‘Pathogènes associés à la Faune Sauvage Océan Indien (FS-OI)’, the ‘Leptospirose Ocean Indien (LeptOI)’, the ‘Paramyxovirus Océan Indien (ParamyxOI)’, and the ‘Partenariat Mozambique-Réunion dans la recherche en santé: pour une approche intégrée d’étude des maladies infectieuses à risque épidémique (MoZaR)’ programs (Fond Européen de Développement Régional, Programme Opérationnel de Coopération Territoriale). Fieldwork on Mayotte was funded by the ‘Centre National de la Recherche Scientifique’ (Projets Exploratoires Premier Soutien BATMAN). Molecular analyses were financially supported by tutorship institutions of the UMR PIMIT. L.J. is a recipient of a ‘Région Réunion, European Regional Development Fund (FEDER 2014–2020)’ PhD fellowship. B.R.’s post-doctoral fellowship was supported by the ‘Run Emerge’ European Union’s Seventh Framework Program (FP7/2007–2013; Grant agreement NO 263958), the ‘Fonds de Coopération Régionale, Prefecture de La Réunion’, and The Field Museum of Natural History, Chicago, through the Ralph and Marian Falk Medical Research Trust. C.L. is supported by a ‘Chaire mixte: Université de La Réunion – INSERM’. The funding agencies were not involved in the study design, implementation or publishing of this study, and the research presented herein represents the opinions of the authors but not necessarily the opinions of the funding agencies.
Author information
Authors and Affiliations
Contributions
C.L. and L.J. conceived and designed the study. B.R., C.L., D.A.W., E.L., L.J., P.T., S.M.G. and Y.G. collected biological material on Madagascar. S.M.G., R.S. and Y.G. collected biological material on Mauritius. B.R., E.L. and G.L.M., collected biological material on Mayotte. S.M.G., G.L.M., A.D.S. and M.C.S., collected biological material in Mozambique. B.R., D.A.W., E.L. and G.L.M. collected biological material on Reunion Island. E.L., G.L.M., S.J. and Y.G. collected biological material in the Seychelles. L.J. performed the molecular analyses. C.L., D.A.W., and L.J. analyzed the data. C.L. and L.J. wrote the paper. S.M.G., E.S.G., P.M. and S.J. contributed to the project management in Malagasy, Mauritian, Seychelles, Mozambican, and French institutions. All authors edited, read, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Joffrin, L., Goodman, S.M., Wilkinson, D.A. et al. Bat coronavirus phylogeography in the Western Indian Ocean. Sci Rep 10, 6873 (2020). https://doi.org/10.1038/s41598-020-63799-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-63799-7
This article is cited by
-
Bat species assemblage predicts coronavirus prevalence
Nature Communications (2024)
-
Coronavirus sampling and surveillance in bats from 1996–2019: a systematic review and meta-analysis
Nature Microbiology (2023)
-
Tracking the molecular evolution and transmission patterns of SARS-CoV-2 lineage B.1.466.2 in Indonesia based on genomic surveillance data
Virology Journal (2022)
-
Using Utilitarian Plants for Lemur Conservation
International Journal of Primatology (2022)
-
Computational design of a broad-spectrum multi-epitope vaccine candidate against seven strains of human coronaviruses
3 Biotech (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
