
Abstract
Vermamoeba vermiformis is a predominant free-living amoeba in human environments and amongst the most common amoebae that can cause severe infections in humans. It is a niche for numerous amoeba-resisting microorganisms such as bacteria and giant viruses. Differences in the susceptibility to these giant viruses have been observed. V. vermiformis and amoeba-resisting microorganisms share a sympatric lifestyle that can promote exchanges of genetic material. This work analyzed the first draft genome sequence of a V. vermiformis strain (CDC-19) through comparative genomic, transcriptomic and phylogenetic analyses. The genome of V. vermiformis is 59.5 megabase pairs in size, and 22,483 genes were predicted. A high proportion (10% (n = 2,295)) of putative genes encoded proteins showed the highest sequence homology with a bacterial sequence. The expression of these genes was demonstrated for some bacterial homologous genes. In addition, for 30 genes, we detected best BLAST hits with members of the Candidate Phyla Radiation. Moreover, 185 genes (0.8%) best matched with giant viruses, mostly those related to the subfamily Klosneuvirinae (101 genes), in particular Bodo saltans virus (69 genes). Lateral sequence transfers between V. vermiformis and amoeba-resisting microorganisms were strengthened by Sanger sequencing, transcriptomic and phylogenetic analyses. This work provides important insights and genetic data for further studies about this amoeba and its interactions with microorganisms.
Similar content being viewed by others

Introduction
Amoebozoa species are widely distributed in different environments from terrestrial to aquatic ecosystems, where they can play important ecological roles1,2. Members of the family Hartmannellidae are frequently detected along with a few other amoebae belonging to different genera of the taxon Amoebozoa3. Vermamoeba vermiformis, a free-living amoeba of the family Hartmannellidae, formerly named Hartmannella vermiformis, was first isolated in freshwater from the Pigeon Lake, Wisconsin, and the Kankakee River, Indiana (United States)4. V. vermiformis was thereafter commonly found in fresh surface water5, and also in tap water, bottled mineral water, thermal water, and recreational water environments such as fountains and swimming pools6,7,8. Its density in drinking water sources and biofilms is higher than that of Acanthamoeba castellanii9. V. vermiformis has two-stage life, switching between trophozoite and cystic form3. Free-living amoebae (FLA) are commonly in contact with animals including humans. V. vermiformis was found to be the predominant amoeba in human environments10,11, and has been isolated more frequently from different hospital water systems than Acanthamoeba spp.12. This amoeba is of special interest for human health as it is able, along with other Amoebozoa members including Acanthamoeba spp., to cause severe infections such as human keratitis13,14,15. Despite its prevalence in human environments and its pathogenicity in humans, the genome of V. vermiformis had not been sequenced.
FLA are the niche of several amoeba-resisting microorganisms, including bacteria and fungi. They are potential reservoirs for several human pathogens, including Salmonella spp., Escherichia coli, Shigella spp., and Campylobacter spp., which cause disorders in the human intestinal tract10,16,17,18, and Legionella pneumophila, a human pathogen associated with Legionnaires’ disease that can propagate in V. vermiformis19. Indeed, V. vermiformis strain CDC-19 was isolated from a swab sample recovered from a cooling tower in the boiler room of the hospital during a nosocomial legionellosis investigation20. Volatile organic compounds have been identified to be involved in the predator-prey interactions between V. vermiformis and bacteria, with differences according to the protist-prey partners. Bacterial prey such as Dyella sp. and Collimonas sp. were recently found to reduce or conversely stimulate the activity of V. vermiformis, respectively21.
The discovery of the first giant virus, Mimivirus, in the amoeba Acanthamoeba polyphaga in 2003, unveiled an unexpected giant virus diversity in different environments22,23,24,25. In addition to the remarkable sizes of the virions, their genomes were also found to be giant with sizes ranging between about 340 kilobase pairs (kbp) for marseilleviruses and 2,500 kbp for pandoraviruses. These viral genomes had broad gene repertoires reaching more than two thousand genes encoding various functions and many ORFans, and the genetic composition of these viruses far exceeds quantitatively and qualitatively that of known viruses, and rivals that of other small microbes23. Moreover, giant viruses have a high level of genome mosaicism, which is likely linked to their sympatric lifestyle in amoebae with other microorganisms, including bacteria, fungi, and virophages26,27. Indeed, important sequence exchanges have been observed between giant viruses and both species Acanthamoeba castellanii and Acanthamoeba polyphaga28,29,30,31. The majority of described giant viruses have been experimentally isolated from Acanthamoeba spp. These amoebae demonstrated differences in their susceptibility to giant viruses, as for the case of pithoviruses and pandoraviruses that were only isolated from A. castellanii32. Thereafter, different cellular cultures of amoebae other than Acanthamoeba spp. have been infected by these giant viruses33,34. Furthermore, ten additional isolates of a new giant viral lineage named the faustovirus lineage were obtained from V. vermiformis, and their genomes were sequenced. Faustoviruses have icosahedral virions with a diameter of 200–240 nm and are distantly related to the mammalian African swine fever virus35,36. Other members of giant virus families were also obtained by co-culturing with V. vermiformis, such as Kaumoebavirus found in sewage water37, and Orpheovirus IHUMI-LCC2 isolated from a rat stool sample38. Abrahão et al. discovered the first Mimiviridae members, called tupanviruses, that infect both V. vermiformis and A. castellanii39.
Here, for the first time we sequenced the genome of a V. vermiformis strain (CDC-19) that has been used for the isolation of giant viruses. Lateral gene transfers with bacteria and giant viruses were also explored.
Results
Genome structure and characterization of the putative genes of V. vermiformis CDC-19
A total of 41,068,870 and 25,445 reads were obtained by the Illumina MiSeq Nextera XT and the Oxford Nanopore MinION sequencing, respectively, then were used to assemble the V. vermiformis CDC-19 genome. Additionally, 1,584,658 reads were obtained by next-generation RNA sequencing on the Illumina MiSeq instrument. A total of 17,244 and 15 scaffolds were obtained by assembling the MiSeq and MinION sequencing products, respectively. Genome coverage was 43X. The draft genome sequence of V. vermiformis CDC-19 has a size of 59.5 megabase pairs (Mbp). It encompasses 14,852 scaffolds, with a G + C content of 41.7%. The phylogenetic tree based on 18S rRNA shows that V. vermiformis CDC-19 is clustered with other V. vermiformis strains (Supplementary Fig. S1). A total of 22,483 putative genes were predicted. The proportion of putative genes with a size equal to or greater than 100 amino acids (aa) was estimated to be 90.3% (20,299 genes). Out of all the predicted genes, 67.9% (15,266) were non-ORFan genes and 32.1% (7,217) were ORFans (i.e. they have no homologs in the NCBI GenBank protein sequence database (nr)) (Table 1). A total of 12,593 genes (56%) were assigned to COG categories (Supplementary Fig. S2). The main functional categories represented were those corresponding to unknown functions (category S (2,829 genes)); signal transduction mechanisms (category T (1,680 genes)); post-translational modifications, protein turnover, chaperones (category O (1,208 genes)); intracellular trafficking, secretion, and vesicular transport (category U (622 genes)); and defense mechanisms (category V (154 genes)) (Supplementary Fig. S2). V. vermiformis putative genes have an average of 3.5 introns per gene. This is less than for A. castellanii Neff (6.2 introns per gene)29. In contrast, the genes putatively derived from lateral sequence transfers have a lower intron composition. On average, the genes best matching with bacteria and archaea have 2.7 introns per gene, and those best matching with giant viruses have 1.4 intron per gene.
Taxonomical assignments of V. vermiformis CDC-19 genes and identification and analysis of gene trafficking between V. vermiformis CDC-19 and bacteria
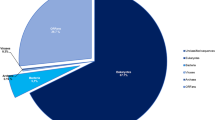
The taxonomical assignment through BLAST searches of genes predicted for V. vermiformis CDC-19 showed that 12,567 (55.9%) of them had best hits with eukaryotes, including 4,457 genes best matching with amoebozoan members (19.8%). Also, a high proportion of amoebal genes best matched with bacterial genes (2,295 genes or 10.2%), while 139 (0.6%) and 188 (0.8%) genes had a best hit with archaea and viruses, respectively (Fig. 1). The functional annotation of the V. vermiformis CDC-19 putative genes revealed a high proportion of homologous sequences from bacteria, equal to 17.8% of the predicted genes (3,993 genes). Of these 3,993 genes, 2,295 genes were maintained after excluding all suspected contaminant scaffolds, as each of these scaffolds harbored a totality of genes best matching with homologous genes from the same bacteria. For these 2,295 genes, the taxonomical assignment showed that Proteobacteria were the most represented with 811 genes (35.3%), followed by Bacteroidetes with 283 genes (12.3%), and Cyanobacteria with 276 genes (12%) (Fig. 2), compared to 35.4%, 10.5% and 15% for Acanthamoeba castellanii strain Neff, respectively29. Among these V. vermiformis CDC-19 genes best matching with bacteria, 626 (27.3%) were involved in undetermined functions; 164 genes (7.2%) were related to carbohydrate transport and metabolism; 125 genes (5.4%) were related to signal transduction mechanisms; and 97 genes (4.2%) were related to cell wall/membrane/envelope biogenesis (Supplementary Table S1). PCR and Sanger sequencing performed with specific primers designed to target 10 genes among those best matching with bacteria from different phyla were all positive, and one of these genes was found to be expressed and encoded a 1-pyrroline-5-carboxylate dehydrogenase (Supplementary Fig. S3; Supplementary Table S2). Expression of other genes homologous to bacterial genes was detected, such as for the homolog of a tandem-95 repeat protein of Solitalea canadensis, which exhibited the highest level of gene expression among genes best matching with bacteria (349 reads). Other examples included expression of genes encoding homologs to an hypothetical protein A7U43_25800 of Mycobacterium sp. YC-RL4 (147 reads), an arylsulfatase regulatory protein of Sphingobium ummariense RL-3 (45 reads), a NADPH dehydrogenase NamA of Chitinophagaceae bacterium PMP191F (37 reads), and a transposase of Salmonella enterica subsp. enterica serovar Heidelberg str. SL476 (31 reads) (Table 2).

Taxonomical distribution of the V. vermiformis CDC-19 predicted proteins.

Phylogenetic diversity and number of reads generated from the V. vermiformis CDC-19 DNA that best matched with bacteria.
Furthermore, 30 genes best matching with members of the Candidate Phyla Radiation (CPR) were detected in the genome of V. vermiformis CDC-19, which is equal to 1.3% of the total set of homologs to bacterial genes. These genes were primarily related to the Parcubacteria group (22 genes), then to the Microgenomates group (5 genes), and to CPR2, Peregrinibacteria and Doudnabacteria (with 1 gene related to each CPR phylum) (Fig. 2; Supplementary Table S3). A majority of the genes best matching with CPR corresponded to hypothetical proteins (23 genes (76.6%)), whereas some were found to be involved in carbohydrate transport and metabolism (1 gene; a 6-phosphogluconolactonase), nucleotide transport and metabolism (1 gene), translation, ribosomal structure and biogenesis (1 gene), cell wall, membrane, and envelope biogenesis (1 gene), and in undetermined functions (3 genes) (Supplementary Table S3). Phylogenetic reconstructions confirmed that these genes underwent sequence transfers between V. vermiformis and bacteria (Fig. 3; Supplementary Fig. S4), and one of them was found to be expressed (Fig. 3a). Moreover, there was at least on example of a DUF4419 domain-containing protein that might have been thereafter transferred to Catovirus CTV1 and Tupanvirus, two giant viruses (Fig. 3b). The same observations were obtained for genes showing sequence similarity with a CPR homolog (Fig. 3c,d).

Phylogenetic reconstructions for four examples of putative lateral sequence transfers implicating V. vermiformis and bacteria. Lateral sequence transfer was inferred from the comparison of V. vermiformis predicted sequences with their best BLAST hits. (a,b) Trees based on two proteins with sequence similarity with a non-CPR bacterial homolog. (c,d) Trees based on two proteins with sequence similarity with a CPR homolog. In dark yellow: V. vermiformis genes. Colors of branches are related to bootstrap values.
Sequence exchanges between V. vermiformis CDC-19 and viruses
Of the 188 genes detected in the genome of V. vermiformis CDC-19 that best matched with viruses, 185 of these best matched with giant viruses. The three other genes best matched with Ralstonia phage phiRSL1 (2 genes) and Synechococcus phage S-SKS1(1 gene), and encode hypothetical proteins. Genes best matching with giant viral genes were mostly related to Megavirales members, such as those best matching with Bodo saltans virus, a member of family Mimiviridae, subfamily Klosneuvirinae (69 genes) (Fig. 4). Other best matches were genes from assembled genomes of putative members of the Klosneuvirinae: their best hits were with genomes of Klosneuvirus KNV1 (16 genes), Catovirus CTV1 (10 genes), Hokovirus HKV1, and Indivirus ILV1 (3 genes each). Other homologs were from two mimivirus isolates, Tupanvirus deep ocean (16 genes) and Tupanvirus soda lake (4 genes), which replicate in A. castellanii and V. vermiformis. Viral sequences from other Megavirales groups than Mimiviridae were also identifed as best hits, such as genes from Orpheovirus IHUMI-LCC2 (34 genes), faustoviruses (16 genes), Kaumoebavirus (4 genes), cedratviruses (2 genes), and Pandoravirus inopinatum (1 gene). In addition, 5 V. vermiformis genes best matched with phycodnavirus genes, 4 of them belonging to Acanthocystis turfacea Chlorella viruses MN0810.1 and WI0606, and one gene best matched with Phaeocystis globosa virus. A homolog was also detected in Canarypox virus and African swine fever virus (Supplementary Table S4). Phylogenies strengthened suspicions of lateral sequence transfer for two genes best matching with giant viruses (Fig. 5). At least one gene of V. vermiformis best matched with viral sequences as well as with CPR and other bacteria (Fig. 5b). A total of 70 of the 185 genes best matching with giant viruses encode ankyrin repeat domain-containing proteins. The majority of these genes (68) were homologs to Bodo saltans virus genes, and the two other genes were homologous to Klosneuvirus KNV1 and Canarypox virus genes. Eighteen genes encode proteins with a DUF4114 domain and were related to Orpheovirus IHUMI-LCC2 (16 genes) and Catovirus CTV1 (2 genes). Gene expression was detected for nine genes best matching with giant viral sequences. Eight genes were related to Orpheovirus IHUMI-LCC2 and notably encode a DUF4114 protein and an E-class cytochrome P450-like protein. Among remaining best matches was a gene of Kaumoebavirus predicted to encode a peroxinectin, which was the first gene encoding cell adhesion ligand and peroxidase molecule cloned from invertebrate blood40 (Table 3). Finally, the rhizomes of V. vermiformis CDC-19 genes best matching with Klosneuvirinae representative sequences demonstrated that sequence exchanges between V. vermiformis CDC-19 and each member of this subfamily were widely distributed on different scaffolds of the amoebal draft genome sequence (Fig. 6a). Similar observations were found for genes best matching with sequences from other Megavirales members, such as Orpheovirus IHUMI-LCC2, faustoviruses, and tupanviruses (Fig. 6b).

Taxonomical origins of predicted genes with a giant virus as best hit.

Phylogenetic reconstructions for two examples (a,b) of putative lateral sequence transfers implicating V. vermiformis and giant viruses. Lateral sequence transfer was inferred from the comparison of V. vermiformis predicted sequences with their best BLAST hits. In dark yellow: V. vermiformis genes. Colors of branches are related to bootstrap values.

Rhizomes representation of the proteins best matching with giant viruses. Taxonomical distribution of V. vermiformis CDC-19 predicted proteins for which best BLAST hits were members of family Mimiviridae (a) and other giant viruses (b).
Discussion
We herein describe for the first time the genome sequencing, composition and characteristics for a strain of the amoeba Vermamoeba vermiformis CDC-19. This draft genome sequence is larger than those of other amoebae such as Naegleria gruberi and Acanthamoeba castellanii Neff, which are estimated to be equal to 41 and 42 Mbp, respectively29,41. It is comprised by 14,852 scaffolds, fewer than previously described for the Acanthamoeba spp. draft genome sequence. One third of predicted genes in this V. vermiformis strain were ORFans, which leaves questions surrounding the repertoire of the genes and their roles. In addition, a large proportion of the non-ORFan genes was found to encode unknown functions based on comparative analyses with COGs. On average, V. vermiformis genes were found to contain 3.5 introns whereas A. castellanii Neff genes harbor 6.2 introns29. The difference between these amoebae may reflect extensive intron losses or gains, and supports the importance of introns in evolution42. The prevalence of introns in genes involved in sequence tranfers with bacteria and giant viruses implies the proposed mechanisms of intron gain subsequently to lateral sequence transfer43.
More than half of V. vermiformis CDC-19 predicted genes have eukaryotic sequences as closest relatives. Approximately 10% of genes have bacterial sequences as best hits, a proportion similar to that described for A. castellanii Neff29. However, the proportion of genes best matching with bacteria was greater than for other described amoebae, as for A. polyphaga (3%)31 or Naegleria gruberi (1%)41. The presence in the V. vermiformis CDC-19 genome of sequences that were predicted to have resulted from exchanges with amoeba-resisting microorganisms, particularly bacteria, was confirmed by Sanger sequencing in all cases when tested for a small set of genes. In addition, transcriptomics showed expression of several genes best matching with bacterial sequences, the highest level of gene expression being observed for a gene encoding a tandem-95 repeat protein. Classically, tandem repeats act as a support for protein-protein interactions, but it has been hypothetized that the gain or loss rates of such sequences might generate genetic diversity and evolutionary adaptation to a pathogen44. The other transcribed genes best matching with bacteria were mainly related to either undetermined functions, or to replication, recombination and repair, and energy production and conversion pathways. As in the study of Clarke et al. on the draft genome sequence of A. castellanii Neff, sequences best matching with genes from members of phyla Proteobacteria, Bacteroidetes and Cyanobacteria were those the most represented in the genome of V. vermiformis CDC-1929. However, the proportion of genes best matching with Bacteroidetes and Cyanobacteria members was slightly greater (2% and 3%, respectively) for V. vermiformis CDC-19, when compared to A. castellanii Neff.
We reported here the first identification in an amoebal genome of sequences best matching with CPR. CPR were recently described as small bacteria that may represent >15% of all bacterial diversity and dozens of phyla45. It is likely that they have been previously overlooked because of their small size, and they have small genomes and an apparent symbiotic lifestyle with bacteria46,47. They have been detected in a wide range of natural systems, including groundwaters and sediments. Sequences from CPR were only recently available in the NCBI database, which prevented their earlier detection. CPR homologs encompassed 1.3% of the gene products best matching with bacteria in the genome of V. vermiformis. These data highlight a yet unexplored gene trafficking between CPR and V. vermiformis.
A set of 188 genes in V. vermiformis CDC-19 was related to sequences from viruses, essentially giant viruses. Their number was smaller, albeit similar, compared to those reported for A. castellanii Neff (261)30 or A. polyphaga draft genomes (262)31. These genes were detected in a large set of 179 scaffolds comprising the draft genome sequence of V. vermiformis CDC-19, suggesting that they are widely distributed along the genome of this amoeba. We demonstrated that the genomes of klosneuviruses, particularly that of Bodo saltans virus, harbored the largest set of such virus-related sequences. This suggests a considerable gene trafficking between this amoeba and Klosneuvirinae members. Among this group, only the Bodo saltans virus was isolated (only the genomes assembled from metagenomic data being available for the other described members) and this was on the kinetoplastid Bodo saltans, a microzooplankton48. Other recently described mimiviruses named tupanviruses can grow on both Acanthamoeba spp. and V. vermiformis37. However, most commonly, the permissivity of known eukaryotic hosts to giant viruses differs considerably according to the host strain or to the viral family or lineage, as previously described for mimiviruses, pandoraviruses, and Bodo saltans virus32,48. The analysis of giant virus homologs in the V. vermiformis genome showed here that the most represented sequences were those of giant viruses that grew in V. vermiformis, including faustovirus isolates and Orpheovirus IHUMI-LCC2, whereas a small proportion included genes from giant viruses isolated from Acanthamoeba spp.. Ankyrin repeats, which are associated with protein-protein interactions, were highly represented among V. vermiformis genes best matching with giant viruses49,50 in addition to DUF4114 domains which are conserved domains that help to adapt to nutrient-depleted conditions by down-regulating protein biosynthesis51. Overall, the phylogenies of genes predicted to have arisen through lateral sequence transfer illustrate the complexity of sequence exchanges between amoebae, bacteria (including CPR), and giant viruses. This result is in line with the recent analysis of Acanthamoeba genomes, suggesting that the sequence flow was not a one way mechanism, and a possible result of their sympatric lifestyle30,31.
Overall, these first V. vermiformis genome-wide genetic data allow for a better understanding of this amoeba and its interactions with microorganisms. They provide insight on an extensive gene trafficking with distinct amoeba-resisting microorganisms, including bacteria and giant viruses. They also suggest as expected that the presence of genes from these microorganisms in cellular genomes are hints that these cells are among their possible hosts. Moreover, the comparison of different amoebal genomes and gene repertoires is an important task that might help us understand the different levels of their susceptibility to giant viruses, and select efficient cellular supports for their isolation.
Materials and Methods
Vermamoeba vermiformis strain CDC-19 culture
Vermamoeba vermiformis strain CDC-19 was isolated from cooling tower water in a hospital during a legionellosis investigation20. This strain was obtained from the American Type Culture Collection database (ATCC). V. vermiformis CDC-19 (ATCC 50237) was grown at 32 °C in 175 cm² culture flasks (Thermo Fisher Scientific, Illkirch, France) containing 75 mL of PYG medium52. When amoebas formed a monolayer, they were detached by tapping the culture flasks then harvested by centrifugation at 1,000 g for 10 min followed by three steps of washing using Page’s modified Neff’s Amoeba Saline medium (2 mM NaCl, 16 μM MgSO4, 27.2 μM CaCl2, 1 mM Na2HPO4, 1 mM KH2PO4). Strain CDC-19 quantification was performed using a KOVA slide cell counting chamber.
Genomic DNA extraction and sequencing of the amoeba V. vermiformis CDC-19
The DNA of V. vermiformis CDC-19 was extracted using the EZ1 DNA Tissue Kit (Cat No: 953034, Qiagen, Hilden, Germany), then purified using the Agencourt AMPure XP beads (1.8x ratio, Beckman Coulter Inc, Fullerton, CA, United States). Genomic DNA was quantified by a Qubit assay with the high sensitivity kit (Life technologies, Carlsbad, CA, USA); the concentration was equal to 2.3 ng/µl. A dilution was performed to provide 1 ng of DNA as input to prepare the paired end library. The «tagmentation» step fragmented and tagged the DNA and limited cycle PCR amplification (12 cycles) completed the tag adapters and introduced dual-index barcodes, in order to allow mixing with other genomic projects. After purification on AMPure XP beads (Beckman Coulter Inc), the libraries were normalized on specific beads according to the Nextera XT DNA sample prep kit protocol (Illumina, San Diego, CA, USA). Normalized libraries were pooled into a single library for sequencing on the MiSeq instrument (Illumina). Automated cluster generation and paired-end sequencing with dual index reads were performed in a 39-hour run with 2 × 250 bp. To improve the assembly, the Oxford Nanopore technology (Oxford Nanopore Technologies Ltd., United Kingdom) was used by 1D genomic DNA sequencing on the MinION device using the SQK-LSK108 kit. The library was constructed from 1.5 µg of genomic DNA without fragmentation and end repair. Adapters were ligated to both ends of genomic DNA. After purification on AMPure XP beads (Beckman Coulter Inc), the library was quantified by a Qubit assay with the high sensitivity kit (Life technologies), and loaded on the flow cell via the SpotON port. Finally, 498 active pores were detected for the sequencing and the workflow WIMP was chosen for sequence analysis. Adapter trimming, quality filtering and error correction of all sequencing raw data analyzed here were performed using the Trimmomatic program (version 0.36)53.
Total RNA preparation and sequencing
The RNA of V. vermiformis CDC-19 was extracted using the RNeasy mini kit (Cat No: 74104, Qiagen). RNaseOUT (Thermo Fisher Scientific, San Jose, CA, USA) was added to the 50 μL volume of eluted RNA, thus preventing RNA degradation. To ensure of the absence of DNA contamination, two cycles of DNase treatment with 30 min of incubation at 37 °C were performed using TURBO DNase (Invitrogen, Carlsbad, CA, USA). Total RNA was purified using the RNeasy MinElute Cleanup Kit (Cat No: 74204, Qiagen) according to the manufacturer’s instructions. cDNA amplicons were obtained using the SuperScript VILO Synthesis Kit (Invitrogen) with random primers. The amplicons were purified using the Agencourt AMPure XP beads (Beckman Coulter, Inc.), then sequenced on the MiSeq instrument using the Nextera XT DNA sample prep kit (Illumina), with a paired-end strategy and a read length of 125 bp. The cDNA was visualized and quantified on a LabChip Bio-analyzer (Agilent Technologies). Fragmentation, tagging and barcoding were performed over 12 PCR amplification cycles. The library was purified on Agencourt AMPure XP beads (Beckman Coulter Inc.), normalized on specific beads, and pooled for sequencing.
Assembly of the V. vermiformis CDC-19 genomic sequences
We assembled the genome of V. vermiformis CDC-19, whose ploidy was estimated to be 4 N5,54 using the A5-miseq pipeline, which included supplementary steps of adapter trimming and quality filtering55. Although the A5 software was classically used for bacterial and haploid organisms, it was also used for polyploid eukaryotic organisms (including Verticillium tricorpus and Verticillium dahliae) and allowed obtaining assemblies of high quality56. The quality assessment of the genome asssembly was performed using the QUAST software57. MinION fastq reads were assembled separately using the SPAdes program58. Thereafter, mapping of both MiSeq and MinION contigs was performed using the CLC Genomics Workbench software (version 7.5) (https://www.qiagenbioinformatics.com/products/clc-genomics-workbench/), followed by manual treatment to detect consensus sequences and gaps filling on the resulting genomic sequences of V. vermiformis CDC-19 using the GapFiller program59. A phylogenetic analysis based on the 18S rRNA gene was performed. For this task, we detected the 18S rRNA gene of V. vermiformis CDC-19 by comparison through BLASTn between the amoebal genome assembly and the published 18 s rRNA sequence of another V. vermiformis strain (KY476315.1), and also searched for similar sequences in the NCBI GenBank nucleotide sequence collection (nt). We then carried out multiple sequence alignments by using the MEGA version 7 software60. Finally, we performed a phylogenetic reconstruction of these nucleotide sequences using MEGA760 and the maximum likelihood (ML) algorithm, with 1,000 replicates for bootstrap determination.
Prediction, expression assessment, taxonomical distribution, and functional annotation of the V. vermiformis CDC-19 putative genes
Prediction of putative genes was implemented using the BRAKER1 program61 based on the genomic sequences and the RNA-seq raw data of V. vermiformis CDC-19. An additional mapping of the RNA-seq reads on the assembled genome was performed by using the HISAT2 software62, with default parameters. The reads aligned on the amoebal genome sequence were analyzed using HTSeq-count software63, with union mode. Predicted genes were estimated as transcribed if covered by at least 5 reads. In addition, we searched for homologous sequences of predicted open reading frames (ORFs) in the NCBI GenBank protein sequence database (nr) using the BLASTp program, with an e-value threshold of 0.001 and default parameters (word size equal to 3, gap costs equal to 11 for the opengap parameter and 1 for the gap extend parameter)64. To ensure the absence of suspected contaminant reads in the genome of V. vermiformis CDC-19, scaffolds harboring a totality of their genes best matching with the same bacteria were excluded. Sequences homologies were also identified using the eggNOG-mapper through searches using DIAMOND in the NCBI COG (Clusters of Orthologous Groups of proteins) database65,66,67. Finally, taxonomical assignments were deduced using the MEGAN6 program68.
Detection of sequence exchanges between V. vermiformis CDC-19 and other microorganisms
The representations as a ‘rhizome’ of the repertoire of genes predicted for V. vermiformis CDC-19 that best matched with sequences from giant viruses were built using the Circos tool (http://circos.ca/). Rhizomes aim to display genome mosaicism. Here, rhizomes of genes were built by BLASTp searches with complete sequences of these genes. The number and taxonomical assignments of V. vermiformis CDC-19 genes best matching with bacterial sequences were determined using the MEGAN6 program68. We randomly extracted the nucleotide sequences of 10 genes best matching with bacteria that were found to be co-localized with other genes of V. vermiformis CDC-19 at different positions in its genome. PCR primer systems were designed in order to target the region that straddles a gene best matching with bacteria and a gene of the amoeba, using the Primer3Plus program69. V. vermiformis CDC-19 DNA was amplified during 35 PCR cycles with the ten primer systems separately and the AmpliTaq Gold 360 Master mix (Applied Biosystems, Foster City, CA, USA; ref. 4398881). PCR products were purified using the Nucleofast 96 PCR clean-up kit (Macherey Nagel, Düren, Germany; ref. 743100). Purified products were sequenced using the BigDye Terminator V1.1 Sequencing Kit (Applied Biosystems; ref. 4336776), with a Sanger sequencing method on an ABI-3130 XL genetic analyser (Applied Biosystems). Finally, phylogenetic analyses were performed to strengthen the evidence of lateral sequence transfer for four genes whose presence was confirmed by PCR and Sanger sequencing and that have a bacterial homolog, two genes that had as top BLASTp hits a CPR homolog and two genes best matching with giant viral homologs. After amino acid sequence alignment with corresponding best hits using the MUSCLE program70, phylogenetic reconstructions were performed using the MEGA6 program, with a Maximum Likelihood method (http://www.megasoftware.net/).
References
Marciano-cabral, F. Advances in free-living amebae research 2003: workshop summary. J. Eukaryot. Microbiol. 50, 507–507 (2003).
Rodríguez-Zaragoza, S. Ecology of free-living amoebae. Crit. Rev. Microbiol. 20, 225–241 (1994).
Delafont, V., Rodier, M.-H., Maisonneuve, E. & Cateau, E. Vermamoeba vermiformis: a free-living amoeba of interest. Microb. ecol. 76, 991–1001 (2018).
Page, F. C. Taxonomic criteria for limax amoebae, with descriptions of 3 new species of Hartmannella and 3 of Vahlkampfia. J. Protozool. 14, 499–521 (1967).
Kuiper, M. W. et al. Quantitative detection of the free-living amoeba Hartmannella vermiformis in surface water by using real-time PCR. Appl. Environ. Microbiol. 72, 5750–5756 (2006).
Nazar, M. et al. Molecular identification of Hartmannella vermiformis and Vannella persistens from man-made recreational water environments, Tehran, Iran. Parasit. Res. 111, 835–839 (2012).
Armand, B., Motazedian, M. H. & Asgari, Q. Isolation and identification of pathogenic free-living amoeba from surface and tap water of Shiraz City using morphological and molecular methods. Parasit. Res. 115, 63–68 (2016).
Di Filippo, M. M. et al. Isolation and molecular characterization of free-living amoebae from different water sources in Italy. Int. J. Environ. Res. Public Health 12, 3417–3427 (2015).
Wang, H., Edwards, M., Falkinham, J. O. & Pruden, A. Molecular survey of the occurrence of legionella spp., mycobacterium spp., pseudomonas aeruginosa, and amoeba hosts in two chloraminated drinking water distribution systems. Appl. Environ. Microbiol. 78, 6285–6294 (2012).
Bradbury, R. S. Free-living amoebae recovered from human stool samples in Strongyloides agar culture. J. Clin. Microbiol. 52, 699–700 (2018).
Coşkun, K. A., Özçelik, S., Tutar, L., Elaldı, N. & Tutar, Y. Isolation and identification of free-Living amoebae from tap water in sivas, Turkey. BioMed Res. Int. 2013 (2013).
Pagnier, I., Valles, C., Raoult, D. & La Scola, B. Isolation of Vermamoeba vermiformis and associated bacteria in hospital water. Microb. Pathog. 80, 14–20 (2015).
Marciano-cabral, F. & Cabral, G. Acanthamoeba spp. as agents of disease in humans. Clin. Microbiol. Rev. 16, 273–307 (2003).
Khan, N. A. Acanthamoeba: biology and increasing importance in human health. FEMS Microbiol. Rev. 30, 564–595 (2006).
Park, J. S. First record of potentially pathogenic amoeba Vermamoeba vermiformis (Lobosea: Gymnamoebia) isolated from a freshwater of Dokdo island in the East Sea, Korea. Anim. Syst. Evol. Divers. 32, 1–8 (2016).
Wildschutte, H. & Lawrence, J. G. Differential Salmonella survival against communities of intestinal amoebae. Microbiology 153, 1781–1789 (2007).
Chekabab, S. M., Daigle, F., Charette, S. J., Dozois, C. M. & Harel, J. Survival of enterohemorrhagic Escherichia coli in the presence of Acanthamoeba castellanii and its dependence on Pho regulon. Microbiologyopen 1, 427–437 (2012).
Jeong, H. J. et al. Acanthamoeba: Could it be an environmental host of Shigella? Exp. Parasitol. 115, 181–186 (2007).
Brieland, J. K. et al. The role of Legionella pneumophila-infected Hartmannella vermiformis as an infectious particle in a murine model of Legionnaires’ disease. Infect. Immun. 65, 5330–5333 (1997).
FIELDS, B. S. et al. Characterization of an axenic strain of Hartmannella vermiformis obtained from an investigation of nosocomial legionellosis. J. Protozool. 37, 581–583 (1990).
Schulz-Bohm, K. et al. The prey’s scent - volatile organic compound mediated interactions between soil bacteria and their protist predators. ISME J. 11, 817–820 (2017).
La Scola, B. et al. A Giant virus in amoebae. Science 299, 2033–2033 (2003).
Raoult, D. et al. The 1.2-megabase genome sequence of Mimivirus. Science 306, 1344–1350 (2004).
Aherfi, S., Colson, P., La Scola, B. & Raoult, D. Giant viruses of amoebas: An Update. Front. Microbiol. 7, 1–14 (2016).
Colson, P., La Scola, B., Levasseur, A., Caetano-Anolles, G. & Raoult, D. Mimivirus: leading the way in the discovery of giant viruses of amoebae. Nat. Rev. Microbiol. 15, 243–254 (2017).
Greub, G. & Raoult, D. Microorganisms resistant to free-living amoebae. Clin. Microbiol. Rev. 17, 413–433 (2004).
Raoult, D. & Boyer, M. Amoebae as genitors and reservoirs of giant viruses. Intervirology 321–329 (2010) https://doi.org/10.1159/000312917.
Bertelli, C. & Greub, G. Lateral gene exchanges shape the genomes of amoeba-resisting microorganisms. Front. Cell. Infect. Microbiol. 2, 1–15 (2012).
Clarke, M. et al. Genome of Acanthamoeba castellanii highlights extensive lateral gene transfer and early evolution of tyrosine kinase signaling. Genome biol. 14, R11 (2013).
Maumus, F. & Blanc, G. Study of gene trafficking between Acanthamoeba and giant viruses suggests an undiscovered family of amoeba-infecting viruses. Genome Biol. Evol. 8, 3351–3363 (2016).
Chelkha, N. et al. A Phylogenomic study of Acanthamoeba polyphaga draft genome sequences suggests genetic exchanges with giant viruses. Front. Microbiol 9, 1–14 (2018).
Dornas, F. P., Khalil, J. Y. B., Pagnier, I. & Raoult, D. Isolation of new Brazilian giant viruses from environmental samples using a panel of protozoa. Front. Microbiol. 6, 1–9 (2015).
Colson, P., La Scola, B. & Raoult, D. Giant viruses of amoebae: a journey through innovative research and paradigm changes. Annu. Rev. Virol. (2017) https://doi.org/10.1146/annurev-virology-101416-041816.
Khalil, J. Y. B., Andreani, J. & La Scola, B. Updating strategies for isolating and discovering giant viruses. Curr. Opin. Microbiol. 31, 80–87 (2016).
Reteno, D. G. et al. Faustovirus, an Asfarvirus-related new lineage of giant viruses infecting amoebae. J. Virol. 89, 6585–6594 (2015).
Benamar, S. et al. Faustoviruses: Comparative genomics of new Megavirales family members. Front. Microbiol. 7, 1–9 (2016).
Bajrai, L. H. et al. Kaumoebavirus, a new virus that clusters with Faustoviruses and Asfarviridae. Viruses 8 (2016).
Andreani, J. et al. Orpheovirus IHUMI-LCC2: A new virus among the giant viruses. Front. Microbiol. 8, 1–11 (2018).
Abrahão, J. et al. Tailed giant Tupanvirus possesses the most complete translational apparatus of the known virosphere. Nat. Commun. 9 (2018).
Johansson, M. W., Lind, M. I., Holmblad, T., Thornqvist, P. O. & Soderhall, K. Peroxinectin, a novel cell adhesion protein from Crayfish Blood. Biochem. Biophys. Res. Commun. 216, 1079–1087 (1995).
Fritz-Laylin, L. K. et al. The genome of Naegleria gruberi illuminates early eukaryotic versatility. Cell 140, 631–642 (2010).
Roy, S. Intron-rich ancestors. Trends. Genet. 22, 468–471 (2006).
Roy, S. W., Irimia, M. & Penny, D. Very little intron gain in Entamoeba histolytica genes laterally transferred from prokaryotes. Mol. Biol. Evol. 23, 1824–1827 (2006).
Schaper, E. & Anisimova, M. The evolution and function of protein tandem repeats in plants. New Phytol. 206, 397–410 (2015).
Luef, B. et al. Diverse uncultivated ultra-small bacterial cells in groundwater. Nat. Commun. 6, 1–8 (2015).
Brown, C. T., Olm, M. R., Thomas, B. C. & Banfield, J. F. Measurement of bacterial replication rates in microbial communities. Nat. Biotechnol. 34, 1256–1263 (2016).
Brown, C. T. et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523, 208–211 (2015).
Deeg, C. M., Chow, C. E. T. & Suttle, C. A. The kinetoplastid-infecting bodo saltans virus (Bsv), a window into the most abundant giant viruses in the sea. eLife 7, 1–22 (2018).
Pagnier, I. et al. Babela massiliensis, a representative of a widespread bacterial phylum with unusual adaptations to parasitism in amoebae. Biol. Direct 10, 1–17 (2015).
Islam, Z., Nagampalli, R. S. K., Fatima, M. T. & Ashraf, G. M. New paradigm in ankyrin repeats: beyond protein-protein interaction module. Int. J. Biol. Macromol. 109, 1164–1173 (2018).
Häuser, R. et al. RsFA (YbeB) proteins are conserved ribosomal silencing factors. PLoS Genet. 8, 1–12 (2012).
Greub, G. & Raoult, D. Crescent bodies of Parachlamydia acanthamoeba and its life cycle within Acanthamoeba polyphaga: an electron micrograph study. Appl. Environ. Microbiol. 68, 3076–3084 (2002).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Moreno, Y. et al. Multiple identification of most important waterborne protozoa in surface water used for irrigation purposes by 18S rRNA amplicon-based metagenomics. Int. J. Hyg. Environ. Health 221, 102–111 (2018).
Coil, D., Jospin, G. & Darling, A. E. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 31, 587–589 (2015).
Seidl, M. F. et al. The genome of the saprophytic fungus Verticillium tricorpus reveals a complex effector repertoire resembling that of its pathogenic relatives. Mol. Plant Microbe Interact. 28, 362–373 (2014).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Nadalin, F., Vezzi, F. & Policriti, A. GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics 13, S8 (2012).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M. & Stanke, M. BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32, 767–769 (2015).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2016).
Anders, S., Pyl, P. T. & Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. mol. biol. 215, 403–10 (1990).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2014).
Tatusov, R. L. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36 (2000).
Huson, D. H., Beier, S., Flade, I., Górska, A. & El-hadidi, M. MEGAN community edition - interactive exploration and analysis of large-scale microbiome sequencing data. PLOS Comput. Biol. 1–12 (2016), https://doi.org/10.1371/journal.pcbi.1004957.
Untergasser, A. et al. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 35, 71–74 (2007).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC bioinformatics 5, 113 (2004).
Acknowledgements
This work was supported by a grant from the French State managed by the National Research Agency under the “Investissements d’avenir (Investments for the Future)” program with the reference ANR-10-IAHU-03 (Méditerranée Infection) and by région Provence Alpes Côte d’Azur and European funding FEDER PRIMI. Nisrine Chelkha was financially supported through a grant from the Infectiopole Sud foundation. Issam Hasni was financially supported through a grant from the Amoéba society.
Author information
Authors and Affiliations
Contributions
N.C., B.L.S., P.C. and A.L. designed the experiments. N.C. and P.C. wrote the manuscript. N.C. and I.H. performed the experiments. All authors analyzed the data and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chelkha, N., Hasni, I., Louazani, A.C. et al. Vermamoeba vermiformis CDC-19 draft genome sequence reveals considerable gene trafficking including with candidate phyla radiation and giant viruses. Sci Rep 10, 5928 (2020). https://doi.org/10.1038/s41598-020-62836-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-62836-9
This article is cited by
-
Giant virus biology and diversity in the era of genome-resolved metagenomics
Nature Reviews Microbiology (2022)
-
The draft genome of Cochliopodium minus reveals a complete meiosis toolkit and provides insight into the evolution of sexual mechanisms in Amoebozoa
Scientific Reports (2022)
-
Microbial warfare in the wild—the impact of protists on the evolution and virulence of bacterial pathogens
International Microbiology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
