
Abstract
An integrative understanding of nuclear events including transcription in normal and cancer cells requires comprehensive and quantitative measurement of protein dynamics that underlie such events. However, the low abundance of most nuclear proteins hampers their detailed functional characterization. We have now comprehensively quantified the abundance of nuclear proteins with the use of proteomics approaches in both normal and transformed human diploid fibroblasts. We found that subunits of the 26S proteasome complex were markedly down-regulated in the nuclear fraction of the transformed cells compared with that of the wild-type cells. The intranuclear proteasome abundance appeared to be inversely related to the rate of cell cycle progression, with restraint of the cell cycle being associated with an increase in the amount of proteasome subunits in the nucleus, suggesting that the nuclear proteasome content is dependent on the cell cycle. Furthermore, chromatin enrichment for proteomics (ChEP) analysis revealed enrichment of the proteasome in the chromatin fraction of quiescent cells and its apparent dissociation from chromatin in transformed cells. Our results thus suggest that translocation of the nuclear proteasome to chromatin may play an important role in control of the cell cycle and oncogenesis through regulation of chromatin-associated transcription factors.
Similar content being viewed by others


Introduction
Most biological processes including development, cell differentiation and proliferation, and homeostasis in mammals are regulated at the level of gene expression. A full understanding of such processes will thus require comprehensive characterization of how the expression of each gene is regulated1. The combination of genomics, proteomics, and other molecular technologies with bioinformatics has recently led to a rapid increase in our knowledge of gene regulatory networks that control gene expression2,3,4. Proteomics is an indispensable technique for characterization of the dynamics of proteins. Given that the abundance of nuclear proteins that contribute to the control of gene expression is generally low, however, the application of proteomics to characterization of the dynamics of such proteins has been limited.
Various methods to enrich nuclear proteins have been developed in an attempt to overcome this limitation. Although subcellular fractionation is a typical approach that has long been applied to separate the cytoplasm and nucleus with the use of hypotonic buffers and detergents, it is not suitable for isolation of proteins that bind to chromatin. Chromatin enrichment for proteomics (ChEP) enriches interphase chromatin and allows quantitative analysis of chromatin binding proteins. This approach is based on the cross-linking of intranuclear proteins to DNA in hypotonic buffer followed by isolation of the protein-DNA complex by centrifugation as a transparent gelatinous pellet5. Another approach to nuclear protein enrichment based on the interaction of such proteins with synthetic DNA containing transcription factor response elements has been developed6. This approach is restricted to in vitro conditions and so evaluates only indirectly protein-DNA binding under physiological conditions. Although several other methods have been developed in recent years to interrogate chromatin binding proteins, a disadvantage of these methods is that non–chromatin-associated proteins cannot be completely eliminated.
The 26S proteasome complex featured in this study is a key component of the ubiquitin-proteasome system (UPS), which is responsible for the catabolism of many proteins in both the cytoplasm and nucleus. The UPS mediates two discrete steps in such catabolism: the covalent attachment of multiple ubiquitin molecules to the protein substrate by a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3), and the degradation of the polyubiquitylated protein by the 26S proteasome complex7,8. In addition to the degradation of cytoplasmic proteins, the 26S proteasome regulates gene expression by controlling the abundance of transcription factors associated with chromatin9,10,11. The dynamics of proteasome localization have been well studied, with the 26S proteasome, which is formed by assembly of 20S and 19S complexes in the cytoplasm, being thought to translocate into the nucleus12. In yeast, the amount of the proteasome in the nucleus is greater in the stationary phase than in the growth phase13,14. On the other hand, the nuclear abundance of the proteasome in human cells is thought to increase in the proliferative phase, although many studies have been performed with cancer cells and the dynamics of the nuclear proteasome in normal human cells remain unknown15. In addition, analysis of the localization dynamics of the proteasome has often been performed with the use of proteasome subunits fused to a fluorescent protein, but whether such fusion influences incorporation of the subunit into the proteasome complex and its function has been unclear. Furthermore, evaluation of proteasome localization dynamics ideally requires a comprehensive analysis of all proteasome subunits, but such an analysis has been technically difficult to perform.
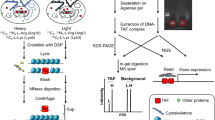
We have now developed a novel nuclear fractionation method to evaluate the network of nuclear proteins responsible for the control of gene expression. In this method, nuclei isolated by cell disruption with a hypotonic buffer are subjected to nucleolytic enzyme treatment and exposed to a solution of high ionic strength in order to allow the extraction and concentration of nuclear proteins without cytoplasmic contamination. The combination of this approach with label-free nontargeted proteomics showed that proteasome subunits disappeared from the nucleus of normal human cells in association with cell transformation. A detailed targeted proteomics analysis of proteasome subunits16 revealed the loss of all subunits in the nucleus of transformed cells. Further analyses suggested that the nuclear proteasome binds to chromatin in a cell cycle–dependent manner and may contribute to gene regulatory networks.
Results
Nuclear proteasome abundance declines in association with oncogenic transformation
We studied TIG-3 normal human diploid fibroblasts. These cells were engineered to stably express the human telomerase catalytic subunit (hTert) either alone or together with the simian virus 40 (SV40) early region, with the resulting cells being designated TIG-3(T) and TIG-3(T + SV40) and representing immortalized and transformed cells, respectively. To evaluate the dynamics of nuclear proteins that directly control gene expression, we developed a novel nuclear fractionation method and performed label-free quantitative proteomics analysis (Fig. 1a). Wild-type (WT) TIG-3 cells and TIG-3(T + SV40) cells were treated with a hypotonic buffer to allow separation of the nucleus (P fraction) from the cytoplasm (S fraction). The P fraction was treated with a nucleolytic enzyme in a low-salt solution and then centrifuged, and the resulting supernatant (P1 fraction) was collected whereas the pellet was incubated in a high-salt solution and then centrifuged to yield the P2 fraction. The validity of the fractionation was verified by immunoblot analysis of TIG-3(WT) cells (Fig. 1b). Proteins known to be located predominantly in the cytoplasm such as LDHA, HSP90, α-tubulin, and calnexin were enriched in the S fraction compared with the P1 and P2 fractions. Proteins that are present in the nucleoplasm or that are loosely associated with chromatin but liberated from chromatin by nuclease treatment, represented by the transcription factors c-Jun and E2F1, were detected in the P1 fraction. On the other hand, proteins that are strongly associated with chromatin such as histone H1 were recovered in the P2 fraction after exposure of the P1 fraction to the solution of high ionic strength. These results thus demonstrated the effectiveness of the fractionation method for the separation of nuclear proteins associated with chromatin.

Proteomics analysis shows that the nuclear abundance of the proteasome is reduced in association with oncogenic transformation. (a) Strategy for cell transformation and subcellular fractionation. TIG-3(WT) cells were transformed by the introduction of hTert and the early region of SV40. Cellular proteins of both TIG-3(WT) cells and the transformed cells, designated TIG-3(T + SV40), were separated into three fractions corresponding to the cytoplasm (S), the nucleoplasm and proteins loosely associated with chromatin (P1), and proteins tightly associated with chromatin (P2). Each fraction was analyzed by liquid chromatography and tandem mass spectrometry (LC-MS/MS). (b) Validation of subcellular fractionation. Whole cell extract (WCE) and subcellular fractions of TIG-3(WT) cells were subjected to immunoblot (IB) analysis with antibodies to LDHA, HSP90, α-tubulin, and calnexin as cytoplasmic marker proteins; to c-Jun and E2F1 as nucleoplasmic marker proteins; to histone H1 as a chromatin marker protein; and to the proteasome subunits PSMA2 and PSMD1. (c) Label-free proteomics analysis of P1 and P2 fractions. The log2[fold change] for protein abundance in TIG-3(T + SV40) cells relative to TIG-3(WT) cells and the –log10[q-value] are shown as Volcano plots. The threshold for determining differential expression is indicated by the dashed lines (q-value of ≤0.05). Proteasomal proteins are shown in red. (d) KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway–based enrichment analysis of the P1 fraction with Fisher’s exact test. The top three significantly (P < 0.05) up-regulated or down-regulated KEGG pathways in TIG-3(T + SV40) cells relative to TIG-3(WT) cells are shown. (e) Absolute quantification of proteasome subunits by iMPAQT analysis. The S and P1 fractions of TIG-3(WT), TIG-3(T), and TIG-3(T + SV40) cells were analyzed for the abundance of proteasome subunits of both the 20S and 19S complexes. Data are means ± s.d. for six independent biological replicates. *P < 0.05, **P < 0.01 compared with TIG-3(WT); #P < 0.05, ##P < 0.01 compared with TIG-3(T) (one-way ANOVA followed by Bonferroni’s post hoc test).
The P1 and P2 fractions were then subjected to label-free proteomics analysis. Volcano plot analysis and KEGG pathway analysis17 of the quantitative proteomics data revealed that the abundance of multiple 26S proteasome subunits in the P1 fraction was reduced in TIG-3(T + SV40) cells compared with TIG-3(WT) cells (Fig. 1c,d), whereas that of most such subunits in the P2 fraction did not differ between the two cell types. The reduced abundance of proteins associated with the endoplasmic reticulum (ER) apparent in the P1 fraction of TIG-3(T + SV40) cells (Fig. 1d) might be attributable to differential contamination of the P1 fraction with cytoplasmic proteins, given that a residual amount of the ER protein calnexin was detected in the P1 fraction of TIG-3(WT) cells by immunoblot analysis (Fig. 1b). The 26S proteasome is composed of 33 subunits, and we prepared synthetic proteins corresponding to 30 of the subunits by stable isotope labeling with [13C6/15N2]Lys and [13C6/15N4]Arg and then performed absolute quantification of these subunits in the S and P1 fractions of TIG-3(WT), TIG-3(T), and TIG-3(T + SV40) cells by application of our recently developed large-scale targeted proteomics platform, designated iMPAQT (in vitro proteome–assisted MRM [multiple reaction monitoring] for protein absolute quantification)16. Precise and absolute quantification of 26S proteasome subunits by the iMPAQT system confirmed a marked decrease in the abundance of almost all analyzed subunits in the P1 fraction of TIG-3(T + SV40) cells compared with that of TIG-3(WT) cells (Fig. 1e). Such depletion of proteasome subunits was not apparent in the P1 fraction of TIG-3(T) cells.
We next examined the nuclear localization of the proteasome in the normal and transformed cells by immunofluorescence analysis with antibodies to α-proteasome subunits (Fig. 2a). The intensity of the nuclear immunofluorescence signal normalized by the area of 4′,6-diamidino-2-phenylindole (DAPI) staining of DNA was plotted as a frequency distribution, and cells with a value greater than the median of the distribution for TIG-3(WT) cells were defined as positive for nuclear localization of the proteasome. The ratio of nuclear proteasome–positive cells to total cells was significantly reduced for TIG-3(T + SV40) cells compared with TIG-3(WT) or TIG-3(T) cells (Fig. 2b). Immunoblot analysis confirmed that the amounts of two representative proteasome subunits, PSMA2 (α2 subunit of the 20S complex) and PSMD1 (non-ATPase regulatory subunit 1 of the 19S complex), were markedly reduced in the P1 fraction of TIG-3(T + SV40) cells compared with that of TIG-3(WT) or TIG-3(T) cells (Fig. 2c,d).

Immunofluorescence and immunoblot analyses confirming that the nuclear localization of the proteasome is suppressed by oncogenic transformation. (a) Immunofluorescence staining of α-proteasomal subunits (red) in TIG-3(WT), TIG-3(T), and TIG-3(T + SV40) cells fixed after cytoplasm removal. DNA (blue) was counterstained with DAPI (upper panel). Scale bars, 10 µm. Histograms show the frequency distribution for normalized immunofluorescence signal intensity (A.U., arbitrary units) in the nucleus, with the median value for TIG-3(WT) cells being indicated with a dashed red line (lower panel). (b) Box plot of the percentage of cells with proteasome fluorescence intensity greater than the median of the distribution for TIG-3(WT) cells determined as in (a). Data are for four independent biological replicates. *P < 0.05 (one-way ANOVA followed by Bonferroni’s post hoc test). (c) Immunoblot analysis of PSMA2 and PSMD1 in the S and P1 fractions of TIG-3(WT), TIG-3(T), and TIG-3(T + SV40) cells. HSP90 and α-tubulin were examined as markers for the S fraction, and c-Jun as a marker for the P1 fraction. (d) Quantitative analysis of band intensities for PSMA2 and PSMD1 in experiments similar to that in (c). Data are from six independent biological replicates. **P < 0.01 (one-way ANOVA followed by Bonferroni’s post hoc test). Boxes and whiskers show the median, the lower and upper quartiles, and the minimum and maximum values. R software version 3.6.2 (https://www.r-project.org) was used to draw the plots in (b) and (d).
We also measured proteasome activity in the S and P1 fractions of parental, immortalized, and transformed fibroblasts by determining the degradation rate for specific fluorogenic peptide substrates targeted by chymotrypsin-like (Fig. 3a), caspase-like (Fig. 3b), and trypsin-like (Fig. 3c) activities of the proteasome. The proteasome activity with each substrate in the P1 fraction was reduced for TIG-3(T + SV40) cells compared with TIG-3(WT) or TIG-3(T) cells, whereas the activity in the S fraction did not differ significantly among TIG-3(WT), TIG-3(T), and TIG-3(T + SV40) cells. These results suggested that the activity of the proteasome changed in parallel with its abundance. We confirmed that two proteasome inhibitors, MG-132 and epoxomicin18,19, blocked the chymotrypsin-like (Supplementary Fig. 1), caspase-like (Supplementary Fig. 2), and trypsin-like (Supplementary Fig. 3) activities in both P1 and S fractions of TIG-3(WT), TIG-3(T), and TIG-3(T + SV40) cells, suggesting that these activities were indeed attributable to the proteasome.

Nuclear proteasome activity changes with transformation in parallel with its abundance. Chymotrypsin-like (a), caspase-like (b), and trypsin-like (c) proteasome activities in S and P1 fractions of TIG-3(WT), TIG-3(T), and TIG-3(T + SV40) cells were determined on the basis of digestion of specific fluorogenic peptides. Representative reaction time courses and quantitation of activity based on the relative slope for 0 to 30 min for five independent biological replicates are shown. **P < 0.01 (one-way ANOVA followed by Bonferroni’s post hoc test). Boxes and whiskers show the median, the lower and upper quartiles, and the minimum and maximum values. R software version 3.6.2 (https://www.r-project.org) was used to draw the box plots.
Large T, but not small T, alters the nuclear abundance of the proteasome
The early region of the SV40 genome encodes both large T antigen (LT) and small T antigen (ST), which are generated by alternative splicing20. The introduction of this region of SV40 into cells has long been adopted as a model of cell transformation and early tumorigenesis21,22. LT binds to and inactivates the tumor suppressor proteins p53 and retinoblastoma protein (Rb), resulting in the promotion of cell cycle progression23. On the other hand, ST binds to protein phosphatase 2 A and inhibit its serine-threonine phosphatase activity24. To examine whether LT or ST affects the nuclear localization of the proteasome, we established TIG-3(T) cells expressing LT or ST, which were designated TIG-3(T + SV40LT) and TIG-3(T + SV40ST), respectively (Fig. 4a). Immunofluorescence analysis revealed that, compared with TIG-3(T) cells, the ratio of nuclear proteasome–positive cells to total cells was significantly reduced for TIG-3(T + SV40LT) cells to an extent similar to that apparent for TIG-3(T + SV40) cells, whereas no such a change was detected for TIG-3(T + SV40ST) cells (Fig. 4b,c). Similar results were obtained by immunoblot analysis of PSMA2 and PSMD1 in the P1 fraction (Fig. 4d,e). These findings thus indicated that the change in the nuclear localization of the proteasome apparent in TIG-3(T + SV40) cells is attributable to LT rather than to ST.

SV40 LT, but not ST, alters the nuclear abundance of the proteasome. (a) Immunoblot analysis of SV40 early region expression in whole cell extracts of TIG-3(T), TIG-3(T + SV40), TIG-3(T + SV40LT), and TIG-3(T + SV40ST) cells. The antibodies detect the NH2-terminal region of both LT and ST. (b) Immunofluorescence staining of α-proteasomal subunits (red) in cells as in (a). The cells were fixed after cytoplasm removal and before staining. DNA (blue) was counterstained with DAPI (upper panel). Scale bars, 20 µm. Histograms show the frequency distribution for normalized immunofluorescence signal intensity in the nucleus, with the median value for TIG-3(T) cells being indicated with the dashed red line (lower panel). (c) Box plot for the percentage of cells with a proteasome fluorescence intensity greater than the median of the distribution for TIG-3(T) cells determined as in (b). Data are for three independent biological replicates. *P < 0.05 (one-way ANOVA followed by Bonferroni’s post hoc test). (d) Immunoblot analysis of PSMA2 and PSMD1 in the S and P1 fractions of TIG-3(T), TIG-3(T + SV40), TIG-3(T + SV40LT), and TIG-3(T + SV40ST) cells. HSP90 and α-tubulin were examined as controls for the S fraction, and c-Jun as a control for the P1 fraction. (e) Quantitative analysis of band intensities for PSMA2 and PSMD1 in experiments similar to that in (d). Data are for five independent biological replicates. **P < 0.01 (one-way ANOVA followed by Bonferroni’s post hoc test). Boxes and whiskers show the median, the lower and upper quartiles, and the minimum and maximum values. R software version 3.6.2 (https://www.r-project.org) was used to draw the plots in (c) and (e).
Nuclear proteasome abundance changes with the cell cycle
The cell cycle is accelerated in cells transformed with SV40, with LT expression being correlated with the rate of cell cycle progression25. We confirmed that the proportion of cells in S phase was increased for TIG-3(T + SV40) or TIG-3(T + SV40LT) cells compared with TIG-3(T) cells (Fig. 5a,b). To characterize cell cycle dynamics in detail, we applied a double thymidine block in TIG-3(WT), TIG-3(T), and TIG-3(T + SV40) cells (Fig. 5c). Whereas most TIG-3(T + SV40) cells progressed synchronously to M phase via S phase, TIG-3(WT) and TIG-3(T) cells included populations that either progressed to M phase via S phase (cycling cells) or which remained in G1 phase (noncycling cells). Given that TIG-3(WT) and TIG-3(T) cells also showed a biphasic frequency distribution for nuclear proteasome abundance, whereas TIG-3(T + SV40) cells did not include cells with a high nuclear proteasome content (Fig. 2a,b), we hypothesized that changes in cell cycle status might be related to the nuclear localization of the proteasome.

Nuclear proteasome abundance is increased in response to cell cycle arrest at G0-G1. (a) Cell cycle analysis in TIG-3(T), TIG-3(T + SV40), TIG-3(T + SV40LT), and TIG-3(T + SV40ST) cells. Cells were labeled with bromodeoxyuridine (BrdU) for 90 min, fixed, and stained with fluorescein isothiocyanate (FITC)–conjugated antibodies to BrdU. Nuclear DNA was stained with propidium iodide, and the cells were then analyzed by flow cytometry. (b) Box plot of the proportion of proliferative cells (cells in S phase) in experiments similar to that in (a). Data are for six independent biological replicates. **P < 0.01 (one-way ANOVA followed by Bonferroni’s post hoc test). (c) Analysis of cell cycle progression by double thymidine block. TIG-3(WT), TIG-3(T), and TIG-3(T + SV40) cells were arrested at the G1-S transition by exposure to excess thymidine and then released for the indicated times. They were then stained with propidium iodide for flow cytometric analysis of DNA content. Data from the asynchronous cells are also shown at the top. (d) Cell cycle analysis by the colcemid-challenge test. Asynchronous TIG-3(WT) cells and TIG-3(WT) cells remaining (colcemid-resistant cells) after long-term treatment with colcemid and shake-off of those arrested at G2-M phase were analyzed for cell cycle profile as in (a). (e) Box plot of the proportion of BrdU-positive cells in experiments similar to that in (d). Data are for three independent biological replicates. **P < 0.01 (Student’s t test). (f) Immunoblot analysis of PSMA2 and PSMD1 in S and P1 fractions of asynchronous and colcemid-resistant TIG-3(WT) cells. HSP90 and α-tubulin were examined as controls for the S fraction, and c-Jun as a control for the P1 fraction. (g) The band intensities for PSMA2 and PSMD1 in experiments similar to that in (f) were quantified. Data are for nine independent biological replicates. *P < 0.05, **P < 0.01 (Student’s t test). Boxes and whiskers show the median, the lower and upper quartiles, and the minimum and maximum values. R software version 3.6.2 (https://www.r-project.org) was used to draw the plots in (b), (e), and (g).
To examine whether nuclear proteasome abundance is increased in quiescent cells, we performed a colcemid-challenge test on TIG-3(WT) cells in order to remove cycling cells26. The cells were thus cultured in the presence of the mitotic inhibitor colcemid for 12 h on each of 5 days, after which those that had passed through mitosis were “shaken off.” The cells that had not passed through mitosis remained in the dish (colcemid-resistant cells), with this procedure thus resulting in enrichment of noncycling cells. We confirmed that the proportion of cells in S phase was greatly reduced among colcemid-resistant TIG-3(WT) cells compared with corresponding asynchronous cells (Fig. 5d,e). Immunoblot analysis revealed that the abundance of proteasome subunits in the P1 fraction was significantly greater for the colcemid-resistant cells than for the asynchronous cells (Fig. 5f,g). Collectively, our results thus suggested that the nuclear abundance of the proteasome is related to cell cycle status.
To directly demonstrate the increase in the nuclear abundance of the proteasome in quiescent cells, we performed immunofluorescence analysis for both the proteasome and the proliferation marker Ki-67 (Fig. 6a). For TIG-3(WT) cells, most Ki-67+ cells (proliferating cells) showed a low nuclear abundance of the proteasome, whereas Ki-67– cells (quiescent cells) were positive for nuclear proteasome immunofluorescence. On the other hand, most TIG-3(T + SV40) cells were positive for Ki-67 (cycling) and showed few nuclear proteasome signals (Fig. 6b). These results thus supported the notion that the nuclear abundance of the proteasome is regulated in a cell cycle–dependent manner.

Immunofluorescence analysis of cell cycle–dependent proteasome localization. (a) Immunofluorescence staining of α-proteasomal subunits (red) and Ki-67 (green) in TIG-3(WT) and TIG-3(T + SV40) cells. DNA (blue) was counterstained with DAPI (upper panel). Scale bars, 20 µm. Scatter plots for the intensity of nuclear proteasome staining versus loge[Ki-67 staining intensity] are also shown (lower panel). (b) Box plot for the percentage of cells with high proteasome content in Ki-67− cells (quiescent cells) determined as in (a). Data are for three independent biological replicates. **P < 0.01 (Student’s t test). Boxes and whiskers show the median, the lower and upper quartiles, and the minimum and maximum values. R software version 3.6.2 (https://www.r-project.org) was used to draw the plot in (b).
Nuclear proteasomes localize to chromatin in a cell cycle–dependent manner
Nuclear proteasomes control gene expression by mediating the degradation of transcription factors associated with chromatin9,10. We therefore performed ChEP analysis of TIG-3(WT) and TIG-3(T + SV40) cells to determine whether the association of the proteasome with chromatin is dependent on the cell cycle (Fig. 7a). Such analysis revealed that the amount of the proteasome bound to chromatin was reduced in TIG-3(T + SV40) cells compared with TIG-3(WT) cells (Fig. 7b,c). Furthermore, the levels of PSMA2 and PSMD1 associated with chromatin were significantly higher in colcemid-resistant TIG-3(WT) cells than in corresponding asynchronous cells (Fig. 7d,e). These results thus suggested that nuclear proteasomes are bound to chromatin in quiescent cells but are liberated from chromatin on reentry into the cell cycle.

The nuclear proteasome is enriched in the chromatin fraction. (a) Scheme for isolation of the chromatin-binding protein fraction by ChEP. (b) Immunoblot analysis of PSMA2 and PSMD1 in the chromatin-enriched fraction of TIG-3(WT) or TIG-3(T + SV40) cells. c-Jun was examined as a positive control for the chromatin-enriched fraction (left panel). Total proteins in the whole cell extract (WCE) and the ChEP fraction were also revealed by Coomassie brilliant blue staining of the polyacrylamide gel (right panel). Note that histones (indicated by the asterisk) are enriched in the ChEP fraction. (c) The abundance of PSMA2 and PSMD1 in the ChEP fraction was quantified in experiments similar to that in (b). Data were normalized by histone abundance and are for six independent biological replicates. *P < 0.05, **P < 0.01 (Student’s t test). (d) Immunoblot analysis of PSMA2 and PSMD1 in the chromatin-enriched fraction of asynchronous and colcemid-resistant (quiescent) TIG-3(WT) cells (left panel). Total proteins in the whole cell extract and in the ChEP fraction were also revealed by Coomassie brilliant blue staining of the polyacrylamide gel (right panel). (e) Quantitative analysis of PSMA2 and PSMD1 in the ChEP fraction for experiments similar to that in (d). Data are from six independent biological replicates. *P < 0.05, **P < 0.01 (Student’s t test). Boxes and whiskers show the median, the lower and upper quartiles, and the minimum and maximum values. R software version 3.6.2 (https://www.r-project.org) was used to draw the plots in (c) and (e).
To confirm the generality of our findings with TIG-3 human fibroblasts, we performed similar experiments with NIH 3T3 mouse fibroblasts. The cell cycle of NIH 3T3 cells was arrested by confluent culture and serum deprivation for 96 h27. We confirmed that the proportion of cells in S phase was greatly reduced among such arrested NIH 3T3 cells compared with corresponding asynchronous cells (Fig. 8a,b). Immunoblot analysis revealed that the abundance of the proteasome in the P1 fraction was significantly greater for the arrested cells than for the asynchronous cells (Fig. 8c,d).

Nuclear proteasome abundance is regulated in a cell cycle–dependent manner in NIH 3T3 cells. (a) Cell cycle analysis of NIH 3T3 cells in asynchronous culture or after induction of quiescence by confluent culture and serum deprivation. Cells were labeled with BrdU for 30 min, fixed, and stained with FITC-conjugated antibodies to BrdU. Nuclear DNA was stained with propidium iodide, and the cells were then analyzed by flow cytometry. (b) Box plot of the proportion of proliferative cells (cells in S phase) for experiments similar to that in (a). Data are for three independent biological replicates. **P < 0.01 (Student’s t test). (c) Immunoblot analysis of PSMA2 in the S and P1 fractions of NIH 3T3 cells maintained as in (a). HSP90 and α-tubulin were examined as controls for the S fraction, and c-Jun as a control for the P1 fraction. (d) Quantitative analysis of PSMA2 in the P1 fraction for experiments similar to that in (c). Data are for three independent biological replicates. *P < 0.05 (Student’s t test). (e) Immunoblot analysis of SV40 early region expression in NIH 3T3(WT), NIH 3T3(SV40), NIH 3T3(SV40LT), and NIH 3T3(SV40ST) cells. (f) Immunoblot analysis of PSMA2 in the S and P1 fractions of NIH 3T3(WT), NIH 3T3(SV40), NIH 3T3(SV40LT), and NIH 3T3(SV40ST) cells maintained as in (a). (g) Quantitative analysis of band intensities for PSMA2 in the P1 fraction of cells after induction of quiescence by confluent culture and serum deprivation for experiments similar to that in (f). Data are for three independent biological replicates. **P < 0.01 (one-way ANOVA followed by Bonferroni’s post hoc test). Boxes and whiskers show the median, the lower and upper quartiles, and the minimum and maximum values. R software version 3.6.2 (https://www.r-project.org) was used to draw the plots in (b), (d), and (g).
NIH 3T3 cells were also engineered to stably express the SV40 early region, LT, or ST, with the resulting cells being designated NIH 3T3(SV40), NIH 3T3(SV40LT), and NIH 3T3(SV40ST), respectively (Fig. 8e). Immunoblot analysis revealed that, compared with parental cells, the abundance of the proteasome in the P1 fraction was significantly reduced for NIH 3T3(SV40) and NIH 3T3(SV40LT) cells but not for NIH 3T3(SV40ST) cells (Fig. 8f,g). Together, these results thus suggested that the nuclear abundance of the proteasome is related to cell cycle status not only in human TIG-3 cells but also in mouse NIH 3T3 cells.
Discussion
We have here developed a novel nuclear fractionation method that identifies nuclear proteins associated with chromatin by combining conventional subcellular fractionation with targeting of the chromatin binding proteome, and which allows a comprehensive characterization of nuclear proteins that contribute to gene regulatory networks. With this new method, we have shown that the nuclear abundance of the proteasome changes in a cell cycle–dependent manner.
The validity of a fractionation method is key to its application to analysis of the intracellular localization of molecules. We assessed the effectiveness of our method by immunoblot analysis of proteins in each of the obtained fractions as well as by immunofluorescence analysis. Given that the cytoplasmic proteins examined were largely absent from the P1 fraction, contamination appears to be less of a problem for our method than for conventional fractionation. In any case, to eliminate the effects of such potential cytoplasmic contamination of the P1 fraction, we performed targeted proteomics analysis.
Many proteins function as components of large complexes and thereby contribute to intracellular processes including regulation of gene expression28. In the present study, we applied our recently developed targeted proteomics approach, iMPAQT16, to the absolute quantification of almost all subunits of the 26S proteasome complex. This approach is able to simultaneously evaluate the abundance of all these subunits in a single sample. This technology should also prove to be applicable to other large protein complexes such as the ribosome or spliceosome, thereby allowing estimation of the stoichiometry of the subunits within each entire complex.
The UPS regulates transcriptional activity in various situations9,29,30, with the localization of the proteasome being a key determinant of such regulation15,31. Recent studies of proteasome localization have adopted live cell imaging techniques that allow tracing of the movement of proteasomal subunits tagged with fluorescent labels such as green fluorescent protein13. These techniques have revealed that, in Saccharomyces cerevisiae, proteasomes are included within proteasome storage granules (PSGs)32,33 that serve as storage compartments in a cell cycle–dependent manner. In the stationary phase, PSGs are initially localized on the nucleoplasmic side of the nuclear envelope as spherical organelles without a membrane, whereas they eventually translocate to the cytoplasm in cells that remain in stationary phase over the long term. On exit from long-term stationary phase, PSGs rapidly dissociate and the proteasomes are transported to the nucleus within minutes31,32. Similar phenomena in mammals have been detected only in melanoma cells and hippocampal neurons to date13. Although our fractionation-based proteomics approach did not distinguish between PSGs and chromatin-bound proteasomes in normal human fibroblasts, immunofluorescence analysis revealed that proteasomes appeared to be dispersed throughout the nucleus with no signs of PSG-like structures in these cells. However, it is difficult to formally exclude the possibility that detergent treatment in the immunostaining procedure precluded the detection of PSGs.
It has long been unclear whether degradation of transcriptional regulators by the UPS promotes or represses overall transcriptional activity29,34,35. High transcriptional activity is likely associated with an increase in proteasome activity, however, a phenomenon referred to as transcriptional license. For example, in the mammalian circadian clock, UPS-mediated degradation of the transcriptional repressors Cryptochrome (CRY) and Period (PER) activates transcription of the genes for the clock proteins CLOCK and BMAL136,37. Importantly, the proteasome degrades different substrates in quiescent and proliferating cells. In the present study, we found that, in normal human fibroblasts, chromatin-bound proteasomes accumulate in the noncycling state but decrease in abundance in the cycling state. The identification of specific substrates of the nuclear proteasome in the quiescent phase awaits future study.
Many proteins that contribute to cell cycle progression such as cyclins as well as cyclin-dependent kinases and their inhibitors, as well as the tumor suppressor protein p53, signaling-related proteins such as IκBα, transcriptional factors, and misfolded and malfunctioning proteins, are targets of the UPS30. For maintenance of quiescence, proteasomes actively degrade many substrates including proteins related to the Notch signaling pathway and the transcriptional regulator STAT538,39. We previously showed that c-Myc undergoes degradation by the UPS in a manner dependent on the ubiquitin ligase SCFFbxw7 in order to maintain stem cells in the quiescent state40,41,42. SCFFbxw7 also regulates apoptosis by targeting MCL1 for ubiquitylation and destruction43. Proteasomes are recruited to chromatin and degrade the transcriptional repressor ATF3 during the response to DNA damage40. This degradation of ATF3 results in the recruitment of RNA polymerase II to gene promoters and activation of transcription11. Ubiquitylation of RAG-2 mediated by SCFSkp2 at the G1-to-S transition links degradation of the V(D)J recombinase to the cell cycle44. The nuclear proteasome thus maintains protein homeostasis in the nucleus by degrading specific substrates in response to changes in environmental and cellular conditions45,46. However, it is likely that many such specific substrates remain to be identified.
SV40 LT binds to and inactivates the tumor suppressor proteins p53 and Rb, resulting in the promotion of cell cycle progression23, and in induction of restriction point failure47. In mammals, temporary cell cycle arrest and entry into quiescence occur before cells reach the environmental factor–dependent restriction point of G1 phase48,49,50. We now show that cells transformed with SV40 and therefore unable to maintain quiescence as a result of the inactivation of p53 and Rb function manifested a reduction in the abundance of chromatin-bound proteasomes. This effect is likely a result of active cell cycling in the transformed cells rather than a direct consequence of loss of a specific function of p53 or Rb. On the other hand, the nuclear abundance of the proteasome was previously found to be increased in tumor cells compared with normal cells51,52. Our experimental model in the present study is based on the expression of hTert and SV40 LT, which inactivates p53 and Rb function. The transformed cells may thus reflect the initial phase of carcinogenesis53,54,55, whereas the previous studies were based on clinical cancer specimens or cells established from cancer that might be expected to reflect a more advanced state of malignancy. It will thus be of interest to determine the nuclear abundance of the proteasome in more highly malignant cells expressing c-Myc or mutant Ras in addition to hTert and SV40 LT.
The mechanism by which nuclear proteasome abundance changes in association with cell cycle status or cell transformation remains to be determined. From the opposite perspective, it remains unclear precisely how dissociation of the proteasome from chromatin might contribute to cell cycle progression or carcinogenesis. The binding of the proteasome to chromatin would be expected to regulate gene expression through the degradation of transcription factors, but identification of the cellular processes regulated in such a manner awaits further study. If release of the proteasome from chromatin during cell cycle progression is essential for cancer growth and metastasis, characterization of the underlying mechanism may provide a basis for the development of new anticancer drugs.
Methods
Cell culture, transfection, and infection
Cell culture, transfection, and infection were performed as previously described16,56,57. TIG-3 human embryonic lung diploid fibroblasts (Japanese Collection of Research Bioresources) were cultured under an atmosphere of 5% CO2 at 37 °C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Life Technologies) and antibiotics57. NIH 3T3 cells were cultured under the same conditions in DMEM supplemented with 10% fetal calf serum (Life Technologies) and antibiotics58. We authenticated both TIG-3 and NIH 3T3 cells and tested them for mycoplasma contamination. For retroviral infection, the mouse ecotropic retrovirus receptor was introduced into TIG-3 cells with the use of an amphotropic virus produced by HEK293T cells transfected with both pCX4-EcoVR and pGP/pE-ampho (Takara Bio)16. TIG-3 cells expressing the mouse ecotropic retrovirus receptor were infected with a retrovirus encoding hTert56. The resulting TIG-3(T) cells were infected with a retrovirus for the SV40 early region, SV40 LT, or SV40 ST and subjected to selection to yield TIG-3(T + SV40), TIG-3(T + SV40LT), or TIG-3(T + SV40ST) cells, respectively. Corresponding NIH 3T3 cells were established in the same way.
Cell fractionation and sample preparation
Cells (1 × 107) were suspended in 1.0 ml of a hypotonic solution containing 10 mM HEPES-NaOH (pH 7.6), 10 mM KCl, 1.5 mM MgCl2, 10% glycerol, 0.34 M sucrose, 1 mM phenylmethylsulfonyl fluoride, 3 μM aprotinin, and 20 μM leupeptin (all chemicals were from Sigma-Aldrich unless stated otherwise), and they were maintained on ice for 30 min before the addition of Nonidet P-40 (NP-40) and RNase A (Wako) to final concentrations of 0.5% and 0.1%, respectively, and incubation at room temperature for 15 min. The cell suspension was then centrifuged at 800 × g for 5 min at 4 °C, and the resulting supernatant was collected as the S fraction. The pellet was suspended in hypotonic buffer containing 0.5% NP-40, and the suspension was centrifuged again at 800 × g for 5 min. The resulting pellet was suspended in 0.2 ml of a low-salt buffer containing 25 mM Tris-HCl (pH 7.4), 2 mM MgCl2, 0.1% NP-40, and 0.5% Benzonase nuclease (Millipore) and incubated at 37 °C for 30 min before centrifugation at 2300 × g for 5 min at 4 °C. The resulting supernatant was collected as the P1 fraction, and the pellet was suspended in 0.2 ml of a solution containing 2 M KCl, 25 mM Tris-HCl (pH 7.4), and 0.5% Triton X-100 and maintained on ice for 30 min before centrifugation at 20,000 × g for 5 min at 4 °C. The resulting supernatant was collected as the P2 fraction.
For label-free quantification and MRM analysis, SDS was added to a final concentration of 1% to each fraction and each mixture was incubated at 95 °C for 10 min. The protein concentration of the samples was determined with the BCA assay (Bio-Rad). Cysteine residues were blocked by incubation of the samples with 2 mM tris(2-carboxyethyl)phosphine hydrochloride (Thermo Fisher Scientific) for 30 min at 37 °C followed by alkylation with 10 mM 2-iodoacetamide for 30 min at room temperature. The samples were then digested with trypsin (5 μg/ml, Thermo Fisher Scientific) for 14 h at 37 °C.
Label-free quantification of proteins by LC-MS/MS analysis
Peptides were acidified by the addition of trifluoroacetic acid to a final concentration of 1% and were then subjected to LC-MS/MS analysis in DDA mode with a QExactive instrument coupled with a nano-LC system (AdvanceLC, Michrom). Up to the top 10 most abundant ions with a charge of 2+ or 3+ were selected from the survey scan with an isolation window of 1.6 m/z units and were fragmented by higher-energy collision dissociation with a normalized collision energy of 27. Acquired data were analyzed with MaxQuant (version 1.2.0.11) and the human IPI database (version 3.68; 87,083 entries)59.
MRM analysis
Recombinant proteasome subunits were generated by in vitro transcription and translation with the use of a wheat germ extract and corresponding human cDNA clones. The purified proteins were digested with trypsin and subjected to LC-MS/MS analysis, and MS/MS spectra assigned to peptides with more than six amino acids were selected from the iMPAQT-knowledge database and converted into MRM transitions16. High-responding proteotypic peptide sequences were concatenated into several synthetic artificial protein sequences, and corresponding cDNAs were synthesized (Eurofins Genomics). Artificial protein sequences and corresponding MRM assays are listed in Supplementary Table 1. To obtain isotopically labeled recombinant proteins with maximum labeling efficiency, we generated expression strains by P1 transduction of Escherichia coli BL21(DE3) that was auxotrophic for arginine and lysine and was derived from argA and lysA deletion mutants in the Keio Collection of single-gene knockouts60. Recombinant proteins were expressed as His6-tagged molecules in the auxotrophic cells cultured in the presence of [13C6/15N2]Lys and [13C6/15N4]Arg and were purified with Ni-resin. The vector sequences for production of the recombinant proteins are listed in Supplementary Table 2. Both pENTR2B containing each vector sequence and pET21b-QcodeAA-DEST are recombined by the LR reaction in the bacterial cells. MRM analysis was performed with a QTRAP5500 instrument (SCIEX) operated in positive-ion mode as described previously16.
Immunoblot analysis
Immunoblot analysis was performed as described previously61,62,63. Primary antibodies included those to PSMA2 (Cell Signaling Technology, 2455), to PSMD1 (Abcam, 140682), to LDHA (Cell Signaling Technology, 3582), to c-Jun (Cell Signaling Technology, 9165), to histone H1 (Merck Millipore, 05–457), to E2F1 (Cell Signaling Technology, 3742), to SV40 LT and ST (Santa Cruz Biotechnology, sc-148), to HSP90 (BD Transduction Laboratories, 610419), and to α-tubulin (Thermo Fisher Scientific, 13–8000), and to calnexin (Medical and Biological Laboratories, M178–3). Horseradish peroxidase–conjugated goat antibodies to mouse or rabbit immunoglobulin G were obtained from Promega62, and ECL reagents were from Thermo Fisher Scientific. Immune complexes were quantified with a ChemiDoc Imaging System (Bio-Rad). Uncropped blot images are provided in Supplementary Figures 4 to 7.
Immunofluorescence analysis
Immunofluorescence analysis was performed as previously described64,65,66,67, with a minor modification. After cytoplasm was removed from cells by exposure to phosphate-buffered saline (PBS) containing 0.5% Triton X-100 and 4% polyethylene glycol, nuclei were fixed for 30 min at room temperature with 4% paraformaldehyde in PBS, incubated first with antibodies to α subunits of the 20S proteasome (Enzo Life Sciences, HP810) and to Ki-67 (Lab Vision Corporation, SP6) and then with Alexa Fluor 546– or Alexa Fluor 488–conjugated secondary antibodies (Thermo Fisher Scientific) in PBS containing 0.5% Triton X-100, 2% bovine serum albumin, 0.2% Tween 20, 10% glycerol, and 0.05% NaN3, and then covered with a drop of Vectashield Mounting Medium with DAPI (Vector Laboratories) before examination with a confocal fluorescence microscope (LSM700, Carl Zeiss)64. Proteasome fluorescence intensity relative to the nuclear area was quantified with the use of ImageJ software (NIH).
Measurement of proteasomal activity
The in vitro assay of 26S proteasome activities was performed as previously described68. ATP and dithiothreitol were added to S and P1 fractions at final concentrations of 2 and 1 mM, respectively, and the resulting samples (200 μl, containing ~15 µg of total protein) were transferred to the wells of a 96-well microtiter plate (Optiplate-96, PerkinElmer) and mixed with 10 µl of fluorogenic substrate. The substrates included Z-Leu-Leu-Glu-AMC (final concentration of 75 μM, Enzo) for measurement of caspase-like activity, Suc-Leu-Leu-Val-Tyr-AMC (final concentration of 75 μM, Enzo) for measurement of chymotrypsin-like activity, and Boc-Leu-Arg-Arg-AMC (final concentration of 2.5 μM, Enzo) for measurement of trypsin-like activity. Fluorescence (excitation at 380 nm, emission at 460 nm) was monitored every 5 min for 1 h at 37 °C with a microplate fluorometer (EnSpire, PerkinElmer). Proteasome inhibitors were added to S and P1 fractions 10 min before the assay. MG-132 was obtained from Peptide Institute, and epoxomicin was from Selleck Chemicals. MG-132 was used at a concentration of 25 µM. Epoxomicin was used at a concentration of 1 nM for inhibition of chymotrypsin-like activity and at 100 nM for inhibition of caspase-like and trypsin-like activities. The slope of the initial linear portion of each reaction (0 to 30 min after reaction onset) was determined for comparison of proteasome activities.
Experimental model for cell cycle arrest
An experimental model for cell cycle arrest was performed as described previously69,70. Cells were synchronized at G1-S with a double thymidine block. They were thus incubated in medium containing 2 mM thymidine for 18 h, released for 8 h, and exposed again to 2 mM thymidine for 16 h. The cells were collected at 0 h (G1-S phase), 4 h (S phase), 8 h (S-G2 phase), or 12 h (G2-M phase) after the first or second release.
Colcemid-challenge test
A colcemid-challenge test were performed as previously described26. In brief, TIG-3 cells were treated with 2 mM thymidine for 18 h, released for 8 h, and incubated with 40 nM colcemid (N-desacetyl-N-methylocolchicine, Thermo Fisher Scientific) for 12 h, after which mitotic cells were removed by shake-off treatment. To ensure complete removal of mitotic cells, we repeated the shake-off process five times (40 nM colcemid for 12 h, release of cells for 12 h). The remaining cells were studied as colcemid-resistant cells.
Serum deprivation and density-dependent growth inhibition
NIH 3T3 cells were rendered quiescent as previously described27. The cells were thus cultured in the presence of 0.5% calf serum for 96 h after achieving confluence.
ChEP analysis
The protocol for cross-linking and purification of protein-DNA complexes was described previously5,71. In brief, cells (1 × 107) were fixed by incubation for 10 min at 37 °C with 5 ml of 1% formaldehyde in PBS, after which glycine was added to a final concentration of 0.25 M and the cells were incubated for an additional 10 min at room temperature. The cells were harvested and resuspended in 1 ml of an ice-cold lysis buffer containing 25 mM Tris-HCl (pH 7.4), 0.1% Triton X-100, 85 mM KCl, and Roche protease inhibitors (one tablet per 10 ml of buffer solution)5, and the resulting lysates were centrifuged to obtain a nuclear pellet. The pellet was washed with 250 μl of lysis buffer supplemented with 0.4% RNase A (Wako). For removal of noncovalently bound proteins, the nuclei were suspended in 250 μl of a solution containing 50 mM Tris-HCl (pH 7.4), 10 mM EDTA, and 4% SDS before the addition of 3 volumes of a solution containing 10 mM Tris-HCl (pH 7.4), 1 mM EDTA, and 8 M urea. The nuclei were then isolated by centrifugation, suspended in 250 µl of a solution containing 10 mM Tris-HCl (pH 7.4), 1 mM EDTA, 25 mM NaCl, and 10% glycerol, and subjected to ultrasonic treatment, and the supernatant obtained after subsequent centrifugation was collected as the ChEP sample containing sheared, cross-linked chromatin.
Cell cycle analysis by flow cytometry
Sample preparation and analysis were performed as described previously72,73,74. For determination of BrdU incorporation, cells were exposed to BrdU (10 μM) for 90 min, separated by centrifugation, suspended in PBS, and fixed with 70% ethanol at −20 °C73. They were again separated by centrifugation, incubated with FITC-conjugated mouse antibodies to BrdU (BD Transduction Laboratories, 556028) for 1 h at room temperature, and washed, after which DNA was stained with propidium iodide (10 μg/ml) diluted in PBS containing 0.1% Triton X-100 and RNase A (0.2 mg/ml). The samples were analyzed with a FACSCalibur flow cytometer and FlowJo v10 software (Becton Dickinson). The percentage of cells in S phase was calculated by fitting the propidium iodide signal to a cell cycle distribution with the Watson pragmatic model approach.
Statistical analysis
Data were compared with Student’s t test or by one-way analysis of variance (ANOVA), with P values being adjusted by the Bonferroni method for multiple comparisons. All statistical analysis was performed with R software. A P value of <0.05 was considered statistically significant.
Data availability
The MS data have been deposited with the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the JPOST partner repository under the data set identifiers PXD016259 (label-free quantification) and PXD016260 (MRM analysis).
References
Simicevic, J. & Deplancke, B. Transcription factor proteomics - Tools, applications, and challenges. Proteomics 17 (2017).
Gerstein, M. B. et al. Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100 (2012).
Ernst, J. et al. Genome-scale high-resolution mapping of activating and repressive nucleotides in regulatory regions. Nat. Biotechnol. 34, 1180–1190 (2016).
Landry, C. R., Levy, E. D., Abd Rabbo, D., Tarassov, K. & Michnick, S. W. Extracting insight from noisy cellular networks. Cell 155, 983–989 (2013).
Kustatscher, G., Wills, K. L., Furlan, C. & Rappsilber, J. Chromatin enrichment for proteomics. Nat. Protoc. 9, 2090–2099 (2014).
Ding, C. et al. Proteome-wide profiling of activated transcription factors with a concatenated tandem array of transcription factor response elements. Proc. Natl. Acad. Sci. USA 110, 6771–6776 (2013).
Hershko, A. Ubiquitin: roles in protein modification and breakdown. Cell 34, 11–12 (1983).
Nakayama, K. I. & Nakayama, K. Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer 6, 369–381 (2006).
Muratani, M. & Tansey, W. P. How the ubiquitin-proteasome system controls transcription. Nat. Rev. Mol. Cell Biol. 4, 192–201 (2003).
McCann, T. S. & Tansey, W. P. Functions of the proteasome on chromatin. Biomolecules 4, 1026–1044 (2014).
Epanchintsev, A. et al. Cockayne’s syndrome A and B proteins regulate transcription arrest after genotoxic stress by promoting ATF3 degradation. Mol. Cell 68, 1054–1066 (2017).
Pack, C. G. et al. Quantitative live-cell imaging reveals spatio-temporal dynamics and cytoplasmic assembly of the 26S proteasome. Nat. Commun. 5, 3396 (2014).
Enenkel, C. Proteasome dynamics. BBA-Mol. Cell Res. 1843, 39–46 (2014).
Wendler, P. & Enenkel, C. Nuclear transport of yeast proteasomes. Front. Mol. Biosci. 6, 6–34 (2019).
Chowdhury, M. & Enenkel, C. Intracellular Dynamics of the ubiquitin-proteasome-system. F1000Res. 4, 367 (2015).
Matsumoto, M. et al. A large-scale targeted proteomics assay resource based on an in vitro human proteome. Nat. Methods 14, 251–258 (2017).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Meng, L. et al. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci. USA 96, 10403–10408 (1999).
Schwarz, K. et al. The selective proteasome inhibitors lactacystin and epoxomicin can be used to either up- or down-regulate antigen presentation at nontoxic doses. J. Immunol. 164, 6147–6157 (2000).
Fanning, E. & Knippers, R. Structure and Function of Simian Virus-40 Large Tumor-Antigen. Annu. Rev. Biochem. 61, 55–85 (1992).
Land, H., Parada, L. F. & Weinberg, R. A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304, 596–602 (1983).
Danielsson, F. et al. Majority of differentially expressed genes are down-regulated during malignant transformation in a four-stage model. Proc. Natl. Acad. Sci. USA 112, 6853–6858 (2015).
Ahuja, D., Saenz-Robles, M. T. & Pipas, J. M. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 24, 7729–7745 (2005).
Yang, S. I. et al. Control of protein phosphatase 2A by simian virus 40 small-t antigen. Mol. Cell. Biol. 11, 1988–1995 (1991).
Manfredi, J. J. & Prives, C. The Transforming Activity of Simian-Virus-40 Large Tumor-Antigen. Bba-Rev. Cancer 1198, 65–83 (1994).
Wang, R. X. W., MacDuffie, E. & Santaguida, S. Generation and isolation of cell cycle-arrested cells with complex karyotypes. Jove-J. Vis. Exp. 134 (2018).
Rodier, G. et al. p107 inhibits G1 to S phase progression by downregulating expression of the F-box protein Skp2. J. Cell Biol. 168, 55–66 (2005).
Gingras, A. C., Aebersold, R. & Raught, B. Advances in protein complex analysis using mass spectrometry. J. Physiol-London 563, 11–21 (2005).
Ee, G. & Lehming, N. How the ubiquitin proteasome system regulates the regulators of transcription. Transcription 3, 235–239 (2012).
Zhu, Q. et al. The ubiquitin-proteasome system regulates p53-mediated transcription at p21waf1 promoter. Oncogene 26, 4199–4208 (2007).
Burcoglu, J., Zhao, L. & Enenkel, C. Nuclear import of yeast proteasomes. Cells 4, 387–405 (2015).
Laporte, D., Salin, B., Daignan-Fornier, B. & Sagot, I. Reversible cytoplasmic localization of the proteasome in quiescent yeast cells. J. Cell. Biol. 181, 737–745 (2008).
Peters, L. Z., Karmon, O., Miodownik, S. & Ben-Aroya, S. Proteasome storage granules are transiently associated with the insoluble protein deposit in Saccharomyces cerevisiae. J. Cell. Sci. 129, 1190–1197 (2016).
Nalley, K., Johnston, S. A. & Kodadek, T. Proteolytic turnover of the Gal4 transcription factor is not required for function in vivo. Nature 442, 1054–1057 (2006).
Collins, G. A., Lipford, J. R., Deshaies, R. J. & Tansey, W. P. Gal4 turnover and transcription activation. Nature 461 (2009).
Kume, K. et al. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell 98, 193–205 (1999).
Shearman, L. P. et al. Interacting molecular loops in the mammalian circadian clock. Science 288, 1013–1019 (2000).
Strikoudis, A., Guillamot, M. & Aifantis, I. Regulation of stem cell function by protein ubiquitylation. EMBO Rep. 15, 365–382 (2014).
Mohammad, K., Dakik, P., Medkour, Y., Mitrofanova, D. & Titorenko, V. I. Quiescence entry, maintenance, and exit in adult stem cells. Int. J. Mol. Sci. 20 (2019).
Yada, M. et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 23, 2116–2125 (2004).
Matsuoka, S. et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev. 22, 986–991 (2008).
Takeishi, S. et al. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell. 23, 347–361 (2013).
Inuzuka, H. et al. SCFFBW7 regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471, 104–U128 (2011).
Jiang, H. et al. Ubiquitylation of RAG-2 by Skp2-SCF links destruction of the V(D)J recombinase to the cell cycle. Mol. Cell. 18, 699–709 (2005).
Nielsen, S. V., Poulsen, E. G., Rebula, C. A. & Hartmann-Petersen, R. Protein quality control in the nucleus. Biomolecules 4, 646–661 (2014).
Enam, C., Geffen, Y., Ravid, T. & Gardner, R. G. Protein quality control degradation in the nucleus. Annu. Rev. Biochem. 87, 725–749 (2018).
Moser, J., Miller, I., Carter, D. & Spencer, S. L. Control of the restriction point by Rb and p21. Proc. Natl. Acad. Sci. USA 115, Eb219–Eb227 (2018).
Pardee, A. B. A restriction point for control of normal animal cell proliferation. Proc. Natl. Acad. Sci. USA 71, 1286–1290 (1974).
Foster, D. A., Yellen, P., Xu, L. & Saqcena, M. Regulation of G1 cell cycle progression: Distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer 1, 1124–1131 (2010).
Yang, H. W., Chung, M., Kudo, T. & Meyer, T. Competing memories of mitogen and p53 signalling control cell-cycle entry. Nature 549, 404–408 (2017).
Kanayama, H. et al. Changes in expressions of proteasome and ubiquitin genes in human renal cancer cells. Cancer Res. 51, 6677–6685 (1991).
Tsvetkov, P. et al. Oncogenic addiction to high 26S proteasome level. Cell Death Dis. 9, 773 (2018).
Kandoth, C. et al. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013).
Muller, P. A. J. & Vousden, K. H. Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 25, 304–317 (2014).
Maddison, L. A., Sutherland, B. W., Barrios, R. J. & Greenberg, N. M. Conditional deletion of Rb causes early stage prostate cancer. Cancer Res. 64, 6018–6025 (2004).
Oshikawa, K., Matsumoto, M., Oyamada, K. & Nakayama, K. I. Proteome-wide Identification of Ubiquitylation Sites by Conjugation of Engineered Lysine-less Ubiquitin. J. Proteome Res. 11, 796–807 (2012).
Nakatsumi, H., Oka, T., Higa, T., Shirane, M. & Nakayama, K. I. Nuclear-cytoplasmic shuttling protein PP2A(B56) contributes to mTORC1-dependent dephosphorylation of FOXK1. Genes Cells 23, 599–605 (2018).
Hatakeyama, S., Watanabe, M., Fujii, Y. & Nakayama, K. I. Targeted destruction of c-Myc by an engineered ubiquitin ligase suppresses cell transformation and tumor formation. Cancer Res. 65, 7874–7879 (2005).
Schwanhausser, B. et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011).
Matic, I. et al. Absolute SILAC-compatible expression strain allows Sumo-2 copy number determination in clinical samples. J. Proteome Res. 10, 4869–4875 (2011).
Foster, J. S., Fernando, R. I., Ishida, N., Nakayama, K. I. & Wimalasena, J. Estrogens down-regulate p27Kip1 in breast cancer cells through Skp2 and through nuclear export mediated by the ERK pathway. J. Biol. Chem. 278, 41355–41366 (2003).
Sugiyama, S., Yumimoto, K., Inoue, I. & Nakayama, K. I. SCF(Fbxw7) ubiquitylates KLF7 for degradation in a manner dependent on GSK-3-mediated phosphorylation. Genes Cells 24, 354–365 (2019).
Katayama, Y. et al. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 537, 675–679 (2016).
Minamishima, Y. A., Nakayama, K. & Nakayama, K. I. Recovery of liver mass without proliferation of hepatocytes after partial hepatectomy in skp2-deficient mice. Cancer Res. 62, 995–999 (2002).
Matsuzaki, F., Shirane, M., Matsumoto, M. & Nakayama, K. I. Protrudin serves as an adaptor molecule that connects KIF5 and its cargoes in vesicular transport during process formation. Mol. Biol. Cell 22, 4602–4620 (2011).
Nagahama, H. et al. Spatial and temporal expression patterns of the cyclin-dependent kinase (CDK) inhibitors p27Kip1 and p57Kip2 during mouse development. Anat. Embryol. (Berl.) 203, 77–87 (2001).
Takahashi, H., Hatakeyama, S., Saitoh, H. & Nakayama, K. I. Noncovalent SUMO-1 binding activity of thymine DNA glycosylase (TDG) is required for its SUMO-1 modification and colocalization with the promyelocytic leukemia protein. J. Biol. Chem. 280, 5611–5621 (2005).
Vilchez, D. et al. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature 489, 304–308 (2012).
Nakayama, K. et al. Impaired degradation of inhibitory subunit of NF-κB (IκB) and β-catenin as a result of targeted disruption of the β-TrCP1 gene. Proc. Natl. Acad. Sci. USA 100, 8752–8757 (2003).
Becher, I. et al. Pervasive protein thermal stability variation during the cell cycle. Cell 173, 1495–1507 (2018).
Beauchemin, M. & Morse, D. A proteomic portrait of dinoflagellate chromatin reveals abundant RNA-binding proteins. Chromosoma 127, 29–43 (2018).
Matsumoto, A., Takeishi, S. & Nakayama, K. I. p57 regulates T-cell development and prevents lymphomagenesis by balancing p53 activity and pre-TCR signaling. Blood 123, 3429–3439 (2014).
Imaki, H. et al. Cell cycle-dependent regulation of the Skp2 promoter by GA-binding protein. Cancer Res. 63, 4607–4613 (2003).
Parcellier, A. et al. HSP27 favors ubiquitination and proteasomal degradation of p27Kip1 and helps S-phase re-entry in stressed cells. FASEB J. 20, 1179–1181 (2006).
Acknowledgements
We thank A. Nakayama, K. Tsunematsu, E. Koba, T. Hoshino and Y. A. Minamishima for technical assistance; A. Ohta for help with preparation of the manuscript; and M. Kanehisa for permission to use the KEGG pathway database. This study was supported in part by a KAKENHI grant (18H05215) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Author information
Authors and Affiliations
Contributions
Y.K., M.M. and A.M. designed experiments and prepared the manuscript. M.M. and K.O. assisted with molecular biology experiments. M.M. and A.H. performed LC-MS/MS and MRM analysis. M.M. and T.T. developed the informatics pipeline. Y.K. performed all other experiments and data analysis. K.I.N. coordinated the study and wrote the manuscript. All authors discussed the data and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kito, Y., Matsumoto, M., Hatano, A. et al. Cell cycle–dependent localization of the proteasome to chromatin. Sci Rep 10, 5801 (2020). https://doi.org/10.1038/s41598-020-62697-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-62697-2
This article is cited by
-
PSMD8 can serve as potential biomarker and therapeutic target of the PSMD family in ovarian cancer: based on bioinformatics analysis and in vitro validation
BMC Cancer (2023)
-
Chromatin enrichment for proteomics in plants (ChEP-P) implicates the histone reader ALFIN-LIKE 6 in jasmonate signalling
BMC Genomics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
