
Abstract
Optimal clinical decision-making depends on identification of clinically relevant organisms present in a sample. Standard microbiological culture may fail to identify unusual or fastidious organisms and can misrepresent relative abundance of sample constituents. Culture-independent methods have improved our ability to deconvolute polymicrobial patient samples. We used next-generation 16S rRNA gene sequencing (NGS16S) to determine how often cultivatable organisms in complex polymicrobial samples are not reported by standard culture. Twenty consecutive bronchoalveolar lavage (BAL) samples were plated to standard and additional media; bacteria were identified by NGS16S analysis of DNA extracted directly from samples or from washed culture plates. 96% of organisms identified were cultivable, but only 21% were reported by standard culture, indicating that standard work-up provides an incomplete assessment of microbial constituents. Direct NGS16S correlated well with standard culture, identifying the same predominant organism in 50% of samples. When predominant organisms differed, NGS16S most often detected anaerobes, whose growth is unsupported by standard culture conditions for this specimen. NGS16S identified more organisms per sample and allowed identification of fastidious organisms, while culture was better at capturing organisms when bacterial load was low, and allowed incidental recovery of non-bacterial pathogens. Molecular and culture-based methods together detect more organisms than either method alone.
Similar content being viewed by others

Introduction
Culture is a “complex and difficult art”1. As the mainstay of the modern clinical microbiology laboratory, isolated growth of individual organisms is required for antimicrobial susceptibility and virulence testing, epidemiological investigations, and genome sequencing. Nevertheless, standard culture often fails to identify a causative pathogen when unusual or fastidious organisms are present, or after antimicrobial therapy has been initiated2,3,4,5,6,7,8,9. The developing field of culturomics has enabled the isolation of hundreds of new microorganisms, previously considered uncultivable, using a variety of growth conditions and extended incubation times3,6,10,11,12. For example, the addition of the antioxidant uric acid enables the aerobic growth of many organisms thought to be strictly anaerobic13. Therefore, we hypothesized that simple changes to routine culture conditions (for example, including additional types of growth media) could expand the repertoire of recoverable organisms in the clinical laboratory.
Next generation 16S rRNA gene sequencing (NGS16S) can be utilized for deconvolution of polymicrobial clinical samples that are difficult or impossible to resolve by standard molecular methods. Using synthetic polymicrobial samples of defined composition, we have shown that NGS16S analysis more accurately catalogs the bacterial contents of polymicrobial samples than standard culture14. However, this technology is expensive and requires technical expertise, limiting its routine use in the clinical laboratory. Here, we used NGS16S analysis of BAL samples, a readily accessible polymicrobial sample type, to evaluate the ability of culture to accurately catalog the microbial constituents of complex clinical samples. First, we analyzed DNA extracted directly from patient samples to determine the identity and prevalence of organisms for which current culture conditions are sub-optimal. In addition, we expanded standard culture by including four additional culture conditions, and evaluated washes of culture media plates by NGS16S analysis to determine the frequency with which cultivable organisms are not reported after standard work-up. We found that NGS16S identifies more organisms per sample and allows identification of fastidious organisms, while culture is better at capturing organisms when bacterial load is low and allows incidental recovery of non-bacterial pathogens such as yeast or molds. Both methods together detected more organisms in clinical samples than either method alone.
Results
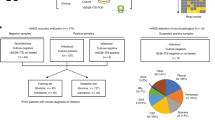
Twenty consecutive BAL samples were collected from in-house oncology or transplant patients. Two samples (BAL03 and BAL06) were from the same patient; all other samples were collected from different individuals. Specimens were analyzed as outlined in Fig. 1. Standard clinical microbiological culture and NGS16S sequencing of DNA extracted directly from clinical specimens (direct NGS16S) was performed by the UWMC clinical microbiology laboratory as described in the methods. No bacteria were reported by standard microbiological work-up for six samples (BAL 06, 07, 11, 12, 15 and 18; Table 1). No reportable organisms were detected by direct NGS16S analysis for four samples (BAL 07, 11, 15 and 18; Table 2, Supplementary Table S1). An equal volume of each specimen was also plated to study culture media (BR, CNA, LKV, and SSA, see methods) and incubated anaerobically for seven days. Standard and study culture plates were washed with PBS and organisms present were identified by NGS16S analysis (Fig. 1).

Study Schematic. The constituents of each BAL sample were surveyed by standard microbiological culture (BA, CA, MAC, incubated at 37 °C for 3 days) and NGS16S sequencing of DNA extracted directly from the sample. Samples were also plated to study media (BR, CNA, LKV and SSA) and incubated anaerobically for 7 days. Individual PBS plate washes were analyzed by NGS16S sequencing.
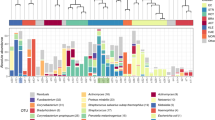
Thirty-nine clinically relevant classifications (CRCs) were identified in this study either by identification of colonies in standard clinical culture workup or by NGS16S sequencing as described in the methods (Table 3). CRCs commonly reported by standard culture of BAL samples from the general patient population were compared to our study CRCs. Six of the 10 most abundant CRCs in our study were anaerobes whose growth is unsupported by standard culture conditions. Common CRCs observed among historical specimens not recovered in our study included enteric Gram-negative rods, Enterococcus species and Pseudomonas species. This is likely due to the small sample size of our study, and sampling restricted to transplant and oncology patients.
Bacteria were defined as present in any given specimen if any one of the following criteria was met: (1) reported in standard culture; (2) read mass detected in one or more culture plate washes above filtering threshold (see methods); (3) reportable by direct NGS16S analysis. If either of the former two criteria were met, the organism was considered cultivable. Eleven of the 39 CRCs identified in this study (28%) were reported from standard culture at least once and 35 (90%) were recovered on at least one culture plate (16 from standard media and 31 on study media, Table 4). Thus, the majority of organisms identified in this study were cultivable. Twenty of the 28 CRCs identified in more than one specimen were anaerobes (Table 4), and anaerobes were the predominant classification by direct NGS16S for 7/20 samples (35%, Table 5).
A total of 188 CRC assignments were made among the 20 specimens, of which 181 (96%) were cultivable (Fig. 2, Table 4). Only 39 (21%) assignments were reported from standard culture and 79 (42%) were reportable by direct NGS16S analysis. Thus, neither method alone was able to provide a complete assessment of the organisms present in the patient samples. Sixty-two (33%) of the assignments were detected in standard culture plate washes and 157 (90%) were detected in study culture plate washes (Fig. 2, Table 4). Together, these data suggest many organisms present in these specimens either (1) have culture requirements not currently met by standard culture conditions and/or (2) were viable on current culture media but failed to be identified by standard culture work-up.

Classification assignment details. The number of CRC assignments attributed to each combination of survey methods (Fig. 1) is shown; dots below each bar indicate methods resulting in detection. Standard media =BA and/or CA; direct NGS16S = NGS16S analysis of DNA extracted directly from clinical samples; study media = BR, CNA, LKV and/or SSA. Figure was generated using UpSetR (10.1093/bioinformatics/btx364).
The CRC assignments fell into four categories based on comparisons of culture report, direct NGS16S analysis, and NGS16S plate wash analysis (Table 4). Two organisms were special cases: Aspergillus, recovered from BAL10, is not detectable by NGS16S analysis (which is restricted to bacterial 16S rRNA), and Tropheryma whipplei, detected by direct NGS analysis in BAL17, requires very specialized growth conditions. Twenty-one assignments of viridans streptococci, Staphylococcus aureus, Neisseria species and Bacillus species constituted a group for which current culture conditions were sufficient for detection and reporting.
The largest category comprised a total of 121 assignments of 26 CRCs for which current culture conditions were insufficient for detection (i.e., not reported in standard culture and/or not detected in BA or CA plate wash by NGS16S analysis). This category represents a majority (26/39) of all CRCs and 6 of the 10 most prevalent classification groups (Tables 4 and 5). Forty-four assignments in this group were reportable by NGS16S. However, the remaining assignments were of low relative abundance: 66 assignments were detected in samples below the 1% relative abundance reporting threshold, and the remaining 11 assignments were not detected by direct NGS16S in any amount (data not shown). Culture was the more reliable detection method for these low abundance assignments (Table 4).
A final category includes a total of 44 assignments of seven CRCs for which there was poor correlation between the CRCs report from standard culture and growth on standard media as measured by NGS16S plate wash analysis (Tables 4 and 6). This discrepancy indicates that organisms capable of growth under standard culture conditions may fail to be identified during culture work-up. Most of these CRCs were in low abundance (0–4.7% relative abundance by direct NGS16S analysis) from specimens containing 10 or more CRCs (Tables 4 and 6). One specimen (BAL10) was overgrown with Aspergillus, making isolation of bacterial colonies difficult. In two specimens (BAL02 and BAL14) the predominant organism was Staphylococcus aureus; the presence of a known pathogen with a distinct colony morphology may have resulted in a less rigorous examination of the plates for additional colony types. Rothia species were frequently reported by standard culture and identified from plate washes of standard culture media. However, this classification was infrequently considered reportable by direct NGS16S (Table 4). An alignment of the universal NGS16S primer sequence against the 16S sequences of Rothia mucilaginosa type strains revealed a single nucleotide mismatch, which could affect relative amplification and account for the lower than expected reporting rate of this species from direct specimens.
Standard culture returned an average of 2.1 CRCs/specimen (range 0–5, Table 2), while plate washes of standard culture media returned an average of 3.2 CRCs (range 0–10, Table 2). The fact that the unreported CRCs present on standard culture media were most frequently identified in samples of high complexity (10 or more organisms, Table 2) indicates the challenges of accurately discerning colony type subpopulations in complex polymicrobial samples on plated media, and suggests that recovery of rare organisms is reduced when more than 5 types are present. Direct NGS16S returned an average of 9.6 CRCs/specimen (range 0 to 22, Table 2) demonstrating the superior ability of this method for deconvoluting polymicrobial samples. Standard culture and direct NGS16S identified the same predominant CRC (or lack of organisms) for 10/20 samples (Table 5).
Given that we estimate 96% of reportable organisms present in samples are cultivable but only 21% are reported, standard conditions are clearly sub-optimal and could potentially be improved. Because the majority of organisms were detectable on one or more study plates, we evaluated the value of supplementing current culture conditions with a single additional medium. First, we compared the number of CRCs detected by plate wash for standard culture media with or without each study medium individually (Fig. 3). Next, we compared the relative abundance per sample of all CRCs on each media type to identify the CRCs supported by each (Fig. 4). All study media provided an increase in the number of organisms detected per specimen; BR provided the largest advantage for the greatest number of specimens (Fig. 3) and supported the greatest number of different organisms (Fig. 4). As predicted, selective media that suppressed the growth of many organisms increased the relative abundance of target organisms (for example LKV and SSA preferentially supported Prevotella species and viridans streptococci, respectively (Fig. 4, Supplementary Fig S1).

Value-added media assessment. The effect of adding a single additional study medium to current standard media for each specimen is shown. The cumulative count of CRC assignments increases as new CRCs are identified. Std = current culture media (BA + CA).

CRC heat map. Relative abundance of each classification group for each of the 20 specimens is shown by media type; relative abundance is indicated by color. DIR = NGS16S sequencing of DNA extracted directly from the sample; plate washes from media as described in Fig. 1.
Discussion
A comprehensive description of the microbial constituents of complex samples has an immediate application in the clinical management of patients when organisms with known pathological properties are identified, or when such organisms can be ruled out. In addition, a more complete profile of the bacterial population present in a specimen may provide additional information as medical knowledge advances: individual organisms or constellations of organisms may serve as biomarkers of disease15,16,17,18,19, or distinct microbial profiles may be associated with different disease states or prognoses20,21,22. While NGS and other emerging technologies can directly expand the resolution of bacterial detection and classification in clinical specimens, there are also opportunities to improve existing methodologies. This study explored possible improvements to routine clinical practices for complex polymicrobial specimens using NGS16S analysis as a benchmark. We chose BAL specimens for our study because they are both readily accessible and likely to be polymicrobial. We restricted our study to samples collected from oncology/transplant patients who are particularly susceptible to lower respiratory infections which are frequently polymicrobial and/or involve organisms not traditionally thought of as pathogens7,23,24.
In general, there was good correlation between NGS16S and standard culture results, with the same predominant organisms identified by both methods in half of the samples (Table 5). In 7 of the cases where different predominant organisms were identified, NGS16S identified anaerobes (Table 5), consistent with the fact that anaerobes are not routinely cultured for this specimen type. Prevotella and Veillonella were frequently recovered (14/20 samples, Table 3) and represented the most abundant CRC in 6/20 samples (Table 5), consistent with previous studies14,25,26,27. Anaerobes were the predominant organism in approximately one third of the samples overall (Table 5). Anaerobes may contribute to pathogenicity in the lung either directly or indirectly via the production of beta-lactamases or other secreted factors or by interactions with other members of the lung microbiota26,28,29,30,31,32,33,34,35. Anaerobes have also been recovered in high abundance in BAL from cystic fibrosis patients with high antibody titers, providing evidence that these organisms can be present in sufficient abundance and duration to trigger host immune responses29,36,37. Although not traditionally thought of as respiratory pathogens, these data suggest that anaerobes may be a significant constituent of the lung microbiome and further studies on their contribution to respiratory pathogenesis are warranted.
Although anaerobes were the organisms most frequently missed by standard culture, other cultivable organisms, many of which are members of the oral microbiota, were also overlooked (Tables 4 and 6). The clinical significance of this finding is undetermined, although underappreciated contributions to disease of “normal” microbiota, are documented in the literature32,38. The lungs are not, in fact, sterile39, and the lung microbiota most likely originates from micro-aspiration of the oral microbiota39,40. Thus, identification of such organisms is not unexpected. Although incidental contamination from oral microbiota during sample collection is possible, many recent studies indicate that this is unlikely to be a significant source of organisms identified from BAL specimens39,40,41,42,43,44. Clinical context is also an important factor when interpreting the significance of any given organism present in a sample. The list of canonical pathogens for the same specimen type may differ based on patient population, and expanding this list to include non-conventional organisms may improve patient care in some circumstances. Our study highlights the fact that cultivatable organisms, often present in major abundance, are frequently missed by standard culture and supports the idea that until a comprehensive catalog of complex samples is routinely attainable, the list of clinically relevant organisms for a sample type cannot be definitively defined. One can’t evaluate the importance of what one doesn’t know is there.
Although optimal culture conditions to recover all organisms present in a BAL is likely to be patient specific11, we identified BR as the best single value-added media, supporting the growth of the largest number of organisms for most specimens (Figs. 3–4). However, selective media was often better than BR for the recovery of certain organisms. For example, Prevotella, the second most frequently isolated CRC (Table 3), was predominant on LKV plates (Fig. 4, Supplementary Fig. S1). This illustrates a fact well known to microbiologists: no single culture medium meets all needs. By design, selective media support the growth of target organisms only; rich media supports the growth of many organisms that could outcompete slow growers or obscure the presence of small colony types, especially when in low abundance14. The physical isolation and differentiation of individual colonies on a culture plate becomes increasingly difficult as the bacterial load or the number of colony types in the sample increases6,14. This is illustrated by failure to identify all cases of currently “growable” organisms like Haemophilus which was detected by NGS16S on standard culture plates (Table 4); this phenomenon has been observed in other studies3,6,18. In addition, culture plates with a predominance of normal microbiota or a known pathogen such as Staphylococcus aureus, may be less carefully scrutinized by laboratory personnel, increasing the likelihood that rare and/or small colony types are overlooked. Our study indicates that five colony types on a single culture plate is the functional limit. Together these data suggest that while the addition of BR to routine culture set-up will broaden the spectrum of recoverable organisms for this sample type to include anaerobes, isolation of any but the most predominant colony types may still be challenging.
Although similar information can theoretically be obtained by either culture or direct NGS16S14, molecular methods allow direct identification of organisms that may require prior knowledge of specialized culture conditions, increasing the ability to detect unusual organisms. This was the case for BAL17, where Trophyrema whipplei, a highly fastidious organism that can cause acute pneumonia45,46, was detected as the predominant organism. Additionally, direct NGS16S often has a lower turnaround time than culture2, particularly for slower growing organisms or those that need subculture for biochemical testing. Although bioinformatic support is required to analyze the results of molecular testing, generating and sequencing next-generation libraries is often straightforward and requires less training than culture.
Multiple factors influence the ability of various methods to detect organisms present in a clinical sample: the overall bacterial load, the number and relative abundance of individual organisms present, as well as organism-specific growth conditions, colonial morphology and DNA extraction efficiency. Molecular methods do an excellent job of de-convoluting highly polymicrobial samples, especially when bacterial load is high. Culture was more sensitive than NGS16S for capturing low-abundance organisms, particularly when bacterial load is too low for efficient PCR amplification (Table 4), and allows incidental recovery of non-bacterial pathogens such as yeast or molds. Combining molecular and culture-dependent methods increases the sensitivity of detection compared to either method alone11,18,47. Therefore, culture and 16S sequencing should be used together for the most comprehensive evaluation of complex polymicrobial specimens27.
Methods
Sample collection
Twenty consecutive BAL samples were prospectively collected from in-house oncology or transplant patients. Participants were identified based on hospital ordering location and specimen type, without any other selection or eligibility criteria. Use of clinical microbiological specimens was approved by the University of Washington Human Subjects Review Board (approval number 42541). Specimens were fully de-identified after being aliquoted from the material submitted for clinical testing, and as such this study does not constitute human subjects research according to University of Washington Institutional Review Board criteria. All experiments were performed in accordance with relevant guidelines and regulations. A 2 mL aliquot of each BAL sample was frozen immediately after culturing and stored at −80 °C until DNA extraction.
Microbiological culture
Standard microbiological culture was performed by the University of Washington Clinical Microbiology Laboratory, as previously described48. Briefly, samples were plated on 5% sheep blood (BA), MacConkey (MAC) and chocolate (CA), agar plates (standard media), and incubated aerobically at 37 °C for 72 h. An internal review of all organisms reported from BAL by clinical NGS16S analysis in our institution identified organisms for which standard culture conditions may be inadequate; additional media was selected for ability to support the growth of these organisms (study media). Brucella agar (BR, Remel) is a general purpose anaerobic medium. Laked Sheep Blood with Kanamycin and Vancomycin Agar (LKV, Hardy Diagnostics) is used for the selective isolation of fastidious and slow growing Gram-negative obligately anaerobic bacteria. Selective Strep Agar (SSA, Hardy Diagnostics) is designed to inhibit Gram-negative bacilli and Staphylococci, thereby allowing the isolation and identification of pathogenic streptococci, including beta-hemolytic streptococci and Streptococcus pneumoniae. Columbia CNA Agar (CNA, Remel) was designed to suppress the growth of most Gram-negative bacteria, thus enriching for Gram-positive bacteria. In addition to standard media, 0.1 mL of each specimen was plated to study media and incubated anaerobically at 37 °C for seven days. Photographs were taken of all plates at the end of incubation.
DNA extraction, library preparation, and sequencing
All culture plates except MAC (regardless of visible bacterial growth) were washed with 3 mL sterile PBS and bacterial colonies were released by gently scraping agar surface with a sterile cell scraper. Bacteria from 1 mL of the resulting suspension were collected by centrifugation, resuspended in 0.2 mL MagNA Pure DNA Tissue Lysis Buffer (Roche) and stored at −80 °C until extraction. DNA was extracted from patient samples and plate washes using the QIAamp UCP Pathogen Mini Kit (Qiagen) with mechanical disruption of samples with 1.4-mm ceramic beads followed by enzymatic lysis via proteinase K. Next generation sequencing libraries were prepared and DNA sequencing was performed as previously described14. Briefly, the 16S v1–v2 region was amplified using custom primers incorporating Illumina-compatible sequencing adaptors and a sample-specific 8-bp barcode sequence; paired-end sequencing was performed on an Illumina Miseq using a 500-cycle sequencing kit (version 2) to a minimum read depth of 50,000 reads per sample. Sequence data generated for this study have been submitted to the NCBI Sequence Read Archive (SRA) under accession no. PRJNA555084.
Data analysis
NGS16S analysis of DNA extracted from patient samples (direct NGS16S) was performed by the University of Washington Clinical Microbiology Laboratory. Sequence analysis was performed without knowledge of culture results. Briefly, sample sequences were demultiplexed into paired end sequence fastq files using the Illumina on-board software with barcodes and adapters removed. Sequence variants (SVs) were generated from the paired end sequence fastq files using DADA249. SVs were identified as 16S rRNA by multiple sequence alignment using cmsearch50 using the default settings and a covariance model available from the Infernal web site (http://infernal.janelia.org). SVs were then passed through the decontam software package51 to identify and remove contaminants. To reduce the effects of possible DNA carry over between runs or samples, SVs corresponding to 100 reads or fewer in each sample were excluded. The remaining SVs were used as blast queries against a curated set of 16S rRNA records retrieved from NCBI. Alignments of at least 90% query coverage were grouped taxonomically and classified as previously described14. An Acinetobacter species SV present in 100% of samples analyzed was used as an internal standard to calculate the number of templates for each classification. All reads were classified, and classifications >1% of the total specimen read mass were considered reportable. In three cases, biologically relevant organisms were only slightly below this threshold and were also included: Actinomycyes odontolyticus was included for BAL01 and BAL02 (0.72% and 0.89% raw reads, respectively) and Solobacterium moorei for BAL13 (0.95% raw reads). Plate wash reads were processed as described above, with additional filtering steps: (1) all sequences with fewer templates than the internal standard were removed from each sample as likely reagent background, and (2) the number of templates expected to produce a visible colony was empirically determined (2500 templates) and SVs below this threshold were excluded from further analysis. Five classification assignments that were not excluded by these filtering criteria were manually excluded from analysis as contaminating DNA; these were recovered from plates without corresponding colonies and were near the filtering threshold. On average, 98% of total reads from plates with bacterial growth were analyzed (range 83–99.9%); all reads from no-growth plates were excluded, confirming that filtering was appropriately removing irrelevant sequences. To compare standard clinical lab culture and NGS16S results, organisms were manually combined into clinically relevant classifications (CRCs) at genus level or based on similar taxonomy and/or colonial morphology (e.g. viridans streptococci or coagulase negative staphylococci, etc.). Refer to Supplementary Table S1 for complete classification details.
References
Lagier, J. C. et al. Current and past strategies for bacterial culture in clinical microbiology. Clin. Microbiol. Rev. 28, 208–236, https://doi.org/10.1128/CMR.00110-14 (2015).
Dunlap, D. G. et al. Improved Detection of Culprit Pathogens by Bacterial DNA Sequencing Affects Antibiotic Management Decisions in Severe Pneumonia. Am J Case Rep 19, 1405–1409, https://doi.org/10.12659/ajcr.912055 (2018).
Hilt, E. E. et al. Urine is not sterile: use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. J Clin Microbiol 52, 871–876, https://doi.org/10.1128/jcm.02876-13 (2014).
Kommedal, Ø. et al. Massive parallel sequencing provides new perspectives on bacterial brain abscesses. J Clin Microbiol 52, 1990–1997, https://doi.org/10.1128/JCM.00346-14 (2014).
Tanaka, T. et al. A hidden pitfall in the preparation of agar media undermines microorganism cultivability. Appl Environ Microbiol 80, 7659–7666, https://doi.org/10.1128/AEM.02741-14 (2014).
Price, T. K. et al. The Clinical Urine Culture: Enhanced Techniques Improve Detection of Clinically Relevant Microorganisms. J Clin Microbiol 54, 1216–1222, https://doi.org/10.1128/jcm.00044-16 (2016).
Evans, S. E. & Ost, D. E. Pneumonia in the neutropenic cancer patient. Curr Opin Pulm Med 21, 260–271, https://doi.org/10.1097/mcp.0000000000000156 (2015).
Ajdler-Schaeffler, E. et al. Increased Pathogen Identification in Vascular Graft Infections by the Combined Use of Tissue Cultures and 16S rRNA Gene Polymerase Chain Reaction. Front Med (Lausanne) 5, 169, https://doi.org/10.3389/fmed.2018.00169 (2018).
Rhoads, D. D., Wolcott, R. D., Sun, Y. & Dowd, S. E. Comparison of culture and molecular identification of bacteria in chronic wounds. Int J Mol Sci 13, 2535–2550, https://doi.org/10.3390/ijms13032535 (2012).
Lau, J. T. et al. Capturing the diversity of the human gut microbiota through culture-enriched molecular profiling. Genome Med 8, 72, https://doi.org/10.1186/s13073-016-0327-7 (2016).
Sibley, C. D. et al. Culture enriched molecular profiling of the cystic fibrosis airway microbiome. PLoS One 6, e22702, https://doi.org/10.1371/journal.pone.0022702 (2011).
Lagier, J. C. et al. Culturing the human microbiota and culturomics. Nat Rev Microbiol, 540–550, https://doi.org/10.1038/s41579-018-0041-0 (2018).
Dione, N., Khelaifia, S., La Scola, B., Lagier, J. C. & Raoult, D. A quasi-universal medium to break the aerobic/anaerobic bacterial culture dichotomy in clinical microbiology. Clin Microbiol Infect 22, 53–58, https://doi.org/10.1016/j.cmi.2015.10.032 (2016).
Cummings, L. A. et al. Clinical Next Generation Sequencing Outperforms Standard Microbiological Culture for Characterizing Polymicrobial Samples. Clin Chem 62, 1465–1473, https://doi.org/10.1373/clinchem.2016.258806 (2016).
Mur, L. A., Huws, S. A., Cameron, S. J., Lewis, P. D. & Lewis, K. E. Lung cancer: a new frontier for microbiome research and clinical translation. Ecancermedicalscience 12, 866, https://doi.org/10.3332/ecancer.2018.866 (2018).
Lee, S. H. et al. Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung Cancer 102, 89–95, https://doi.org/10.1016/j.lungcan.2016.10.016 (2016).
Liu, Y. et al. Lung tissue microbial profile in lung cancer is distinct from emphysema. Am J Cancer Res 8, 1775–1787 (2018).
Zemanick, E. T. et al. Airway microbiota across age and disease spectrum in cystic fibrosis. Eur Respir J 50, https://doi.org/10.1183/13993003.00832-2017 (2017).
Cameron, S. J. S. et al. A pilot study using metagenomic sequencing of the sputum microbiome suggests potential bacterial biomarkers for lung cancer. PLoS One 12, e0177062, https://doi.org/10.1371/journal.pone.0177062 (2017).
Huffnagle, G. B., Dickson, R. P. & Lukacs, N. W. The respiratory tract microbiome and lung inflammation: a two-way street. Mucosal Immunol 10, 299–306, https://doi.org/10.1038/mi.2016.108 (2017).
Yamasaki, K. et al. Possible role of anaerobes in the pathogenesis of nontuberculous mycobacterial infection. Respirology 20, 758–765, https://doi.org/10.1111/resp.12536 (2015).
Kinoshita, Y., Niwa, H., Katayama, Y. & Hariu, K. Dominant obligate anaerobes revealed in lower respiratory tract infection in horses by 16S rRNA gene sequencing. J Vet Med Sci 76, 587–591 (2014).
Letourneau, A. R., Issa, N. C. & Baden, L. R. Pneumonia in the immunocompromised host. Curr Opin Pulm Med 20, 272–279, https://doi.org/10.1097/mcp.0000000000000051 (2014).
Wong, J. L. & Evans, S. E. Bacterial pneumonia in cancer patients: novel risk factors and current management. Clin Chest Med 38, 263–277, https://doi.org/10.1016/j.ccm.2016.12.005 (2017).
Howitt, S. H., Blackshaw, D., Fontaine, E., Hassan, I. & Malagon, I. Comparison of traditional microbiological culture and 16S polymerase chain reaction analyses for identification of preoperative airway colonization for patients undergoing lung resection. J Crit Care 46, 84–87, https://doi.org/10.1016/j.jcrc.2018.04.013 (2018).
Rybojad, P., Los, R., Sawicki, M., Tabarkiewicz, J. & Malm, A. Anaerobic bacteria colonizing the lower airways in lung cancer patients. Folia Histochem Cytobiol 49, 263–266 (2011).
Johansson, N. et al. The bacteriology in adult patients with pneumonia and parapneumonic effusions: increased yield with DNA sequencing method. Eur J Clin Microbiol Infect Dis, https://doi.org/10.1007/s10096-018-3426-0 (2018).
Zhou, P., Li, X., Huang, I. H. & Qi, F. Veillonella Catalase Protects the Growth of Fusobacterium nucleatum in Microaerophilic and Streptococcus gordonii-Resident Environments. Appl Environ Microbiol 83, https://doi.org/10.1128/aem.01079-17 (2017).
Ulrich, M. et al. Relative contribution of Prevotella intermedia and Pseudomonas aeruginosa to lung pathology in airways of patients with cystic fibrosis. Thorax 65, 978–984, https://doi.org/10.1136/thx.2010.137745 (2010).
Mirković, B. et al. The Role of Short-Chain Fatty Acids, Produced by Anaerobic Bacteria, in the Cystic Fibrosis Airway. Am J Respir Crit Care Med 192, 1314–1324, https://doi.org/10.1164/rccm.201505-0943OC (2015).
Sherrard, L. J. et al. Production of extended-spectrum β-lactamases and the potential indirect pathogenic role of Prevotella isolates from the cystic fibrosis respiratory microbiota. Int J Antimicrob Agents 47, 140–145, https://doi.org/10.1016/j.ijantimicag.2015.12.004 (2016).
Shinzato, T. & Saito, A. A mechanism of pathogenicity of “Streptococcus milleri group” in pulmonary infection: synergy with an anaerobe. J Med Microbiol 40, 118–123, https://doi.org/10.1099/00222615-40-2-118 (1994).
Kondo, Y. et al. Involvement of PorK, a component of the type IX secretion system, in Prevotella melaninogenica pathogenicity. Microbiol Immunol 62, 554–566, https://doi.org/10.1111/1348-0421.12638 (2018).
Pustelny, C. et al. Contribution of Veillonella parvula to Pseudomonas aeruginosa-mediated pathogenicity in a murine tumor model system. Infect Immun 83, 417–429, https://doi.org/10.1128/IAI.02234-14 (2015).
Duan, K., Dammel, C., Stein, J., Rabin, H. & Surette, M. G. Modulation of Pseudomonas aeruginosa gene expression by host microflora through interspecies communication. Mol Microbiol 50, 1477–1491 (2003).
Tunney, M. M. et al. Detection of anaerobic bacteria in high numbers in sputum from patients with cystic fibrosis. Am J Respir Crit Care Med 177, 995–1001, https://doi.org/10.1164/rccm.200708-1151OC (2008).
Worlitzsch, D. et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest 109, 317–325, https://doi.org/10.1172/JCI13870 (2002).
Maraki, S. & Papadakis, I. S. Rothia mucilaginosa pneumonia: a literature review. Infect Dis (Lond) 47, 125–129, https://doi.org/10.3109/00365548.2014.980843 (2015).
Dickson, R. P. et al. Bacterial Topography of the Healthy Human Lower Respiratory Tract. MBio 8, https://doi.org/10.1128/mBio.02287-16 (2017).
Bassis, C. M. et al. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. MBio 6, e00037, https://doi.org/10.1128/mBio.00037-15 (2015).
Dickson, R. P. et al. Changes in the lung microbiome following lung transplantation include the emergence of two distinct Pseudomonas species with distinct clinical associations. PLoS One 9, e97214, https://doi.org/10.1371/journal.pone.0097214 (2014).
Hogan, D. A. et al. Analysis of Lung Microbiota in Bronchoalveolar Lavage, Protected Brush and Sputum Samples from Subjects with Mild-To-Moderate Cystic Fibrosis Lung Disease. PLoS One 11, e0149998, https://doi.org/10.1371/journal.pone.0149998 (2016).
Segal, L. N. et al. Enrichment of lung microbiome with supraglottic taxa is associated with increased pulmonary inflammation. Microbiome 1, 19, https://doi.org/10.1186/2049-2618-1-19 (2013).
Morris, A. et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med 187, 1067–1075, https://doi.org/10.1164/rccm.201210-1913OC (2013).
Lagier, J. C. et al. Tropheryma whipplei DNA in bronchoalveolar lavage samples: a case control study. Clin Microbiol Infect 22, 875–879, https://doi.org/10.1016/j.cmi.2016.07.010 (2016).
Fenollar, F. et al. First isolation of Tropheryma whipplei from bronchoalveolar fluid and clinical implications. J Infect 65, 275–278, https://doi.org/10.1016/j.jinf.2011.11.026 (2012).
Pearce, M. M. et al. The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. MBio 5, e01283–01214, https://doi.org/10.1128/mBio.01283-14 (2014).
Salipante, S. J. et al. Rapid 16S rRNA next-generation sequencing of polymicrobial clinical samples for diagnosis of complex bacterial infections. PLoS One 8, e65226, https://doi.org/10.1371/journal.pone.0065226 (2013).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583, https://doi.org/10.1038/nmeth.3869 (2016).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A. & Callahan, B. J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226, https://doi.org/10.1186/s40168-018-0605-2 (2018).
Acknowledgements
The authors would like to thank the University of Washington Medical Center Clinical Microbiology Laboratory for their co-operation and assistance.
Author information
Authors and Affiliations
Contributions
L.A.C. and B.T.C. designed the study with input from S.J.S. and N.G.H.; L.A.C. performed all data collection and wrote the manuscript. D.R.H., S.L. R.-B., C.A.R. and N.G.H. performed data analysis. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cummings, L.A., Hoogestraat, D.R., Rassoulian-Barrett, S.L. et al. Comprehensive evaluation of complex polymicrobial specimens using next generation sequencing and standard microbiological culture. Sci Rep 10, 5446 (2020). https://doi.org/10.1038/s41598-020-62424-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-62424-x
This article is cited by
-
In vitro virulence activity of Pseudomonas aeruginosa, enhanced by either Acinetobacter baumannii or Enterococcus faecium through the polymicrobial interactions
Archives of Microbiology (2022)
-
A prevalent and culturable microbiota links ecological balance to clinical stability of the human lung after transplantation
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
