
Abstract
Amphibians evolved in the Devonian period about 400 Mya and represent a transition step in tetrapod evolution. Among amphibians, high-throughput sequencing data are very limited for Caudata, due to their largest genome sizes among terrestrial vertebrates. In this paper we present the transcriptome from the fire bellied newt Cynops orientalis. Data here presented display a high level of completeness, comparable to the fully sequenced genomes available from other amphibians. Moreover, this work focused on genes involved in gametogenesis and sexual development. Surprisingly, the gsdf gene was identified for the first time in a tetrapod species, so far known only from bony fish and basal sarcopterygians. Our analysis failed to isolate fgf24 and foxl3, supporting the possible loss of both genes in the common ancestor of Rhipidistians. In Cynops, the expression analysis of genes described to be sex-related in vertebrates singled out an expected functional role for some genes, while others displayed an unforeseen behavior, confirming the high variability of the sex-related pathway in vertebrates.
Similar content being viewed by others

Introduction
Amphibians represent a group of vertebrates containing over 7,100 species worldwide1. These organisms evolved in the Devonian period about 400 Mya and represent a transition step in tetrapod evolution2. Modern-day amphibians have diverged into three orders with distinct anatomical features: Gymnophiona (caecilians, limbless amphibians), Caudata (salamanders and tritons), and Anura (frogs and toads). Amphibians have many features that make them ideal as animal models because of their physiology, diversity, phylogeny, and amazing capability of tissue regeneration. On the other hand, amphibians represent a case of global biodiversity crisis since they are among the most threatened species in the world3.
The Chinese fire-bellied newt, Cynops orientalis (David, 1873), is a small (6–10.3 cm long) urodele (Salamandridae: Pleurodelinae) distributed in Asia. It shows a color ranging from dark brown to black on the back and with belly from orange to scarlet red with numerous black spots. The sexual dimorphism is evident in size (males are smaller than females) and shape of tail and cloaca. C. orientalis lives in ponds and marshes and prefers cold and calm waters with a bottom of mud. In nature, in the Chinese region of Changsha, the reproduction period spans from March to July, when water temperatures range from 15 to 23 °C, and juveniles reach sexual maturity from 1 to 3 years.
Data obtained by whole genome sequencing (WGS) are a very valuable resource to understand the evolutionary history and biology of species. Despite the importance of amphibians, the genomes of only 13 species have been fully sequenced as of January 2020. The genomes of newts contain a large proportion of repetitive sequence and are the largest ones among terrestrial vertebrates4. These factors currently represent a significant challenge for whole genome assembly5, in particular due to the high computational resources required for handling the large amount of input sequencing data and the technical limitations of most assembly algorithms, optimized for genomes with a size comparable with human or smaller. The only exception was the axolotl genome (~32 Gb)6, which has been produced through a huge joint effort and at high costs, together with the development of ad hoc bioinformatics tools and computational strategies to manage the unusual size of the assembly.
RNA sequencing technologies represent a cost-effective tool for large-scale transcriptome profiling that allows the rapid and complete elucidation of transcript sequence information in a specific tissue, developmental stage or physiological condition. Currently, most of published works in urodeles are focused on limb regeneration7 and very few from the Pleurodelinae8,9,10. Data on the expression and function of genes participating in sexual development and gametogenesis in urodeles and in general in amphibians are very limited11,12,13,14,15,16,17,18. The investigation of these processes in this taxonomic group is extremely interesting for the variety of mechanisms involved in sexual development present in Anura and Caudata and for their phylogenetic position. In general, the fate of the undifferentiated gonad towards male or female development is determined by genetic or environmental mechanisms. However, a great number of studies provides increasing evidence that the master sex determination gene and genes involved in the downstream sex differentiation network are extremely variable even between closely related species19,20. Thus, knowledge on newts can provide insights on the gene network acting in these organisms and its evolution. Indeed, salamanders belong to Caudata for which little information is available compared to the sister group Anura.
In this paper we report the assembly of a Cynops orientalis transcriptome, which is characterized by a high level of completeness and provides one of the most comprehensive molecular resources for amphibians. Moreover, transcriptome data obtained from testes and ovaries were exploited to investigate genes involved in gametogenesis and sexual development. We report for the first time the identification of the important sex determining gsdf gene in tetrapods, so far known only from bony fish, lungfish and coelacanth. In particular, our data represent a useful tool for future studies involving gene expression experiments, comparative transcriptomics, genomics, and metabolomics in amphibians.
Results and Discussion
Sequencing, de novo assembly, and annotation
Upon quality trimming, the total sequencing output was ~422.5 million paired-end reads obtained from male and female gonads, and female liver in three biological replicates (Supplementary Table S1). The non-redundant transcriptome assembly obtained from these three tissues comprised 45,831 contigs, whose size ranged from 201 to 22,390 base pairs, with a mean length of 1,594 bp and N50 length of 3,475 bp, accounting for a total assembly size slightly higher than 78 Mb.
The reference transcriptome displayed a high level of completeness, with just 6.2% tetrapod BUSCOs being absent. At the same time, it contained a very high fraction of contigs containing complete open reading frames, as revealed by the detection of 89.7% complete and just 4.1% fragmented tetrapod BUSCOs. Overall, the quality of the C. orientalis transcriptome, both in terms of number of orthologs present and in terms of complete BUSCOs identified, was significantly higher than most available amphibian reference transcriptomes6,21,22,23. Completeness and fragmentation rates were comparable with the outcome of the most recent high-depth approaches targeting multiple tissues24,25,26 (Fig. 1, Supplementary Table S2).

Comparative qualitative assessment of selected genomic and transcriptomic resources currently available for Amphibia, based on the evaluation of complete, fragmented and missing metazoan BUSCOs from OrthoDB v.9. G: genome; T: transcriptome.
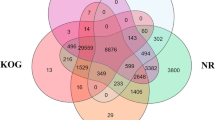
A total of 19,632 contigs contained an ORF longer than 100 codons, and this number closely matched the estimates of the full complement of protein-coding genes of diploid tetrapods (e.g. 20,454 in human, 18,442 in Xenopus tropicalis and 18,595 in the Anolis carolinensis). Overall, 19,961 contigs obtained a positive BLAST hit, which resulted in the annotation of 41.61%, 38.76% and 31.20% sequences with the Gene Ontology, KEGG and eggNOG resources, respectively. The inspection of the most abundant Pfam domains and Gene Ontology terms (Fig. 2) confirmed the comparability of the assembled transcriptome with the complete repertoire of protein-coding genes encoded by tetrapod genomes. Indeed, the most represented annotations closely mirrored the distribution observed in other Amphibia, as exemplified by the Xenopus tropicalis genome27, with a widespread representation of domains with structural function (e.g. ankyrin repeats, WD40 repeats), marked protein-protein interaction properties (e.g. immunoglobulin and fibronectin type III domains) or ion-binding potential (e.g. C2H2 zinc finger motifs). In light of these observations, the C. orientalis transcript collection is one of the most complete available mRNA collections for amphibians and provides a reference for comparative genomics studies of comparable quality to the fully sequenced amphibian genomes of Xenopus laevis28 and Nanorana parkeri29, and just slightly lower than Xenopus tropicalis, Microcaecilia unicolor and Rhinatrema bivittatum27. Newts (Salamandridae, Pleurodelinae) have very large genomes even if compared with amphibian standards (usually > 20 Gb)30. In particular, the nuclear DNA content of C. orientalis has been previously estimated at 44.27 Gb, exceeding the size of axolotl (32 Gb), making the attainment of a complete genome assembly for this species unlikely in the foreseeable future due to the high sequencing costs and computational requirements that would be required to achieve this task.

Top 20 most abundant PFAM domains (panel a), Gene Ontology cellular component terms (panel b), molecular function terms (panel c) and biological process terms (panel d) in the C. orientalis transcriptome.
The Chinese fire-bellied newt transcriptome, with its high level of completeness and high proportion of contigs containing complete open reading frames, most certainly meets the requirements that might potentially allow its use as a temporary genome substitute until further technological advancements will finally allow to obtain a complete reference genome sequence.
While a few other Pleurodelinae transcriptomic studies have been previously published or are currently in progress8,9, the resources available for Cynops spp. are still limited and no high-throughput data is available for C. orientalis. To date, the phylogenetically closest species, which has been the subject of RNA-sequencing is the congeneric Japanese fire-berry newt Cynops pyrrhogaster10. However, this study has been targeted on eyeballs, to study the retinal regeneration process, and therefore it does not provide the opportunity to investigate gene expression profiles from multiple tissues.
General overview on gene expression profiles
The C. orientalis liver, male and female gonads displayed highly distinct gene expression profiles (Fig. 3), which is consistent with the highly specialized function that these organs carry out in newt biology and with observations previously collected in other tetrapods31,32. The three biological replicates did not display any significant variation in pairwise comparisons, as revealed by both heat map (Fig. 3) and PCA graphical representations (Supplementary Fig. S1).

Heat map, providing an overview on gene expression profiles of the nine newt samples. Log10 TPM gene expression values were hierarchically clustered, based on an average method and correlation-based dissimilarity matrix. Only transcripts achieving TPM values > = 200 in at least in one sample are shown.
As expected, the liver, a tissue extensively involved in biosynthetic processes, displayed the lowest diversity of expressed mRNAs. The three samples were characterized by a low Transcriptomic Diversity Index (TDI), between 1.98 and 2.1733, with about 50% of the global transcriptional effort directed towards the production of ~100 highly expressed mRNAs. On the other hand, a broader range of transcripts were expressed in male and female gonads, with TDIs in the range of 2.76–3.24 and 3.45–4.39, respectively (Supplementary Fig. S2). These values to some extent explain the very high completeness of the transcriptome assembly, in spite of its construction from just three adult tissues. The rich transcriptomic landscape of testis could be related to the increased accessibility of chromatin to transcription factors in meiotic and early haploid spermatogenic cells34, which results in promiscuous transcription35,36,37.
Compared with testis, female gonads displayed an even richer transcriptional pattern. This observation might be more intimately linked to the function of amphibian oocytes and with the extensive presence of lampbrush chromosomes in these cells38. Widespread transcription of a broad range of mRNA types, which appear to be up-regulated compared with somatic cells, has been previously reported in many amphibian species. In Xenopus, this high transcriptional activity has been hypothesized to play an important role in preparation for the rapid differentiation of cell lines in early embryonic stages39.
Analysis of gonad transcriptomes
Histological analysis of the gonads confirmed the sex and the gonadal maturation stage of the animals (Fig. 4). Testes had the typical lobular structure of salamanders, and each lobe contained several cysts with spermatogenic cells in different stages of development. The gonads of all male individuals presented a high number of spermatozoa, the cysts displaying a cephalo-caudal gradient with respect to the stage of spermatogenesis40. The ovaries of all females contained oocytes at different stages of oogenesis, including vitellogenic and highly pigmented oocytes41.

Morphology of C. orientalis gonads. Testes of all males (MG1, MG2 and MG3) are mature containing high numbers of spermatozoa. The spermatogonia are located in a more cephalic position and confined to cysts. Scale bars = 100 µm. All female gonads (FG1, FG2 and FG3) are mature presenting germ cells in advanced oogenesis stages. Lumen (L), ovarian cavity (Oc), pre-vitellogenic (Po) oocyte, spermatogonia (arrow), spermatozoa (Sz), and vitellogenic oocyte (Vo). Scale bar = 400 µm.
The most highly expressed genes in female gonads were histones, which cumulatively accounted for ~15% of the transcriptome of this tissue and displayed expression levels >1,000 folds higher than testis (Supplementary Table S3). The high expression levels of many histone types (e.g. H1, H2A, H2B, H3 and H4) is in line with the significant stage-dependent histone modifications occurring during oocyte development in many tetrapods, including amphibians42 and mammals43. Some of the histone isoforms identified closely matched the Xenopus H2AX variants, which have been previously shown to be highly enriched in oocytes and early embryos44, the human oocyte-specific histone H145, and accessory histone linker proteins such as the Xenopus B4 protein46. Due to the accepted role of oocyte-specific histones in maintaining genome stability in the female germ line42,43, these nuclear proteins might hold a particularly important role in organisms with a genome whose size exceeds by >10 folds the average size of mammals.
Other genes abundantly expressed in female gonads included several molecular players involved in the regulation of meiosis and cell cycle (e.g. cyclins and geminin47, as well as in DNA synthesis (e.g. the ribonucleotide reductases), oocyte maturation and early embryonic development. With this regard, the strong expression of perilipins (>1,500 TPM) and glycogenin-1 (>1,000 TPM) is consistent with the important role of these proteins in the formation of oocyte lipid droplets48,49 and glycogen storage50, respectively. The strong female specificity of Cyclin A1 (expression level >1,000 TPM, nearly 3,000 folds higher than testis) is noteworthy, since it displayed a strikingly opposite trend compared to mammals. In human testis cyclin A1 is a master regulator of gametogenesis; and it is only expressed in female gonads in cases of ovarian cancer51.
Male gonads displayed a generalized lack of dominant tissue-specific transcripts, exception made for protamines. However, these were probably not identified due to their extreme low complexity. The majority of the most highly expressed genes were not directly linked with gametogenesis, being rather involved in housekeeping functions (e.g. ribosomal and cytoskeletal proteins), which were usually found expressed at levels 5–10 times higher than ovary (Supplementary Table S4). There were, however, some key exceptions, such as high expression of ornithine decarboxylase antizyme 3, which is linked with polyamine biosynthesis, a fundamental process to ensure sperm motility52. The dual specificity phosphatase DUSP13, which shows skeletal muscle and testis-specificity in human (based on GTEx data, https://www.gtexportal.org/home/) also displayed an expression level >1,000 folds higher in testis than in ovary. Similarly, the tubulin polymerization promoting protein TPPP2, which is expressed in a testis-specific manner in humans, showed this feature also in the newt. The biological significance of other “out of place” transcripts, expressed at much higher levels in testis compared with ovary remains to be fully elucidated.
Genes potentially involved in gonad sex differentiation and gametogenesis in Cynops orientalis: expression analysis and evolutionary considerations
The set of genes involved in sex differentiation and gametogenesis analyzed previously in basal sarcopterygians53 was investigated also in C. orientalis to increase knowledge in early tetrapods. This analysis performed on gonad transcriptomes allowed to retrieve 38 transcripts annotated from genes involved in sexual development (Table 1), which show complete CDSs except for 6 transcripts (Supplementary Table S5).
Evaluation of the expression profiles in gonads revealed 14 genes that are differentially expressed between the two sexes and may play important roles in sex differentiation, gonad maintenance, and gametogenesis (Fig. 5).

Expression levels of gametogenesis and sex-differentiation related genes in C. orientalis gonad transcriptomes. (a) Growth factor and receptor genes I. (b) Growth factor and receptor genes II. (c) Steroidogenic enzyme genes. (d) Transcription factor genes. (e) Meiosis regulation genes. (f) Follistatin and β-catenin. (g) Steroid hormone receptor genes.
The Anti-Müllerian hormone (amh) encoded protein is involved in the regression of the Müllerian ducts, the ontogenetic precursors of the female reproductive tract, in most vertebrates, and acts with its receptor Amhr2 in triggering the development of gonadal tissue54. Besides the function in duct regression, this gene plays a role in spermatogenesis55,56. Among vertebrates, expression of this gene is variable in relation to sex and time of onset. In Xenopus laevis the amh expression has been more readily detectable in mature testes than in undifferentiated gonads57. In C. orientalis the high expression of amh preferentially in males (Fig. 5a) is in line with its known involvement in proliferation and differentiation of spermatogonia55. This observation is in agreement with our histological analyses showing the presence of spermatocytes in seminiferous tubules (Fig. 4). Moreover, the same expression pattern that we find in the Chinese fire-bellied newt has been reported for other vertebrate species such as lungfish and teleosts53. The lack of identification of an orthologous amhr2 sequence in Cynops is likely attributable to absence of transcripts and not a gene loss event since this gene has been found in other amphibians58,59.
Gonadal Soma-Derived Factor (gsdf), as amh, is a member of the TGF-β superfamily and is involved in testis development in fish60. Moreover, in some teleosts it has a role as male sex initiator61 or as master sex determining gene60,62. We have previously reported the presence of an orthologous sequence in the cartilaginous fish Callorhinchus milii and in the two basal sarcopterygian fish, Latimeria menadoensis and Protopterus annectens53,63. The analysis of C. orientalis transcriptome identified a bona fide gsdf sequence (Supplementary Fig. S3) whose orthology was confirmed by phylogenetic analysis (Fig. 6a). This finding represents the first report of a gsdf gene in a tetrapod species (Fig. 6b). The presence of this gene was also investigated in the genomes of Xenopus and Microcaecilia unicolor, belonging to Anura and Gymnophiona, respectively. The identification only in caecilian species (orthology confirmed by phylogenetic analysis), besides salamander, and the absence in reptiles, birds, and mammals indicate that gsdf was lost at least twice in the evolutionary history of tetrapods: in the Anuran lineage and in the common ancestor of Amniota. The expression analysis revealed a male biased sexually dimorphic pattern (Fig. 5a), although with values lower than those identified in basal sarcopterygian fish and teleosts53.

Phylogenetic attribution of GSDF amino acid sequences. (a) Phylogenetic tree of GSDF, AMH and Inhibin α amino acid sequences. Bayesian inference: 1,000,000 generations, sampling every 100, Jones substitution model, stationarity defined as when the average standard deviation of split frequencies approaching 0.0075, burn-in set to 2,500, midpoint rooting. In bold two amphibian GSDF sequences are highlighted. (b) Schematic representation of gsdf evolutionary history in gnathostomes. Red cross indicates gene loss.
Desert hedgehog (dhh) is a member of the Hedgehog gene family, which in vertebrates includes two other homologues, sonic hedgehog (shh) and indian hedgehog (ihh)64. Hedgehog genes encode secreted proteins that influence the growth of several tissues during development. In humans and mice it has been showed that the lack of dhh expression causes infertility and testicular defects65. The activity of this gene persists also into the adult controlling the maintenance of male spermatogenesis and the germline66. Recently, among mammals also a role of DHH in ovarian development in marsupials has been proposed67. The expression pattern observed in C. orientalis (Fig. 5b) is in line with reports in another amphibian, X. laevis13, and confirms a conserved role in male gonad function.
Genes encoding members of the fibroblast growth factors (FGF) and wingless-related MMTV integration site (WNT) families direct the development of several organs. Loss of fgf9 leads to male-to-female sex reversal68, while loss of wnt4 results in partial testis development in XX mice69. In mouse male gonad, the sex determination gene Sry up-regulates fgf9 and represses wnt4. These data suggested that the antagonism between Fgf9 and Wnt4 controls the sex fate of the gonad70. Unexpectedly, fgf9 showed sex bias towards female in Cynops (Fig. 5a), like in lungfish. Similarly, fgf9 was much higher in ovaries than testes of Rana rugosa. In this species, fgf9 was expressed in early gonads of both male and female individuals, displaying no sexual dimorphism at this stage71. In Xenopus laevis, on the other hand, it showed sexual dimorphism towards male around metamorphosis12. Hence, a crucial role of fgf9 for testis development in amphibians still needs to be further investigated.
Regarding the wnt4 gene, the expected female biased expression was observed in Cynops (Fig. 5a). In mammals, WNT4 and RSPO1 through β-catenin stabilization (ctnnb1 gene) lead to female gonadal fate72. Similar stabilization of β-catenin was proposed for X. laevis, where wnt4 and rspo1 were highly expressed in developing ovaries12. However, in Cynops, no difference in expression levels was observed for rspo1 between ovary and testis, while a strong sex-bias towards male was observed for ctnnb1 (Fig. 5f). The correlation between rspo1, wnt4 and ctnnb1 for ovary development seems to be not conserved in vertebrate evolution.
Members of the platelet-derived growth factor (pdgf) family play fundamental roles during several stages of vertebrate development73. In mammals, birds, and reptiles pdgfs and their receptors are involved in testis formation, inducing migration of cells from the mesonephros into the developing gonad74,75. Moreover, analysis of pdgfa-null mice provided evidence for the role of PDGFA and its receptor in Leydig cell development during fetal and adult stage, while pdgfb or pdgfrb knockout led to death during late gestation76. In X. laevis the upregulation of pdgfa and pdgfb genes in testis development supported involvement of the PDGF pathway in testicular organogenesis in anurans12. The high expression of pdgfa and its receptor in Cynops male gonads (Fig. 5b) suggests that the role of this gene in testis could be common to urodeles, not only in early developmental stages, but probably also later in gonad function.
Stroma-cell-derived factor 1 (sdf1) is a member of the CXC-chemokine subfamily and is a physiological ligand for CXCR4, a member of the CXCR subfamily. SDF-1/CXCR4 chemokine signaling, is involved in proper directional migration and survival of primordial germ cells and this has been demonstrated also in X. laevis14. Moreover, recent studies have shown that SDF1 is responsible also for the postnatal maintenance of the spermatogonial stem cells in mouse testis interacting with an alternative receptor, CXCR777. The higher expression of this gene compared to cxcr4 also in Cynops suggests a similar function of SDF1 (Fig. 5b).
Estradiol and dihydrotestosterone (DHT) are steroid hormones that regulate vertebrate reproduction and are synthetized from testosterone. The srd5a1-3 genes encode enzymes involved in the conversion of testosterone to DHT, the most potent androgen in tetrapods, while cytochrome P450 19A1 (cyp19a1 or aromatase) converts testosterone to estradiol. These key enzymes were identified in the transcriptomes of C. orientalis and their expression was analyzed. Steroid-5-Alpha-Reductase Alpha Polypeptides 1 (srd5a1) and 3 (srd5a3) are preferentially expressed in ovaries as well as aromatase, although at lower levels (Fig. 5c). The sexual dimorphic expression of srd5a1 and srd5a3 in C. orientalis gonads is in agreement with previous reports from lungfish53, frogs78, rat79, human80,81, and teleosts78. The expression in ovaries of srd5a1 and srd5a3 implicates the potential production of DHT whose role in amphibian gonads still need to be clarified. However, in females it is known that androgens control reproduction stimulating reproductive events in the ovary. Indeed, it has been proposed that DHT has estrogen-like properties inducing vitellogenin synthesis in hepatocytes and 17β-estradiol production in oocytes78.
Regarding the steroid hormone receptor genes, androgen receptor (ar) is mainly expressed in testis (Fig. 5g) indicating androgen signaling, essential for maintenance of spermatogenesis82. Moreover, in the Japanese frog Rana rugosa AR with its ligand functions has been proposed as male sex-determinant in this amphibian species83. The expression of estrogen receptors (esrs) in male gonads is intriguing despite the absence of expression of aromatase in this tissue (Fig. 5g). This could be due to a delay in expression of esrs compared to that of aromatase. The presence of esr transcripts in males has been observed also in Pleurodeles waltl84 and in the newt Triturus marmoratus85 where a role of estrogens in spermatogenesis has been demonstrated.
Foxl2 (forkhead box L2) is a gene encoding a transcription factor, which in several species of vertebrates including amphibians has been shown to be important for ovary development and for maintaining its fate12,86. Studies demonstrated that FOXL2 acts on the cyp19a1 (aromatase) promoter, leading to synthesis of estrogens87. Despite its low expression in adult gonads of Cynops, foxl2 presented the expected female bias, confirming a conserved role of this gene (Fig. 5d).
Doublesex- and mab-3-related transcriptional factor 1 (dmrt1) is implicated in male sex determination and testis development in diverse metazoan phyla88,89. We found a strongly biased expression of dmrt1 in C. orientalis male gonads in accordance with the expression profile of dmrt1 in most other species53,63,90. As previously reported in basal sarcopterygians also in the newt expression of dmrt3 was not detected (Fig. 6). In mammals, DMRT6 has a role in gametogenic programs coordinating the transition from spermatogonial development to meiosis91. The absence of expression of dmrt6 gene in Cynops does not support a similar role in salamanders (Fig. 5d).
The steroidogenic factor 1 (sf1) and the Wilms tumor 1 gene (wt1) are essential for the differentiation of adrenal glands and gonad development but also for secondary sexual characteristics92. The sf1 gene shows sexually dimorphic expression with higher values in male mouse93, pig94, and in turtles95, while in chicken96 and alligators97 it is higher in developing female gonads. In the American bullfrog Rana catesbeiana sf1 expression decreases with testes formation while in females it increases during ovary development98. In Cynops the expression profile of sf1 is similar to that seen in mammals and turtles and thus opposite to that observed in frogs. Similarly, wt1 presented a strong male bias in Cynops (Fig. 5d). In the frog Rana rugosa, however, no sexual dimorphism of wt1 expression was observed at early stages, but a male strong sex bias was seen in adult gonads71. Hence, the wt1 expression profile is similar in adult vertebrates, indicating a conserved role of this gene throughout evolution.
Transcription factors of the SOX (Sry-type HMG box) gene family are historically known to be important in testicular development and fertility in mammals99. This family contains the HMG box DNA binding domain, which is closely related to that of Sry, the master male sex determination gene in eutherian mammals100. During the sex determination period, Sry is expressed in the somatic gonad of XY individuals, upregulating sox9 and leading to testis formation101. Sox8 was shown to be involved in reinforcing sox9 function and even substituting its role102. In Cynops, both sox genes are weakly expressed in adult gonads of both sexes, with sox9 presenting the expected sex-bias towards males, while sox8 shows similar expression in ovary and testis (Fig. 5d). To date, a conserved role in other vertebrate outside mammals of sox9 as the main actor of testis development is controversial. In some teleosts it was shown to function much more downstream in the sex determination regulatory network103. Expression analyses from Xenopus laevis demonstrate that sox9, together with other male related genes, is upregulated in the developing testis12. Further studies are needed to elucidate a functional role of the sox8 and 9 genes in amphibians.
Other members of the SOX family have been shown to be important in gonad development in vertebrates. Sox5 is highly expressed during spermatogenesis and in fetal gonads of mice, but the exact role is still unknown104,105. Studies in the Japanese medaka, Oryzias latipes, showed that sox5 has a role in regulating germ cell number during sex determination and disruption of this gene leads to XX female-to-male sex reversal106. Interestingly, different from what has been observed in most vertebrates, the expression of sox5 in Cynops was higher in ovary than testis (Fig. 5d), following the pattern observed only in medaka so far53,106.
Sox3 is located on the X chromosome of mammals, and the common notion is that this gene is the precursor of Sry. Sox3 is highly expressed in gonads of mammals, and sox3 ko mice have a disrupted gametogenesis with gonad dysgenesis, but no effect on sex determination was observed107. Notably, in Oryzias dancena sox3 is the Y-linked male sex determination gene, pointing to the potential of this gene to be a master sex regulator108. In Cynops, no significant expression was observed in the gonad of both sexes, similar to other vertebrates, except mammals (Fig. 5d).
In reproduction gametogenesis is one of the most important processes and the morphogen retinoic acid (RA) plays a pivotal role in the regulation of meiosis in mammals109,110, birds111 and urodele amphibians112. The retinaldehyde dehydrogenase (ALDH1A1, ALDH1A2 and ALDH1A3) and the cytochrome P450 26 proteins (CYP26A1, CYP26B1 and CYP26C1) are the key enzymes in RA synthesis and degradation. RA upregulates the stimulated by retinoic acid gene 8 (stra8) required for initiation of meiosis113, although stra8 is absent in several teleost lineages114. In Cynops, no expression of cyp26a1 and cyp26b1 was detected, while aldh1a1, aldh1a2, and stra8 were sexually dimorphic expressed. In particular, these genes show a higher expression in testes than ovaries (Fig. 5e). The expression of stra8 in female gonads suggests that other enzymes are involved in RA synthesis in the ovary of Cynops. The low expression of cyp26a1 is in agreement with its role as meiosis-inhibiting factor109. The cyp26c1 gene has not been identified in the salamander transcriptome, however, given its presence in Xenopus this may be attributable to an absence of expression or a lineage specific gene loss event.
Conclusions
Our analyses demonstrated a high level of completeness of the Cynops orientalis transcriptome, comparable to the in-silico transcriptomes of the fully sequenced genomes available for some amphibians. The huge genome size of C. orientalis estimated at 44.27 Gb represents a significant obstacle for the attainment of a complete genome assembly in the foreseeable future. Therefore, the mRNA collection obtained for the Chinese fire-bellied newt is a valid alternative molecular resource useful for gene expression studies, comparative transcriptomics and large-scale evolutionary analyses.
The analysis of a selected gene set involved in sexual differentiation and gametogenesis performed on the transcriptomes of female and male gonads provided information on gene network evolution and function. Highly interesting is the identification of the gsdf gene in a tetrapod species, so far known only from bony fish and basal sarcopterygians. This finding allows to estimate the dating of the loss of this gene later in the evolution of tetrapods (Fig. 7). Our analysis did not uncover transcripts of fgf24 and foxl3 supporting the possible loss of both genes in the common ancestor of Rhipidistians (Fig. 7).

Summarizing scheme of analyzed sexual development genes in sarcopterygians. – indicates gene loss and ✓ indicates gene presence in sarcopterygians. *Indicates that data in lungfish are referred to transcriptomic resource.
Although some genes, such as the growth factors amh and gsdf or the transcription factors dmrt1 and wt1, showed the expected expression pattern, which suggests a conserved functional role also in Cynops, others displayed a unexpected diverging expression profile confirming the high variability of the sex-related pathways in vertebrates. This motivates and further in depth studies on those genes in order to elucidate the functional role in amphibians.
Material and methods
Specimens of C. orientalis were obtained from a local dealer during the reproductive season. Three females and three males were anesthetized with MS222 at 2 g/l and sacrificed. All experimental procedures were approved by the Italian ethical committee Ministero della Salute (authorization n° 2E1BD.N.LYB) and all methods were performed in accordance with the relevant guidelines and regulations. Female livers, testes, and ovaries were dissected. Total RNA was extracted and used for sequencing using methodologies described in Biscotti et al.31. Pieces of the same gonads used for RNA-seq were fixed in 4% formaldehyde solution for 24 h at 4 °C. The samples were then dehydrated, embedded in paraffin, and sectioned at 5 µm thickness. The sections were counterstained with hematoxylin and eosin (HE) method.
De novo transcriptome assembly and annotation
Raw paired-end reads were subjected to a trimming procedure with Trimmomatic v.0.39115. In detail, trimming was performed by removing Illumina sequencing adapters with the Illuminaclip setting, and low quality bases were detected and removed based on a sliding window approach (size = 4, minimum average quality = 15), also selecting the leading and trailing options to remove low quality read ends. Trimmed read, deposited in the NCBI SRA database under the umbrella BioProject ID PRJNA574599, were then used as an input for a de novo assembly with Trinity v.2.8.3, using default parameters and a minimum allowed contig length of 200 nucleotides116. The complexity of the assembled transcriptome was reduced with the specific aim to remove alternatively spliced isoforms and to build a non-redundant reference sequence database for gene expression studies. This was achieved with EvidentialGene (http://arthropods.eugenes.org/EvidentialGene/), recovering all the contig parts of the okayset and the longest isoform for each of the gene models included in the dropset. In addition, contigs likely to have been originated by fragmentation of longer, scarcely expressed mRNAs, contamination from exogenous sources of DNA or RNA, or by pervasive intergenic transcription, were removed following the procedure described elsewhere31,33,117. Contigs derived from mitochondrial DNA were detected with BLASTN118, based on significant similarity (e-value threshold = 1E-5) against the reference sequences (Genbank IDs: KY399474.1), and removed from the assembly.
The overall quality of the reference transcriptome assembly was evaluated with BUSCO v.3119. The analysis of the Benchmarking single-copy Universally Conserved Orthologs allowed the computation of completeness, duplication and fragmentation rates, with reference to the Tetrapoda OrthoDB v.9 database120. The Transcriptomic Diversity Index (TDI) for each sample was calculated as described in a previous publication33.
Functional annotation was carried out with Trinotate v.3.0.2121. All contigs were translated to putative protein sequences using TransDecoder v.3.0.1 and annotated based on significant BLASTX and BLASTP matches (Altschul et al., 1990118) in the UniProtKB sequence database122, based on an e-value threshold of 1E-5. These homologies were used to associate each contig to cell component, molecular function and biological process Gene Ontology terms123, to eggNOG124 and KEGG125 annotations. Protein sequences were also analyzed with Hmmer v.3.2.1126, searching for conserved domains included in the Pfam 31.0 database127.
Identification and analysis of candidate sexual development genes
Sequences corresponding to key gametogenesis and sexual development-related genes were obtained by tBLASTN sequence homology searches performed against the C. orientalis transcriptomes. The retrieved transcripts were translated using Sequence Translation (https://www.ebi.ac.uk/Tools/st/) and UTRs and CDSs were identified. The orthology of each transcript was confirmed using NCBI BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The sequences have been deposited in GenBank under the accession numbers (MN923216-MN923253).
Orthologous sequences used in the phylogenetic analyses were retrieved from GenBank (http://www.ncbi.nlm.nih.gov/) or ENSEMBL (http://www.ensembl.org/index.html) (for accession numbers see Supplementary Table S6). Multiple alignment of the amino acid sequences was obtained with Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) using default parameters.
Phylogenetic analyses were performed by Bayesian inference using MrBayes (version 3.2)128. Substitution models as defined by posterior probabilities, stationarity, generations, sampling, burnin, and specific tree building parameters and rooting details are specified in the figure legend description.
Gene expression analysis
Trimmed reads for each of the nine biological samples analyzed were imported in the CLC Genomics Workbench v.12 environment (Qiagen, Hilden, Germany), and mapped to the non-redundant reference transcriptome. The alignment between the reads and the reference transcriptome was performed with the RNA-seq mapping tool, setting the length and similarity fraction parameters set to 0.75 and 0.98, respectively. Gene expression levels were computed as Transcripts Per Million (TPM)129, as this metric allows to efficiently compare gene expression levels both within and between samples.
The correct reconstruction of the complete open reading frame of the mRNAs of interest was checked on a case-by-case basis by visually inspecting the uniform coverage by mapped reads. Whenever necessary, the most representative (i.e. most highly expressed) isoform was recovered from the complete redundant transcriptome assembly.
Male and female gonad-specific genes were identified though a statistical analysis based on a negative binomial Generalized Linear Model (GLM)130, which considered in the comparison the three biological replicates for the two samples. The thresholds used for detection of significant differences between the two sexes were Fold Change > = 10 and False Discovery Rate (FDR)-corrected p-value < 1E-5.
The two sets of differentially expressed genes (DEGs) were analyzed with a hypergeometric test on annotations131 to identify significantly enriched GO terms or Pfam domain annotations among the male- and female-specific genes. Significant enrichment was determined for p-values lower than 0.05, paired with a difference between observed and expected values higher than 3.
TPM gene expression values were added 1 unit and log10-transformed before being plotted in a heat map, which clustered the transcripts showing similar expression trends based on Euclidean distance and average linkage criteria.
To enable a comparison of gene expression levels among species, we followed the strategy previously outlined in other publications31,53,132, recalculating gene expression levels on a subset of 1,694 unequivocal single-copy orthologs.
Transcriptomic expression data were validated by real-time quantitative PCRs performed on some candidate genes (for primer sequences see Supplementary Table S7). Reverse transcription was done using Superscript III First-strand Reaction Mix (Thermo Fisher, Invitrogen) and random primers. The reactions were performed using SYBR Green reagent and amplifications were detected with a Applied Biosystem 7900 HT. All results are averages of three independent PCR reactions from cDNA preparations of three males and three females. Transcript levels of target genes were normalized against the fire-bellied newt heterogeneous nuclear ribonucleoprotein D like (hnrpdl) gene (Supplementary Table S8). An additional analysis was performed using another housekeeping gene, the eukaryotic translation initiation factor 2 subunit alpha (eif2s1) confirming the same trend evidenced between males and females.
References
Frost, D. R. et al. The amphibian tree of life. Bull. AMNH 297, 1–291 (2006).
Carroll, R. L. The rise of amphibians: 365 million years of evolution. (ed. Johns Hopkins) (University Press (2009).
Botts, E. A., Erasmus, B. F. N. & Alexander, G. J. Small range size and narrow niche breadth predict range contractions in South African frogs. Glob. Ecol. Biogeogr. 22, 567–576 (2013).
Liedtke, H. C., Gower, D. J., Wilkinson, M. & Gomez-Mestre, I. Macroevolutionary shift in the size of amphibian genomes and the role of life history and climate. Nat. Ecol. Evol. 2, 1792–1799 (2018).
Gerchen, J. F. et al. A single transcriptome of a green toad (Bufo viridis) yields candidate genes for sex determination and -differentiation and non-anonymous population genetic markers. PLoS One 11, e0156419 (2016).
Kuzmin, D. A. et al. Stepwise large genome assembly approach: a case of Siberian larch (Larix sibirica Ledeb). BMC Bioinforma. 20, 37 (2019).
Stocum, D. L. Mechanisms of urodele limb regeneration 4(4), 159–200. Published online 2017 Dec 26. https://doi.org/10.1002/reg2.92 (2017).
Glass, H. C., Melin, A. D. & Vamosi, S. M. De novo transcriptome analysis of dermal tissue from the rough-skinned newt, Taricha granulosa, enables investigation of tetrodotoxin expression. bioRxiv, https://doi.org/10.1101/653238 (2019).
Wielstra, B., McCartney-Melstad, E., Arntzen, J. W., Butlin, R. K. & Shaffer, H. B. Phylogenomics of the adaptive radiation of Triturus newts supports gradual ecological niche expansion towards an incrementally aquatic lifestyle. Mol. Phylogenet Evol. 133, 120–127 (2019).
Nakamura, K. et al. A transcriptome for the study of early processes of retinal regeneration in the adult newt, Cynops pyrrhogaster. PLoS One 9, e109831 (2014).
Piprek, R. P., Kloc, M., Tassan, J. P. & Kubiak, J. Z. Development of Xenopus laevis bipotential gonads into testis or ovary is driven by sex-specific cell-cell interactions, proliferation rate, cell migration and deposition of extracellular matrix. Dev. Biol. 432, 298–310 (2017).
Piprek, R. P., Damulewicz, M., Kloc, M. & Kubiak, J. Z. Transcriptome analysis identifies genes involved in sex determination and development of Xenopus laevis gonads. Differentiation 100, 46–56 (2018).
Piprek, R. P., Damulewicz, M., Tassan, J. P., Kloc, M. & Kubiak, J. Z. Transcriptome profiling reveals male- and female-specific gene expression pattern and novel gene candidates for the control of sex determination and gonad development in Xenopus laevis. Dev. Genes. Evol. 229, 53–72 (2019).
Takeuchi, T., Tanigawa, Y., Minamide, R., Ikenishi, K. & Komiya, T. Analysis of SDF-1/CXCR4 signaling in primordial germ cell migration and survival or differentiation in Xenopus laevis. Mech. Dev. 127, 146–58 (2010).
Urbatzka, R., Lutz, I. & Kloas, W. Aromatase, steroid-5-alpha-reductase type 1 and type 2 mRNA expression in gonads and in brain of Xenopus laevis during ontogeny. Gen. Comp. Endocrinol. 153, 280–288 (2007).
Kuntz, S. et al. Female-enriched and thermosensitive expression of steroidogenic factor-1 during gonadal differentiation in Pleurodeles waltl. J. Mol. Endocrinol. 36, 175–186 (2006).
Che, R., Sun, Y., Wang, R. & Xu, T. Transcriptomic analysis of endangered Chinese salamander: identification of immune, sex and reproduction-related genes and genetic markers. PLoS One 9, e87940 (2014).
Eggert, C. Sex determination: the amphibian models. Reprod. Nutr. Dev. 44, 539–459 (2004).
Herpin, A. & Schartl, M. Plasticity of gene-regulatory networks controlling sex determination: Of masters, slaves, usual suspects, newcomers, and usurpators. EMBO Rep. 16, 1260–1274 (2015).
Kikuchi, K. & Hamaguchi, S. Novel sex-determining genes in fish and sex chromosome evolution. Dev. Dyn. 242, 339–353 (2013).
Huang, L. et al. Comparative transcriptome analyses of seven anurans reveal functions and adaptations of amphibian skin. Sci. Rep. 6, 240969 (2016).
McElroy, K. E. et al. Genome Expression Balance in a Triploid Trihybrid Vertebrate. Genome Biol. Evol. 9, 968–980 (2017).
Zhao, L., Liu, L., Wang, S., Wang, H. & Jiang, J. Transcriptome profiles of metamorphosis in the ornamented pygmy frog Microhyla fissipes clarify the functions of thyroid hormone receptors in metamorphosis. Sci Rep 6 (2016).
Montero-Mendieta, S. et al. A practical guide to build de-novo assemblies for single tissues of non-model organisms: the example of a Neotropical frog. PeerJ 5 (2017).
Nowoshilow, S. et al. The axolotl genome and the evolution of key tissue formation regulators. Nature 554, 50–55 (2018).
Torres-Sánchez, M. et al. Multi-tissue transcriptomes of caecilian amphibians highlight incomplete knowledge of vertebrate gene families. DNA Res. Int. J. Rapid Publ. Rep. Genes. Genomes 26, 13–20 (2019).
Hellsten, U. et al. The genome of the Western clawed frog Xenopus tropicalis. Science 328, 633–636 (2010).
Session, A. M. et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature 538, 336–343 (2016).
Sun, Y. B. et al. Whole-genome sequence of the Tibetan frog Nanorana parkeri and the comparative evolution of tetrapod genomes. Proc. Natl Acad. Sci. USA 112, E1257–1262 (2015).
Litvinchuk, S. N., Rosanov, J. M. & Borkin, L. J. Correlations of geographic distribution and temperature of embryonic development with the nuclear DNA content in the Salamandridae (Urodela, Amphibia). Genome 50, 333–342 (2007).
Biscotti, M. A. et al. The lungfish transcriptome: a glimpse into molecular evolution events at the transition from water to land. Sci. Rep. 6, 21571 (2016).
Pallavicini, A. et al. Analysis of the transcriptome of the Indonesian coelacanth Latimeria menadoensis. BMC Genomics 14, 538 (2013).
Gerdol, M. et al. The purplish bifurcate mussel Mytilisepta virgata gene expression atlas reveals a remarkable tissue functional specialization. BMC Genomics 18, 590 (2017).
Kleene, K. C. A possible meiotic function of the peculiar patterns of gene expression in mammalian spermatogenic cells. Mech. Dev. 106, 3–23 (2001).
Ivell, R. “All that glisters is not gold”—common testis gene transcripts are not always what they seem. Int. J. Androl. 15, 85–92 (1992).
Schmidt, E. E. Transcriptional promiscuity in testes. Curr. Biol. CB 6, 768–769 (1996).
Kleene, K. C. Patterns of translational regulation in the mammalian testis. Mol. Reprod. Dev. 43, 268–281 (1996).
Morgan, G. T. Lampbrush chromosomes and associated bodies: new insights into principles of nuclear structure and function. Chromosome Res. 10, 177–200 (2002).
Simeoni, I., Gilchrist, M. J., Garrett, N., Armisen, J. & Gurdon, J. B. Widespread Transcription in an Amphibian Oocyte Relates to Its Reprogramming Activity on Transplanted Somatic Nuclei. Stem Cell Dev. 21, 181–190 (2012).
Uribe, M. C. & Mejía-Roa, V. Testicular structure and germ cells morphology in salamanders. Spermatogenesis 4(3), e988090 (2015).
Wallace, R. A. & Selman, K. Ultrastructural aspects of oogenesis and oocyte growth in fish and amphibians. J. Electron. Microsc. Tech. 16, 175–201 (1990).
Teranishi, T. et al. Rapid replacement of somatic linker histones with the oocyte-specific linker histone H1foo in nuclear transfer. Dev. Biol. 266, 76–86 (2004).
Gu, L., Wang, Q. & Sun, Q. Y. Histone modifications during mammalian oocyte maturation: dynamics, regulation and functions. Cell Cycle 9, 1942–1950 (2010).
Shechter, D. et al. A distinct H2A.X isoform is enriched in Xenopus laevis eggs and early embryos and is phosphorylated in the absence of a checkpoint. Proc. Natl Acad. Sci. USA 106, 749–754 (2009).
Tanaka, Y., Kato, S., Tanaka, M., Kuji, N. & Yoshimura, Y. Structure and expression of the human oocyte-specific histone H1 gene elucidated by direct RT-nested PCR of a single oocyte. Biochem. Biophys. Res. Commun. 304, 351–357 (2003).
Cho, H. & Wolffe, A. P. Xenopus laevis B4, an intron-containing oocyte-specific linker histone-encoding gene. Gene 143, 233–238 (1994).
McGarry, T. J. & Kirschner, M. W. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 93, 1043–1053 (1998).
Sastre, D. et al. Expression of PLIN2 and PLIN3 during oocyte maturation and early embryo development in cattle. Theriogenology 81, 326–331 (2014).
Yang, X. et al. Identification of perilipin-2 as a lipid droplet protein regulated in oocytes during maturation. Reprod. Fertil. Dev. 22, 1262–1271 (2010).
Dworkin, M. B. & Dworkin-Rastl, E. Glycogen breakdown in cleaving Xenopus embryos is limited by ADP. Mol. Reprod. Dev. 32, 354–362 (1992).
Ochsenreither, S. et al. Cyclin-A1 represents a new immunogenic targetable antigen expressed in acute myeloid leukemia stem cells with characteristics of a cancer-testis antigen. Blood 119, 5492–5501 (2012).
Lefèvre, P. L. C., Palin, M. F. & Murphy, B. D. Polyamines on the reproductive landscape. Endocr. Rev. 32, 694–712 (2011).
Biscotti, M. A. et al. A comparative view on sex differentiation and gametogenesis genes in lungfish and coelacanths. Genome Biol. Evol. 10, 1430–1444 (2018).
Josso, N. & di Clemente, N. Transduction pathway of anti-Müllerian hormone, a sex-specific member of the TGF-beta family. Trends Endocrinol. Metab. 14, 91–97 (2003).
Hu, Q., Guo, W., Gao, Y., Tang, R. & Li, D. Molecular cloning and characterization of amh and dax1 genes and their expression during sex inversion in rice-field eel Monopterus albus. Sci. Rep. 5, 16667 (2015).
Adolfi, M. C., Nakajima, R. T., Nóbrega, R. H. & Schartl, M. Intersex, Hermaphroditism, and gonadal plasticity in vertebrates: evolution of the müllerian duct and Amh/Amhr2 signaling. Annu. Rev. Anim. Biosci. 7, 149–172 (2019).
Piprek, R. P., Pecio, A., Laskowska-Kaszub, K., Kubiak, J. Z. & Szymura, J. M. Sexual dimorphism of AMH, DMRT1 and RSPO1 localization in the developing gonads of six anuran species. Int. J. Dev. Biol. 57, 891–895 (2013).
Jansson, E., Mattsson, A., Goldstone, J. & Berg, C. Sex-dependent expression of anti-Müllerian hormone (amh) and amh receptor 2 during sex organ differentiation and characterization of the Müllerian duct development in Xenopus tropicalis. Gen. Comp. Endocrinol. 229, 132–44 (2016).
Stöck, M. et al. Shedding Light on a Secretive Tertiary urodelean Relict: Hynobiid salamanders (Paradactylodon persicus s.l.) from Iran, Illuminated by Phylogeographic. Developmental Transcriptomic Data. Genes. 10(4), pii: E306 (2019).
Myosho, T. et al. Tracing the emergence of a novel sex-determining gene in medaka, Oryzias luzonensis. Genetics 191, 163–170 (2012).
Zhang, X. et al. Autosomal gsdf acts as a male sex initiator in the fish medaka. Sci. Rep. 6, 19738 (2016).
Zhu, Y., Wang, C., Chen, X. & Guan, G. Identification of gonadal soma-derived factor involvement in Monopterus albus (protogynous rice field eel) sex change. Mol. Biol. Rep. 43, 629–637 (2016).
Forconi, M. et al. Characterization of sex determination and sex differentiation genes in Latimeria. PLoS One 8, e56006 (2013).
Pereira, J. et al. Evolutionary genomics and adaptive evolution of the Hedgehog gene family (Shh, Ihh and Dhh) in vertebrates. PLoS One 9(12), e74132 (2014).
Bitgood, M. J., Shen, L. & McMahon, A. P. Sertoli cell signaling by desert hedgehog regulates the male germline. Curr. Biol. 6, 298–304 (1996).
Szczepny, A., Hime, G. R. & Loveland, K. L. Expression of hedgehog signalling components in adult mouse testis. Dev. Dyn. 235, 3063–3070 (2006).
O’Hara, W. A., Azar, W. J., Behringer, R. R., Renfree, M. B. & Pask, A. J. Desert hedgehog is a mammal-specific gene expressed during testicular and ovarian development in a marsupial. BMC Dev. Biol. 11, 72 (2011).
Colvin, J. S., Green, R. P., Schmahl, J., Capel, B. & Ornitz, D. M. Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell 104, 875–889 (2001).
Jeays-Ward, K. et al. Endothelial and steroidogenic cell migration are regulated by WNT4 in the developing mammalian gonad. Development 130, 3663–3670 (2003).
Kim, Y. et al. Fgf9 and Wnt4 act as antagonistic signals to regulate mammalian sex determination. PLoS Biol. 4, e187 (2006).
Yamamura, Y. et al. Molecular cloning and expression in gonad of Rana rugosa WT1 and Fgf9. Zool. Sci. 22, 1045–50 (2005).
Parma, P. et al. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat. Genet. 38, 1304–1309 (2006).
Andrae, J., Gallini, R. & Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes. Dev. 22, 1276–1312 (2008).
Smith, C. A., McClive, P. J., Hudson, Q. & Sinclair, A. H. Male-specific cell migration into the developing gonad is a conserved process involving PDGF signaling. Dev. Biol. 284, 337–350 (2005).
Rhen, T., Jangula, A., Schroeder, A. & Woodward-Bosh, R. The platelet-derived growth factor signaling system in snapping turtle embryos, Chelydra serpentina: potential role in temperature-dependent sex determination and testis development. Gen. Comp. Endocrinol. 161(3), 335–43 (2009).
Mariani, S., Basciani, S., Arizzi, M., Spera, G. & Gnessi, L. PDGF and the testis. Trends Endocrinol. Metab. 13, 11–17 (2002).
Westernströer, B. et al. Profiling of Cxcl12 receptors, Cxcr4 and Cxcr7 in murine testis development and a spermatogenic depletion model indicates a role for Cxcr7 in controlling Cxcl12 activity. PLoS One 9(12), e112598 (2014).
Martyniuk, C. J., Bissegger, S. & Langlois, V. S. Current perspectives on the androgen 5 alpha-dihydrotestosterone (DHT) and 5 alpha-reductases in teleost fishes and amphibians. Gen. Comp. Endocrinol. 194, 264–274 (2013).
Lephart, E. D., Doody, K. J., McPhaul, M. J. & Simpson, E. R. Inverse relationship between ovarian aromatase cytochrome P450 and 5 alpha-reductase enzyme activities and mRNA levels during the estrous cycle in the rat. J. Steroid Biochem. Mol. Biol. 42, 439–447 (1992).
Haning, R. V. Jr., Tantravahi, U., Zhao, Q., Hackett, R. J. & Canick, J. A. 5alphareductase 1 and 2 expression and activity in human ovarian follicles, stroma and corpus luteum as compared to neonatal foreskin. J. Steroid Biochem. Mol. Biol. 59, 199–204 (1996).
Mahendroo, M. S. & Russell, D. W. Male and female isoenzymes of steroid 5alpha-reductase. Rev. Reprod. 4, 179–183 (1999).
Almeida, F. F. et al. Photoperiod-modulated testis maturation in Atlantic cod (Gadus morhua, L.). Biol. Reprod. 80, 631–640 (2009).
Oike, A. et al. Participation of androgen and its receptor in sex determination of an amphibian species. PLoS One 12(6), e0178067 (2017).
Ko, C. I. et al. Female-enriched expression of ERalpha during gonad differentiation of the urodele amphibian Pleurodeles waltl. Gen. Comp. Endocrinol. 156, 234–245 (2008).
Arenas, M. I. et al. Androgen receptor (AR), estrogen receptor-alpha (ER-alpha) and estrogen receptor-beta (ER-beta) expression in the testis of the newt, Triturus marmoratus marmoratus during the annual cycle. J. Anat. 199, 465–472 (2001).
Bertho, S. et al. Foxl2 and Its Relatives Are Evolutionary Conserved Players in Gonadal Sex Differentiation. Sex. Dev. 10, 111–129 (2016).
Fleming, N. I. et al. Aromatase is a direct target of FOXL2: C134W in granulosa cell tumors via a single highly conserved binding site in the ovarian specific promoter. PLoS One 5, e14389 (2010).
Zhu, L. et al. Sexual dimorphism in diverse metazoans is regulated by a novel class of intertwined zinc fingers. Genes. Dev. 14, 1750–1764 (2000).
Hodgkin, J. The remarkable ubiquity of DM domain factors as regulators of sexual phenotype: ancestry or aptitude? Genes. Dev. 16, 2322–2326 (2002).
Jin, S. B. et al. Comparative transcriptome analysis of testes and ovaries for the discovery of novel genes from Amur sturgeon (Acipenser schrenckii). Genet. Mol. Res. 14, 18913–18927 (2015).
Zhang, T., Murphy, M. W., Gearhart, M. D., Bardwell, V. J. & Zarkower, D. The mammalian Doublesex homolog DMRT6 coordinates the transition between mitotic and meiotic developmental programs during spermatogenesis. Development 141, 3662–3671 (2014).
Piprek, R. P., Kloc, M. & Kubiak, J. Z. Early development of the gonads: origin and differentiation of the somatic cells of the genital ridges. Results Probl. Cell Differ. 58, 1–22 (2016).
Hatano, O. et al. Sex dependent expression of a transcription factor, Ad4BP, regulating steroidogenic P-450 genes in the gonads during prenatal and postnatal rat development. Development 120, 2787–2797 (1994).
Pilon, N., Behdjani, R., Daneau, I., Lussier, J. G. & Silversides, D. W. Porcine steroidogenic factor-1 gene (pSF-1) expression and analysis of embryonic pig gonads during sexual differentiation. Endocrinology 139, 3803–3812 (1998).
Wibbels, T., Cowan, J. & LeBoeuf, R. Temperature-dependent sex determination in the red-eared slider turtle, Trachemys scripta. J. Exp. Zool. 281, 409–416 (1998).
Smith, C. A., Smith, M. J. & Sinclair, A. H. Expression of chicken steroidogenic factor-1 during gonadal sex differentiation. Gen. Comp. Endocrinol. 113, 187–196 (1999).
Western, P. S., Harvey, L. A., Gray, J. M. & Sinclair, A. H. Temperature-dependent sex determination in the American alligator: expression of SF1, WT1 and DAX1 during gonadogenesis. Gene 241, 223–232 (2000).
Mayer, L. P., Overstreet, S. L., Dyer, C. A. & Propper, C. R. Sexually dimorphic expression of steroidogenic factor 1 (SF-1) in developing gonads of the American bullfrog, Rana catesbeiana. Gen. Comp. Endocrinol. 127, 40–47 (2002).
Jiang, T., Hou, C. C., She, Z. Y. & Yang, W. X. The SOX gene family: function and regulation in testis determination and male fertility maintenance. Mol. Biol. Rep. 40, 2187–2194 (2013).
Koopman, P., Münsterberg, A., Capel, B., Vivian, N. & Lovell-Badge, R. Expression of a candidate sex-determining gene during mouse testis differentiation. Nature 348, 450–452 (1990).
Sekido, R., Bar, I., Narváez, V., Penny, G. & Lovell-Badge, R. SOX9 is up-regulated by the transient expression of SRY specifically in Sertoli cell precursors. Dev. Biol. 274(2), 271–9 (2004).
O’Bryan, M. K. et al. Sox8 is a critical regulator of adult Sertoli cell function and male fertility. Dev. Biol. 316, 359–70 (2008).
Nakamura, S. et al. Analysis of medaka sox9 orthologue reveals a conserved role in germ cell maintenance. PLoS One 7, e29982 (2002).
Denny, P., Swift, S., Connor, F. & Ashworth, A. An SRY-related gene expressed during spermatogenesis in the mouse encodes a sequence-specific DNA-binding protein. EMBO J. 11, 3705–3712 (1992).
Daigle, M., Roumaud, P. & Martin, L. J. Expressions of Sox9, Sox5, and Sox13 transcription factors in mice testis during postnatal development. Mol. Cell Biochem. 407, 209–221 (2015).
Schartl, M. et al. Sox5 is involved in germ-cell regulation and sex determination in medaka following co-option of nested transposable elements. BMC Biol. 16(1), 16 (2018).
Weiss, J. et al. Sox3 is required for gonadal function, but not sex determination, in males and females. Mol. Cell Biol. 23, 8084–8091 (2003).
Takehana, Y. et al. Co-option of Sox3 as the male-determining factor on the Y chromosome in the fish Oryzias dancena. Nat. Commun. 5, 4157 (2014).
Bowles, J. et al. Retinoid signaling determines germ cell fate in mice. Science 312, 596–600 (2006).
Koubova, J. et al. Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc. Natl Acad. Sci. USA 103, 2474–2479 (2006).
Yu, M. et al. RALDH2, the enzyme for retinoic acid synthesis, mediates meiosis initiation in germ cells of the female embryonic chickens. Amino Acids 44, 405–412 (2013).
Wallacides, A., Chesnel, A., Chardard, D., Flament, S. & Dumond, H. Evidence for a conserved role of retinoic acid in urodele amphibian meiosis onset. Dev. Dyn. 238, 1389–1398 (2009).
Zhou, Q. et al. Expression of stimulated by retinoic acid gene 8 (Stra8) and maturation of murine gonocytes and spermatogonia induced by retinoic acid in vitro. Biol. Reprod. Mar. 78, 537–45 (2008).
Pasquier, J. et al. Gene evolution and gene expression after whole genome duplication in fish: the PhyloFish database. BMC Genomics 17, 368 (2016).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinforma. Oxf. Engl. 30, 2114–2120 (2014).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644–652 (2011).
Carniel, F. C. et al. New features of desiccation tolerance in the lichen photobiont Trebouxia gelatinosa are revealed by a transcriptomic approach. Plant. Mol. Biol. 91, 319–339 (2016).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinforma. Oxf. Engl. 31, 3210–3212 (2015).
Zdobnov, E. M. et al. OrthoDB v9.1: cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 45, D744–D749 (2017).
Bryant, D. M. et al. A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 18, 762–776 (2017).
UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 (2019).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Powell, S. et al. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res gkt1253 (2013).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, 29–37 (2011).
Punta, M. et al. The Pfam protein families database. Nucleic Acids Res. 40, D290–D301 (2012).
Huelsenbeck, J. P., Ronquist, F., Nielsen, R. & Bollback, J. P. Bayesian inference of phylogeny and its impact on evolutionary biology. Science 294, 2310–2314 (2001).
Wagner, G. P., Kin, K. & Lynch, V. J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. Theor. Den. Biowissenschaften 131, 281–285 (2012).
McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012).
Falcon, S. & Gentleman, R. Hypergeometric Testing Used for Gene Set Enrichment Analysis, in: Bioconductor Case Studies, Use R! Springer New York, pp. 207–220 (2008).
Biscotti, M. A. et al. The small noncoding RNA processing machinery of two living fossil species, lungfish and coelacanth, gives new insights into the evolution of the Argonaute protein family. Genome Biol. Evol. 9, 438–453 (2017).
Acknowledgements
This work was supported by a grant from Ministero della Ricerca e dell’Istruzione, Project numbers: 46, 2018; 232-130, 2019 and by grants supplied to M.S. by Texas A&M University and University of Würzburg. This publication was funded by the German Research Foundation (DFG) and the University of Würzburg in the funding program Open Access Publishing. We thank Dr. Samuele Greco, Dr. Sergio Palmitessa, and Dr. Andrea Visintin (Dipartimento di Scienze della Vita, Università di Trieste, Italy) for their help in transcriptomic data and gene expression analyses.
Author information
Authors and Affiliations
Contributions
M.A.B. and F.C. were involved in the identification and characterization of sex genes, phylogenetic analyses, interpreted the data and wrote the paper. M.B. contributed to interpretation of the data and wrote the paper. A.P. and M.G. subjected in transcriptomic data and in gene expression analyses and wrote the paper. A.C. and M.S. conceived the study, interpreted the data and wrote the paper. M.C.A. did the histological analyses, contributed to the data interpretation and wrote the paper. All authors have given final approval for the version to be published.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Biscotti, M.A., Carducci, F., Barucca, M. et al. The transcriptome of the newt Cynops orientalis provides new insights into evolution and function of sexual gene networks in sarcopterygians. Sci Rep 10, 5445 (2020). https://doi.org/10.1038/s41598-020-62408-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-62408-x
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
