
Abstract
Primary production by phytoplankton represents a major pathway whereby atmospheric CO2 is sequestered in the ocean, but this requires iron, which is in scarce supply. As over 99% of iron is complexed to organic ligands, which increase iron solubility and microbial availability, understanding the processes governing ligand dynamics is of fundamental importance. Ligands within humic-like substances have long been considered important for iron complexation, but their role has never been explained in an oceanographically consistent manner. Here we show iron co-varying with electroactive humic substances at multiple open ocean sites, with the ratio of iron to humics increasing with depth. Our results agree with humic ligands composing a large fraction of the iron-binding ligand pool throughout the water column. We demonstrate how maximum dissolved iron concentrations could be limited by the concentration and binding capacity of humic ligands, and provide a summary of the key processes that could influence these parameters. If this relationship is globally representative, humics could impose a concentration threshold that buffers the deep ocean iron inventory. This study highlights the dearth of humic data, and the immediate need to measure electroactive humics, dissolved iron and iron-binding ligands simultaneously from surface to depth, across different ocean basins.
Similar content being viewed by others


Introduction
Iron is a key micronutrient to marine microorganisms. Low concentrations of dissolved iron limit primary production in up to 40% of the ocean1. Over long timescales, the deep ocean iron inventory is important for global ocean primary productivity2, as it can fuel dissolved iron supplied to the surface through winter mixing and diapycnal diffusion. Since ligand complexation is crucial for maintaining iron in solution3, fully quantifying the concentration and characteristics of the ligand pool is an essential task. For example, varying the concentrations of iron binding ligands in biogeochemical models has a significant impact on atmospheric CO2 calculations4. However, the processes controlling the distribution and iron binding capacity of this pool are difficult to constrain. In order to identify the processes regulating iron complexation, it is necessary to consider the controls on key ligand groups, as well as on the wider dissolved organic carbon (DOC) pool to which they contribute. Although many ligand types exist, they are typically separated into classes defined by the strength of their complexes with iron, measured by competition against artificial ligands of known binding constants5. This gives an average concentration and binding strength of one or more ligand classes, but not their identity, which must be inferred by comparison to known compounds. Siderophores represent the strongest natural ligands, released by specialised microorganisms to acquire iron6. Although a siderophore-like ligand class is often detected5, as of yet, direct measurements have only found siderophores to complex less than 10% of iron (though their contribution could be higher)7,8. Weaker ligands also play a predominant role in iron cycling and biological uptake9. Around 30% of the marine DOC pool is composed of labile polysaccharides10, some of which can form weak complexes with iron11. The further decomposition of cell-derived products contributes to a longer-lived DOC pool known as humic-like substances, which compose around 50% of DOC10 and of which a smaller fraction (around 5% of DOC) can bind to iron12.
When only a single ligand class is detected, it is usually of intermediate binding strength throughout the water column5. Humic ligands form iron complexes of intermediate strength13,14, bracketed by stability constants of the stronger siderophores and weaker polysaccharides. All major sources of iron to the water column also supply humic-like material15,16,17,18, with terrestrial-derived humics often dominating iron complexation in estuarine and coastal waters13,14. Furthermore, the stability of iron-humic complexes allows iron to be transported long distances by ocean currents19. However, despite many indications that humic ligands play a key role in the iron cycle, it has thus far proven difficult to explain the relationship between humic substances, iron and the iron ligand pool in ocean waters. This is because humics are themselves a heterogeneous mixture of soluble and colloidal-sized compounds of various origins14 and iron-binding behaviour20,21. Here, we demonstrate an intrinsic relationship between dissolved iron and humic ligands throughout the water column across multiple open ocean sites. We attempt to further refine the hypothesis for the proposed geochemical control of iron in seawater13 by summarising the key processes that may influence this relationship, which have not previously been considered as controls on oceanic dissolved iron distributions.
Results and Discussion
Variability in iron binding by marine humics
Sampling locations include the North and Northwest Atlantic, the Southern Ocean and the Northeast Pacific (Fig. 1). Sites range from iron-limited, high nutrient-low chlorophyll regions of the open ocean to iron-replete areas influenced by sea ice and terrestrial inputs. Dissolved metal-binding humic substances were measured electrochemically (eHS) and compared to dissolved iron concentrations. These measurements are distinct from fluorescence-based measurements, and assume that the electroactive humic fraction is representative of the bulk of humic ligands for metals. The highest electroactive humic concentrations were found nearest the coast, in the Northwest Atlantic (reaching 189 μg/L). In open ocean samples, we found similar concentration ranges in all ocean basins (Atlantic 12–116 μg/L, Pacific 18–54 μg/L, Southern Ocean 18–81 μg/L, Supplementary Table S1), with the highest concentrations at intermediate depths. In the North Atlantic, iron-binding ligands and fluorescent dissolved organic matter (FDOM) were also measured.

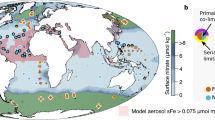
Map of sample locations. Surface samples (5 m depth) are from the Bermuda AE1714 cruise sampled from the Towfish whilst steaming. Depth profiles: Stations G1, G13, G38, G44, G69 and G77 in the North Atlantic are from the GEOVIDE cruise (GA01); P26 in the Northeast Pacific is from the August 2012–13 Line P cruise; KR2, KE4 and KA3 are from the KEOPS2 study in the Southern Ocean (GIpr01). Figure generated using Matlab software.
Humic substances are inherently heterogeneous. Composed of a combination of both humic and fulvic acids, which are defined operationally based on extraction protocols, the number and strength of iron binding sites varies20,21. Increased aromaticity in humics has been linked to increased iron binding22. Terrestrial humics are largely derived from highly aromatic lignins and tannins only found in vascular plants23. In contrast, marine humics are mostly derived from plankton and are carboxyl rich and aliphatic, with lower aromatic content24,25. While the scarcity of lignin in aquatic systems may account for some discrepancies between terrestrial and marine humics, microbially produced compounds (precursors to humics, such as amino acids, lipids and polysaccharides) from bacteria and algae in both systems may account for some similarities26. Since the iron binding capacity of humics is based on numerous factors, here we account for the full range of published iron binding capacities for terrestrial standards from the Suwannee River, using an envelope that encompasses the maximum (Suwannee River Humic Acid, SRHA) and minimum (Suwannee River Fulvic Acid, SRFA) reported values13,21,27 as a first approximation (Fig. 2). While marine humics could well incorporate a wider range, the mean iron-binding capacity of our samples (Supplementary Table S1), as well as in the Pacific Ocean13 and Mediterranean Sea12, indicate a similar range to terrestrial standards, lending confidence to this assumption.

The concentrations of dissolved iron (blue line) and iron-binding ligands (green circles, where available), with an envelope (red) for electroactive humic substances (eHS), encompassing the maximum and minimum iron-binding capacities reported for terrestrial IHSS standards13,27. (a) Station P26 in the Northeast Pacific (Line P); (b) Surface samples (~5 m) Northwest Atlantic (Bermuda AE1714), with salinity included below; (c–g) Stations G1-G77 depth profiles from the North Atlantic (GEOVIDE GA01). Error bars show standard deviation, which for eHS is included within the envelope. Spaces in the eHS boundaries show eHS sampling points. Figure generated using Matlab software.
We find broad correspondence between dissolved iron and humic ligands throughout the water column (Fig. 3, ρ = 0.5, p < 0.0001, n = 105, where ρ is the Spearman’s correlation coefficient). However, the range of iron-binding ligands that exist in marine waters, such as low concentrations of siderophores saturated with iron7, and the variable iron-binding capacity of humic material, hinder straightforward comparisons. To observe if any broad behavioural trends could be detected and explained, we compared the ratio of dissolved iron to humics (DFe/eHS) across all samples. In surface samples where the organic matter pool is highly dynamic, encompassing coastal to open ocean regions, DFe/eHS covered the full range of iron binding capacities reported for terrestrial standards13,27. In 49% of samples shallower than 200 m, DFe/eHS values near or below the lower binding capacity suggested undersaturation of humics with iron. There is a loss of dissolved iron due to scavenging and biological uptake in upper waters. In addition, the humic pool in upper waters is largely aliphatic28, and thus has reduced iron-binding capacities. While freshwater inputs can supply aromatic terrestrial humics, photodegradation destroys terrestrial-derived ligands but is a source of aliphatic humics29. Humic material in aerosols17 and marine-derived organic matter25 are also predominantly aliphatic. In a study focussed in the upper 100–200 m in Arctic waters, electroactive humics were found to be on average 62% saturated with iron (range 14–90%), when assuming that all humic ligands had the lowest reported iron binding capacity27.

The relationship between the concentrations of electroactive humic substances and dissolved iron from this study, coloured by depth (ρ = 0.5, p < 0.0001, n = 105, where ρ is the Spearman’s correlation coefficient). The upper line demonstrates the maximum iron binding capacity for the measured humic concentrations (32 ± 2.2 nM Fe/mg SRHA), and the lower line shows the lower binding capacity for the equivalent concentration (14.6 ± 0.7 nM Fe/mg SRFA), from reported values for terrestrial IHSS standards13,27. These have slopes of 0.33 and 0.73 ± 0.43 as SRHA and SRFA iron-binding equivalents respectively, not shown. The four points with a dashed circle are influenced by sediment, based on transmissiometry data. Error bars represent the standard deviation. Figure generated using Matlab software.
In our samples from the upper 1000 m of the Northeast Pacific and in all samples from the Southern Ocean, where most humic-like material comes from the recent microbial degradation of sinking autochthonous organic matter30, the DFe/eHS ratio was below or equivalent to the lowest reported binding capacity of terrestrial humics (Supplementary Fig. S1, Table S1). The humics in these samples thus have low iron binding capacities, or are not saturated with iron; iron concentrations in these samples were generally lower than in our Atlantic samples. Iron saturation of humic ligands has been found to increase with depth7, as dissolved iron is supplied to the water column from the bacterial remineralisation of sinking organic matter3. In samples deeper than 1000 m in the Pacific, which account for old waters31, the DFe/eHS ratio increased respective to surface waters, whereas in the Southern Ocean, DFe/eHS remained low.
In our samples from the North Atlantic, which experienced greater terrestrial influence32 and reached deeper sampling depths, DFe/eHS ratios exceeded the lower binding capacity in over 50% of samples. In around 25% of samples, DFe/eHS reached the maximum reported binding capacity of terrestrial humic substances (Fig. 3). Terrestrial humics arrive in the marine environment pre-aged and highly aromatic, particularly those derived from peatlands common in the Arctic. A considerable 30% of lignin phenols discharged by Arctic rivers is exported to the North Atlantic33 suggesting some fraction of the humic pool in our North Atlantic samples is likely terrigenous and aromatic32. Deep waters reflect an accumulation of aged humic material of higher aromaticity28,29, linked to the persistence of terrestrial humics32,34, the selective decomposition of organic matter35, and the hypothesized aging process of humification24,28,34. The increased aromaticity of humics in deep and terrestrially-influenced waters, leading to higher iron binding capacities22, could explain the higher DFe/eHS we observe in parts of the North Atlantic.
Indeed, we find that the binding capacity of humic ligands could define an upper limit for dissolved iron concentrations in the ocean interior (Fig. 3). We find that dissolved iron concentrations do not significantly exceed the maximum potential iron binding capacity of humics in any sample, with many values falling along the upper limit. Our results suggest that a combination of the concentration and binding capacity of humic ligands may control bulk dissolved iron distributions in some parts of the global ocean. Finally, we find lower DFe/eHS near the sediment. Bottom waters are influenced by local microbial communities in sediments supplying fresh aliphatic material15, which, along with the increased scavenging of iron onto resuspended particulate material36, result in low DFe/eHS. The processes influencing the aromaticity and iron binding capacity of humic ligands are summarised in Fig. 4.

Our proposed schematic of the processes influencing the supply and iron-binding nature of humic substances in seawater. Above represents the continuum of iron binding by humics linked to aromaticity22, along with the contributors of humic-like material to ocean waters. Below, our hypothesis for the potential contribution of humics to iron complexation and the iron ligand pool in two scenarios: iron limited regions (left) and terrestrially influenced regions (right). Figure generated using Adobe Illustrator CC software.
Metal-binding humic substances: marine and terrestrial influences
As is the case for the wider humic pool, this study finds electroactive humic ligands to be ubiquitous in seawater. Higher concentrations at low salinities demonstrate a terrestrial source, as would be expected. However, elevated concentrations in mesopelagic waters and relatively consistent values at open ocean locations also agree with a marine source and a refractory component. Humic substances are both produced and degraded by bacterial respiration37, modifying the optical signature of the humic pool in the thermocline34. A proxy for the bacterial remineralisation of organic matter is the apparent oxygen utilisation (AOU). Dissolved iron has been found to correlate with both humic-like fluorescence and the AOU in intermediate and deep waters of the central Pacific38. However, it is currently unclear how qualitative fluorescence measurements compare to quantitative electrochemical measurements of humic substances. Only a small fraction of the overall humic pool binds metals, and this may not correspond to the fluorescent fraction. In our samples from the Pacific, dissolved iron showed some agreement with AOU (slope = 0.02 ± 0.14, R2 = 0.63, p < 0.01, n = 9, data not shown), though the depth profile highlighted inconsistencies. The DFe/eHS ratio and high AOU in the Pacific was indicative of bacterial remineralisation supplying fulvic-like ligands (Supplementary Fig. S1), however, we found no relationship between electroactive humic concentrations and the AOU.
In the North Atlantic, AOU did not correlate with dissolved iron, electroactive humics, humic-like fluorescence or iron ligands. The North Atlantic has a high terrestrial humic influence, which, along with frequent ventilation, masks the relationship between bacterial remineralisation and organic matter concentrations30,39 and thus the relationship with dissolved iron40. Our FDOM data agreed with a strong contribution of terrestrial humic-like fluorescence across our North Atlantic samples (Supplementary Table S1). We found no correlation of electroactive humic concentrations with total, terrestrial or marine humic-like fluorescence, suggesting that the fluorescent components of humic material do not always correspond to the metal-binding components. However, metal-binding humics originate from multiple sources, and as the fluorescent properties of marine and terrestrial humics share some common features, their respective contributions are not well distinguished39.
Ligand studies in the Atlantic have similarly not found any correlation of the dominant L1 and L2 ligand classes with AOU41, which is also likely in part to be linked to the combination of terrestrial inputs and frequent ventilation30,39. Our findings agree with other electrochemical humic and AOU comparisons in the East Atlantic, while a negative correlation was found in the Mediterranean12. It was concluded that the negative correlation between electroactive humics and the AOU could be the result of increased degradation of marine-derived humics induced by the enhanced respiration rate of DOC in the Mediterranean, resulting in a humic sink (Dulaquais et al. 2018 and references therein). The authors drew attention to the possibility of two pools of humics: one marine-derived pool produced at the surface and available for bacterial degradation, and a second terrestrial-derived pool trapped in the deep sea, which is truly recalcitrant.
Terrestrially-influenced regions experience plumes of elevated dissolved iron concentrations, transported offshore in humic complexes19. Peatland sources in particular supply highly aromatic humics. A schematic of our proposed humic contribution to iron binding in iron limited regions compared to waters with a terrestrial signature (Fig. 4) is in agreement with general trends for ligand concentrations and binding strengths5. This schematic represents a first approach at highlighting the potential implications of humic aromaticity and variable iron binding capacities on iron complexation in ocean waters. However, it is likely that not all terrestrial inputs are equal, with the iron-binding capacity dependent on the contribution of pre-aged, aromatic components, and the protection of this fraction from degradation during transport. For example, humics from Arctic rivers derived from highly aromatic peatlands and transported during periods of low light intensity may be expected to have higher iron loading than humics from large tropical rivers, where photodegradation and microbial activity reduces the aromaticity of the humic pool during transport. The variability in freshwater iron supply linked to both the concentration and quality of organic matter22 deserves further investigation.
When considering that a large fraction of organic matter in the deep ocean is aged and allochthonous10, our results suggest that the persistence of terrestrial humics in the deep ocean may be pivotal in the global ocean iron cycle. However, there are many sources of both iron and humic material to seawater and it is likely that aged humics in the deep ocean are derived from a combination of terrestrial34, marine37 and even hydrothermal sources16. Regardless of origin, the apparent upper boundary for dissolved iron concentrations suggests that the concentration and quality of humics could limit bulk dissolved iron concentrations in intermediate and deep waters of the North Atlantic. Our data from the Pacific and Southern Ocean demonstrate a relationship exists between iron and humics in other basins, but a lack of deep samples mean the extent of the upper boundary on iron concentrations remains to be verified in future investigations.
Humic contribution to the iron ligand pool
As the iron binding capacity of the humic ligands can vary, and as ligand measurements do not distinguish between different compounds of similar binding strengths, constraining the contribution of humics to the iron ligand pool is challenging. In an effort to compare the relationship between dissolved iron and electroactive humics to the typical relationship with the overall ligand pool, we plotted iron ligand data against iron concentrations from an array of published studies, encompassing multiple basins and methods (Fig. 5). Similar plots have been presented previously, including recently in the Pacific42, showing a relationship consistent with our figure. It is common to find ligand concentrations in excess of dissolved iron, particularly in surface waters where iron depletion coincides with ligand production. However, the maximum dissolved iron concentrations are almost always limited by the concentration of available ligands, as the poor solubility of iron in seawater results in negligible concentrations of uncomplexed iron3.

Black shapes show the relationship between the concentrations of dissolved iron (DFe) and humic substances (eHS, from this study, filled, and published values12, open) compared to the typical relationship between DFe and the ligand pool (L, open coloured shapes, this study and literature-derived values, slope 0.76 ± 1.3, not shown. See Methods for references). The line represents 1:1 complexation of DFe with the concentration of total ligand or eHS expressed as nanomolar iron binding equivalent, which for eHS is based on the maximum reported potential binding capacity (32 nM Fe per mg/L HA13). Figure generated using Matlab software.
We compare this established relationship to that of iron and electroactive humics (our study and published values12) and find strong similarities. In fact, where dissolved iron exceeds the ligand concentration in the Mediterranean it was acknowledged that the contribution of electroactive humics to the ligand pool may have been underestimated43. We also find similar basin-specific features, with concentrations of ligands and electroactive humics in greater excess of iron in the Pacific and Southern Ocean compared to in the Atlantic. We find very good agreement between the slopes of dissolved iron against electroactive humics compared to dissolved iron against published ligand data, 0.73 ± 0.43 and 0.76 ± 0.43 respectively (when using the lower humic iron binding capacity, likely most representative of the bulk of our samples). Similar to previous studies12,44,45, we found electroactive humics to correlate with the ligand pool quite well, although samples with data for both parameters were limited. However, all studies find electroactive humic distributions to better agree with dissolved iron directly than with ligand measurements. The ligand pool is dynamic and its composition is unlikely to be constant regionally or with depth. The seawater pH, the molecular size and composition of marine organic matter and the presence of other iron-binding ligands, as well as competition between different metals for common ligands, will all influence the amount of iron complexed by any specific ligand group44,46. Furthermore, ligand measurements depend on the detection window and the artificial ligand used, some of which may underestimate humic contributions to the ligand pool45 and overestimate the amount of metal complexed by dissolved organic matter47, while the effect of buffering the pH during measurements may dampen the influence of subtle pH changes on metal complexation48.
Nevertheless, valuable information can be obtained from studying the overall ligand pool. In our North Atlantic samples we find a ligand class of intermediate to strong binding strength throughout the water column (Supplementary Table S1), with the strongest ligands typically at the surface, similar to values reported previously in this region49 and in the Southern Ocean50. Although we did not have ligand samples for the Pacific, other studies have found the intermediate ligand class dominating iron complexation at depth42. In our samples where both parameters were measured, a conservative estimate suggests electroactive humics could account for 23–58% of the ligand pool, to as high as 100% at the highest binding capacity. These values are similar to previous studies in the Mediterranean and Eastern Atlantic, where electroactive humics were found to account for 30–50% of the ligand pool12, while they could account for almost all of the ligand pool in the Arctic45, with both studies using the lower binding capacity.
In some cases, a siderophore-like class is reported to dominate iron complexation throughout the water column41. Recent ligand characterisation techniques in samples off the Californian Coast found the iron ligand pool to largely consist of humic-like material in combination with low concentrations of siderophores, with iron saturation of humics increasing with depth7. Thus unsurprisingly, the humic contribution to iron complexation is dependent on the concentration of other, stronger ligand groups, varying regionally and with depth. We find metal-binding humics are ubiquitous and persistent throughout the water column in three ocean basins, corresponding with dissolved iron distributions vertically and laterally. While ligand groups such as siderophores are important for controlling iron bioavailability, humic ligands may control overall dissolved iron distributions, particularly in the deep ocean. The ability of humic ligands to cap dissolved iron concentrations will ultimately be influenced by an array of factors, such as the presence of stronger iron-binding ligands, and the availability of other metals that may compete with iron for humic complexation44,46. Even so, we find the maximum dissolved iron concentrations at depth coinciding with the maximum reported binding capacities reported for humic isolates. Processes that control the production, degradation and iron-binding capacity of humic substances may thus be particularly important for controlling dissolved iron distributions in the deep ocean.
Broader impacts
In light of the central role that marine iron availability may have played in regulating the biological pump during the Pleistocene51, a corollary hypothesis is that changes in the delivery of humic ligands and the processes regulating the iron binding capacity (as presented in Fig. 4) may have had an impact on climate. Glacial periods are characterised by increased soil erosion, lower sea level and continental shelf shrinkage, increasing terrestrial organic matter supply to the deep ocean52. Glacial-interglacial changes in fluvial and aeolian transport of terrestrial humics to the ocean may thus have played a key role in controlling the marine iron inventory18,53,54. However, we know very little about the supply of terrestrial ligands to ocean waters. Currently we do not fully understand the dominant controls on ligands such as humics in the ocean, and whether they persist long enough to influence the iron supply to iron limited regions, or influence its bioavailability to microorganisms. Hence it may be important to better constrain these processes and improve their representation in earth system models if we are to understand and predict climate and climate change.
Conclusions
Here we present the novel hypothesis that variation in both the concentration and binding capacity of humic material in ocean waters could explain key variations in iron distributions, buffering the dissolved iron inventory of the deep ocean. There is mounting evidence that humics play a significant role in dissolved iron distributions globally. There is therefore a requirement to improve our understanding of this poorly characterised pool in order to constrain the controls on dissolved iron distributions in ocean waters. This study highlights the lack of open ocean data, and an immediate need to simultaneously measure humic, ligand and dissolved iron concentrations throughout the water column in different ocean basins. We recommend studies also consider the effect of physicochemical conditions such as pH on these measurements, and incorporate complementary techniques for measuring and characterising DOC content, in order to better establish the controls on iron complexation in ocean waters.
Methods
Sampling
Depth profiles from the North Atlantic were sampled during the GEOVIDE cruise on board the N/O Pourquoi Pas? (15 May–30 June 2014, GEOTRACES GA01), in 12 L Teflon-coated GO-FLO (General Oceanics) bottles attached to a 24-bottle powder-coated modified trace metal clean rosette system55,56. West Atlantic surface samples were collected using a modified trace metal clean Towfish57 pumping seawater directly into a laminar flow hood bubble, during transit of the AE1714 cruise from Bermuda on board the RV Atlantic Explorer (July-August 2017). Samples from the Northeast Pacific were collected at station P26 of the Line P Time Series transect from the Canadian Coast Guard Ship John P. Tully (14–30 August 2012, cruise 2012–13) into 12 L Teflon-coated GO-FLO (General Oceanics) bottles attached to a 12-bottle powder-coated modified trace metal clean rosette system55,56. Samples from the Southern Ocean were collected during the KEOPS2 cruise on board the N/O Marion Dufresne (8 October-30 November 2012, GEOTRACES GIpr01) in 10 L externally closing Teflon-lined Niskin-1010X bottles, mounted on a polyurethane powder-coated aluminium frame (TMR, model 1018, General Oceanics)58.
Water taken for analysis of dissolved trace metals was either taken from the filtrate of particulate samples (collected on polyethersulfone filters, 0.45 μm Supor) or filtered through a 0.2 μm capsule filter (Sartorius Sartobran 300). Seawater was collected in acid-cleaned 60 mL or 125 mL LDPE bottles after rinsing three times with about 20 mL of seawater. Samples for dissolved iron were acidified to ~pH 1.7 with 2‰ (v/v) HCl (Ultrapur, Merck) in the laminar flow hood. The sample bottles were then double bagged and stored at ambient temperature in the dark before analysis on shore. Samples for ligands and humic substances were double bagged and frozen immediately at −20 °C. Before analysis, the seawater samples were thawed in the dark at 4 °C and measured within 3 days of defrosting. Samples were swirled gently and left to come to room temperature (20 °C) before preparation for analysis.
Equipment and reagents
Water used for rinsing and dilution of reagents was purified by reverse osmosis (Millipore) and deionisation (Milli-Q, MQ hereafter). Sample bottles (LDPE) were cleaned according to GEOTRACES protocols59,60 and stored in MQ for at least a week after acid-cleaning.
All voltammetric measurements used cathodic stripping voltammetry (CSV) in differential pulse mode. The voltammetric apparatus consisted of a µ-Autolab III potentiostat (Ecochemie, Netherlands) connected to a 663 VA stand (Metrohm) with hanging mercury drop electrode (HMDE). The reference electrode was Ag/AgCl with a 3 M KCl salt bridge and a glassy carbon counter electrode. Solutions were stirred with a rotating polytetrafluoroethylene (PTFE) rod. PTFE voltammetric cells used were cleaned using 0.1 M HCl (Suprapur Merck) and rinsed with MQ water. Sample (10 mL) was purged with nitrogen for up to 5 minutes to remove dissolved oxygen prior to analysis.
Dissolved iron (DFe)
The DFe concentrations from the GEOVIDE (North Atlantic) and Bermuda AE1714 (West Atlantic) were determined by using an online Inductively Coupled Plasma Mass Spectrometry (ICP-MS), at Pôle Spectrométrie Océan (France). The spectrometer was coupled to an ESI SeaFAST pico system to measure dissolved trace metals with a method analytically similar to that of Lagerström et al.61. Samples collected during KEOPS2 (Southern Ocean) were also analysed by SF-ICP-MS but with a different resin62,63. Full details on sampling and measurement are provided by Tonnard et al. (2018) and Quéroué et al. (2015) respectively. DFe samples from the Pacific were analysed by ICP-MS/MS as described by Jackson et al.64.
Humic substances (HS)
We measured the concentrations of electroactive metal-binding HS (eHS) using cathodic stripping voltammetry of their complexes with copper65. The copper method was favoured as, unlike iron, dissolved copper can be present in excess of HS. Measurements also do not suffer the same interferences from common marine compounds such as glutathione, as the peak is at a different potential. The concentrations of eHS using the copper technique agree very well with HS concentrations from UV spectrophotometry65 and with voltammetric detection of iron-humic complexes directly44 in estuarine waters. For this study we also compared the concentrations measured using the copper method65 to the recently updated iron method13,27 in open ocean waters, surface and deep, and found they agree very well (Supplementary Fig. S2).
Samples from the Atlantic and Southern Ocean were measured with an EPPS buffer (N-(2-hydroxyethyl)piperazine-N′; -2-propanesulfonic acid in 1M NH4; 100 μL addition to 10 mL seawater buffered the pH to 8.05) and 30 nM added copper (spectrophotometry standard, pH 2). A deposition potential of +0.05 V was used, usually for a deposition time of 60 s, with a 1s jump to −0.2 V and background subtraction (subtraction of a 1s scan), to improve the baseline and reduce interference from free copper. Samples from the Atlantic were measured using standard additions of the International Humic Substances Society (IHSS) Suwannee River humic acid (SRHA) standard (II 2S101H), and from the Southern Ocean using the IHSS fulvic acid (SRFA) standard (II 2S101F). We performed tests in UV-digested seawater with added known amounts of HS, and found the HA and FA standards to give identical results (as found previously65), since the electrochemical methods measure total humic substances and cannot distinguish between HA and FA. Concentrations of eHS for the Pacific samples were already published for copper66 and we converted the values to their iron-binding equivalents. The concentration of eHS is measured in mg/L, and the iron-binding equivalent then calculated by multiplication with reported binding capacities. The maximum and minimum Fe-HS binding capacities used for the eHS envelope were derived from published values for IHSS standards: isolated HA bind around 32 nM iron per mg/L13, while FA have a reported binding capacity of 14.6–19 nM iron per mg/L13,21,27. The lower limit of the envelope was calculated by multiplying the measured eHS concentration by the FA binding capacity, while the upper limit was calculated by multiplication with the HA binding capacity, with upper and lower errors included within the width of the envelope (Eqs. 1 and 2). This envelope provides an assumption of the amount of iron that can be bound by these eHS concentrations as a first approximation based on terrestrial standards, but the range could be wider.
Iron-binding ligands
We performed iron speciation measurements using CSV with 10 μM 2-(2-Thiazolylazo)-p-cresol (TAC) as the competing ligand11. Briefly, 100 μL EPPS buffer was added to 10 mL sample in acid-cleane d PTFE pots (10–12), followed by increasing iron additions (iron spectrophotometry standard, pH 2), usually of 0, 0. 2, 0.4, 0.6, 0.8, 1, 2, 4, 6, 8, 10 nM iron. These were left to stand for one hour before addition of the TAC artificial ligand (final concentration 10 μM), and were then left to equilibrate overnight. The CSV measurement was at a deposition potential of −0.36 V for 180s, followed by a 10s equilibration. At the end of the titration, some samples had fresh additions of 2, 4 and 6 nM iron (final concentrations 12, 14 and 16 nM iron) added to the cell after measurement of the final (+10 nM Fe) pot; this was in order to ensure that the ligands were fully complexed and to check the calculation of the sensitivity, but were not used in the data fitting. Data fitting was performed using the ‘complete fitting’ procedure in independent ProMCC software67. These measurements provide a ligand concentration (L) and conditional stability constant (log KFe’) based on the detection window, set by the artificial ligand concentration; this method may miss the contribution of ligands outside of the set detection window, and can underestimate the contribution of humic substances68, therefore the values presented represent the minimum concentration of the ligand pool. For the comparison of published concentrations of dissolved iron and iron-binding ligands across multiple ocean basins shown in Fig. 511,12,41,45,49,50,69,70,71,72,73,74,75,76, when more than one ligand class was distinguished, the total ligand concentration was used.
Optical measurements
Absorbance was measured from 200 to 800 nm on a Varian 300 UV spectrophotometer in 10 cm long quartz cells (Starna Cells, Inc.). MQ water was used as a blank for optical measurements; after blank subtraction, absorbance values (Aλ) were then converted to Napierian absorption coefficients (aλ)77:
where L is the pathlength, in meters. Fluorescence emission (Em) spectra (300 to 600 nm) were measured at multiple excitation (Ex) wavelengths (240 to 600 nm, in 5 nm increments) on a Varian Eclipse fluorescence spectrometer. Integration time was 0.2 s and excitation and emission slit widths were set to 5 nm. MQ water blanks and quinine sulfate standardization of the detector were conducted each analytical day; results were quantified in Raman units (R. U.). Prior to data analysis, corrections for absorption inner-filter effects and instrument bias were conducted using in-house Matlab code. Emission spectra were concatenated into excitation emission matrices (EEMs) for visualization as contour plots. Values for the various “Coble” peaks (Table 1) were extracted from the EEMs using regional integration over reported wavebands78. We used the ratio of different peaks to describe the fulvic and humic nature of HS fluorescence79. The marine HS were estimated as the sum of fluorescence under peaks M and N divided by the sum of peaks A, C, M, and N. The terrestrial-like were estimated as the sum of fluorescence of peaks A and C divided by the sum of peaks A, C, M, and N.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information Files).
References
Moore, J. K., Doney, S. C., Glover, D. M. & Fung, I. Y. Iron cycling and nutrient-limitation patterns in surface waters of the World Ocean. Deep Sea Research Part II: Topical Studies in Oceanography 49, 463–507 (2001).
Tagliabue, A., Aumont, O. & Bopp, L. The impact of different external sources of iron on the global carbon cycle. Geophysical Research Letters 41, 920–926 (2014).
Bruland, K. W., Middag, R. & Lohan, M. C. In Treatise on Geochemistry Vol. 8 (ed. Holland, H. D. & Turekian, K.) 19–51 (Elsevier, 2014).
Tagliabue, A. et al. How well do global ocean biogeochemistry models simulate dissolved iron distributions? Global Biogeochemical Cycles 30, 149–174 (2016).
Gledhill, M. & Buck, K. N. The organic complexation of iron in the marine environment: a review. Front Microbiol 3 (2012).
Hopkinson, B. M. & Morel, F. M. M. The role of siderophores in iron acquisition by photosynthetic marine microorganisms. BioMetals 22, 659–669 (2009).
Boiteau, R. M. et al. Patterns of iron and siderophore distributions across the California Current System. Limnology and Oceanography 64, 376–389 (2019).
Bundy, R. M. et al. Distinct siderophores contribute to iron cycling in the mesopelagic at station ALOHA. Frontiers in Marine Science 5 (2018).
Hassler, C. S. et al. Iron associated with exopolymeric substances is highly bioavailable to oceanic phytoplankton. Marine Chemistry 173, 136–147 (2015).
Zigah, P. K. et al. Allochthonous sources and dynamic cycling of ocean dissolved organic carbon revealed by carbon isotopes. Geophysical Research Letters 44, 2407–2415 (2017).
Croot, P. L. & Johansson, M. Determination of iron speciation by cathodic stripping voltammetry in seawater using the competing ligand 2‐(2‐Thiazolylazo)‐p‐cresol (TAC). Electroanalysis 12, 565–576 (2000).
Dulaquais, G. et al. The biogeochemistry of electroactive humic substances and its connection to iron chemistry in the North East Atlantic and the Western Mediterranean Sea. Journal of Geophysical Research: Oceans (2018).
Laglera, L. M. & van den Berg, C. M. G. Evidence for geochemical control of iron by humic substances in seawater. Limnology and Oceanography 54, 610–619 (2009).
Batchelli, S., Muller, F. L. L., Chang, K.-C. & Lee, C.-L. Evidence for strong but dynamic iron–humic colloidal associations in humic-rich coastal waters. Environmental Science & Technology 44, 8485–8490 (2010).
Zhang, Y., Du, J., Ding, X. & Zhang, F. Comparison study of sedimentary humic substances isolated from contrasting coastal marine environments by chemical and spectroscopic analysis. Environmental Earth Sciences 75, 378 (2016).
Sarma, N. S. et al. Hydrothermal alteration promotes humic acid formation in sediments: a case study of the Central Indian Ocean Basin. Journal of Geophysical Research: Oceans 123, 110–130 (2018).
Calace, N., Cantafora, E., Mirante, S., Petronio, B. M. & Pietroletti, M. Transport and modification of humic substances present in Antarctic snow and ancient ice. Journal of Environmental Monitoring 7, 1320–1325 (2005).
Gelencsér, A. et al. On the possible origin of humic matter in fine continental aerosol. Journal of Geophysical Research: Atmospheres 107, ACH 2-1-ACH 2-6 (2002).
Hioki, N. et al. Laterally spreading iron, humic-like dissolved organic matter and nutrients in cold, dense subsurface water of the Arctic Ocean. Scientific Reports 4, 6775 (2014).
Kinniburgh, D. G. et al. Ion binding to natural organic matter: competition, heterogeneity, stoichiometry and thermodynamic consistency. Colloids and Surfaces A: Physicochemical and Engineering Aspects 151, 147–166 (1999).
Krachler, R. et al. River-derived humic substances as iron chelators in seawater. Marine Chemistry 174, 85–93 (2015).
Kikuchi, T. et al. Correlations between aromaticity of dissolved organic matter and trace metal concentrations in natural and effluent waters: A case study in the Sagami River Basin, Japan. Science of The Total Environment 576, 36–45 (2017).
Bailey, G. W. Life after death: Lignin‐humic relationships reexamined. Critical Reviews in Environmental Science and Technology 26, 95–153 (1996).
Harvey, G. R., Boran, D. A., Chesal, L. A. & Tokar, J. M. The structure of marine fulvic and humic acids. Marine Chemistry 12, 119–132 (1983).
Hertkorn, N. et al. Characterization of a major refractory component of marine dissolved organic matter. Geochimica et Cosmochimica Acta 70, 2990–3010 (2006).
Hatcher, P. G., Maciel, G. E. & Dennis, L. W. Aliphatic structure of humic acids; a clue to their origin. Organic Geochemistry 3, 43–48 (1981).
Sukekava, C., Downes, J., Slagter, H. A., Gerringa, L. J. A. & Laglera, L. M. Determination of the contribution of humic substances to iron complexation in seawater by catalytic cathodic stripping voltammetry. Talanta 189, 359–364 (2018).
Stuermer, D. H. & Harvey, G. R. The isolation of humic substances and alcohol-soluble organic matter from seawater. Deep Sea Research 24, 303–309 (1977).
Timko, S. et al. Depth-dependent photodegradation of marine dissolved organic matter. Frontiers in Marine Science 2 (2015).
Nelson, N. B., Siegel, D. A., Carlson, C. A. & Swan, C. M. Tracing global biogeochemical cycles and meridional overturning circulation using chromophoric dissolved organic matter. Geophysical Research Letters 37 (2010).
Williams, P. M. & Druffel, E. R. M. Radiocarbon in dissolved organic matter in the central North Pacific Ocean. Nature 330, 246–248 (1987).
Álvarez-Salgado, X. A. et al. New insights on the mineralization of dissolved organic matter in central, intermediate, and deep water masses of the northeast North Atlantic. Limnology and Oceanography 58, 681–696 (2013).
Benner, R., Louchouarn, P. & Amon, R. M. W. Terrigenous dissolved organic matter in the Arctic Ocean and its transport to surface and deep waters of the North Atlantic. Global Biogeochemical Cycles 19 (2005).
Nelson, N. B. & Gauglitz, J. M. Optical signatures of dissolved organic matter transformation in the global ocean. Frontiers in Marine Science 2 (2016).
Kothawala, D. N., von Wachenfeldt, E., Koehler, B. & Tranvik, L. J. Selective loss and preservation of lake water dissolved organic matter fluorescence during long-term dark incubations. Science of The Total Environment 433, 238–246 (2012).
Rijkenberg, M. J. A., Slagter, H. A., Rutgers van der Loeff, M., van Ooijen, J. & Gerringa, L. J. A. Dissolved Fe in the deep and upper Arctic Ocean with a focus on Fe limitation in the Nansen Basin. Frontiers in Marine Science 5 (2018).
Romera-Castillo, C., Sarmento, H., Alvarez-Salgado, X. A., Gasol, J. M. & Marrase, C. Net production and consumption of fluorescent colored dissolved organic matter by natural bacterial assemblages growing on marine phytoplankton exudates. Appl Environ Microb 77, 7490–7498 (2011).
Kitayama, S. et al. Controls on iron distributions in the deep water column of the North Pacific Ocean: Iron(III) hydroxide solubility and marine humic-type dissolved organic matter. Journal of Geophysical Research-Oceans 114 (2009).
Stedmon, C. A. & Nelson, N. B. In Biogeochemistry of Marine Dissolved Organic Matter (Second Edition) (eds Dennis A. Hansell & Craig A. Carlson) 481–508 (Academic Press, 2015).
Heller, M. I., Gaiero, D. M. & Croot, P. L. Basin scale survey of marine humic fluorescence in the Atlantic: Relationship to iron solubility and H2O2. Global Biogeochemical Cycles 27, 88–100 (2013).
Buck, K. N., Sohst, B. & Sedwick, P. N. The organic complexation of dissolved iron along the U.S. GEOTRACES (GA03) North Atlantic Section. Deep Sea Research Part II: Topical Studies in Oceanography 116, 152–165 (2015).
Buck, K. N., Sedwick, P. N., Sohst, B. & Carlson, C. A. Organic complexation of iron in the eastern tropical South Pacific: Results from US GEOTRACES Eastern Pacific Zonal Transect (GEOTRACES cruise GP16). Marine Chemistry 201, 229–241 (2018).
Gerringa, L. J. A. et al. Dissolved Fe and Fe-binding organic ligands in the Mediterranean Sea – GEOTRACES G04. Marine Chemistry 194, 100–113 (2017).
Abualhaija, M. M., Whitby, H. & van den Berg, C. M. G. Competition between copper and iron for humic ligands in estuarine waters. Marine Chemistry 172, 46–56 (2015).
Slagter, H. A. et al. Organic Fe speciation in the Eurasian Basins of the Arctic Ocean and its relation to terrestrial DOM. Marine Chemistry 197, 11–25 (2017).
Hiemstra, T. & van Riemsdijk, W. H. Biogeochemical speciation of Fe in ocean water. Marine Chemistry 102, 181–197, https://doi.org/10.1016/j.marchem.2006.03.008 (2006).
Tipping, E., Rey-Castro, C., Bryan, S. E. & Hamilton-Taylor, J. Al(III) and Fe(III) binding by humic substances in freshwaters, and implications for trace metal speciation. Geochimica et Cosmochimica Acta 66, 3211–3224, https://doi.org/10.1016/S0016-7037(02)00930-4 (2002).
Avendaño, L., Gledhill, M., Achterberg, E. P., Rérolle, V. M. C. & Schlosser, C. Influence of ocean acidification on the organic complexation of iron and copper in Northwest European shelf seas; a combined observational and model study. Frontiers in Marine Science 3, https://doi.org/10.3389/fmars.2016.00058 (2016).
Boye, M. et al. The chemical speciation of iron in the north-east Atlantic Ocean. Deep Sea Research Part I: Oceanographic Research Papers 53, 667–683 (2006).
Gerringa, L. J. A. et al. Fe-binding dissolved organic ligands near the Kerguelen Archipelago in the Southern Ocean (Indian sector). Deep-Sea Res Pt Ii 55, 606–621 (2008).
Martin, J. H. Glacial-Interglacial CO2 Change: The Iron Hypothesis. Paleoceanography 5, 1–13 (1990).
Schlünz, B., Schneider, R. R., Müller, P. J., Showers, W. J. & Wefer, G. Terrestrial organic carbon accumulation on the Amazon deep sea fan during the last glacial sea level low stand. Chemical Geology 159, 263–281, https://doi.org/10.1016/S0009-2541(99)00041-8 (1999).
Burdige, D. J. Burial of terrestrial organic matter in marine sediments: A re-assessment. Global Biogeochemical Cycles 19 (2005).
Shaffer, G. & Lambert, F. In and out of glacial extremes by way of dust−climate feedbacks. Proceedings of the National Academy of Sciences 115, 2026–2031 (2018).
Cutter, G. A. & Bruland, K. W. Rapid and noncontaminating sampling system for trace elements in global ocean surveys. Limnology and Oceanography: Methods 10, 425–436 (2012).
Measures, C. I., Landing, W. M., Brown, M. T. & Buck, C. S. A commercially available rosette system for trace metal-clean sampling. Limnology and Oceanography-Methods 6, 384–394 (2008).
Bruland, K. W., Rue, E. L., Smith, G. J. & DiTullio, G. R. Iron, macronutrients and diatom blooms in the Peru upwelling regime: brown and blue waters of Peru. Marine Chemistry 93, 81–103 (2005).
Bowie, A. R. et al. Biogeochemical iron budgets of the Southern Ocean south of Australia: Decoupling of iron and nutrient cycles in the subantarctic zone by the summertime supply. Global Biogeochemical Cycles 23 (2009).
Cutter, G. et al. Sampling and sample-handling protocols for GEOTRACES cruises (2010).
Buck, K. N. et al. The organic complexation of iron and copper: an intercomparison of competitive ligand exchange-adsorptive cathodic stripping voltammetry (CLE-ACSV) techniques. Limnology and Oceanography-Methods 10, 496–515 (2012).
Lagerström, M. E. et al. Automated on-line flow-injection ICP-MS determination of trace metals (Mn, Fe, Co, Ni, Cu and Zn) in open ocean seawater: Application to the GEOTRACES program. Marine Chemistry 155, 71–80 (2013).
Tonnard, M. et al. Dissolved iron in the North Atlantic Ocean and Labrador Sea along the GEOVIDE section (GEOTRACES section GA01). Biogeosciences 17(4), 917-943 (2020).
Quéroué, F. et al. High variability in dissolved iron concentrations in the vicinity of the Kerguelen Islands (Southern Ocean). Biogeosciences 12, 3869–3883 (2015).
Jackson, S. L., Spence, J., Janssen, D. J., Ross, A. R. S. & Cullen, J. T. Determination of Mn, Fe, Ni, Cu, Zn, Cd and Pb in seawater using offline extraction and triple quadrupole ICP-MS/MS. Journal of Analytical Atomic Spectrometry 33, 304–313 (2018).
Whitby, H. & van den Berg, C. M. G. Evidence for copper-binding humic substances in seawater. Marine Chemistry 173, 282–290 (2015).
Whitby, H., Posacka, A. M., Maldonado, M. T. & van den Berg, C. M. G. Copper-binding ligands in the NE Pacific. Marine Chemistry 204, 36–48 (2018).
Omanovic, D., Gamier, C. & Pizeta, I. ProMCC: An all-in-one tool for trace metal complexation studies. Marine Chemistry 173, 25–39 (2015).
Laglera, L. M., Battaglia, G. & van den Berg, C. M. G. Effect of humic substances on the iron speciation in natural waters by CLE/CSV. Marine Chemistry 127, 134–143 (2011).
Powell, R. T. & Donat, J. R. Organic complexation and speciation of iron in the South and Equatorial Atlantic. Deep Sea Research Part II: Topical Studies in Oceanography 48, 2877–2893 (2001).
Witter, A. E., Lewis, B. L. & Luther, G. W. III. Iron speciation in the Arabian Sea. Deep Sea Research Part II: Topical Studies in Oceanography 47, 1517–1539 (2000).
Boye, M. et al. Horizontal gradient of the chemical speciation of iron in surface waters of the northeast Atlantic Ocean. Marine Chemistry 80, 129–143 (2003).
Boye, M. et al. Organic complexation of iron in the Southern Ocean. Deep Sea Research Part I: Oceanographic Research Papers 48, 1477–1497 (2001).
Rue, E. L. & Bruland, K. W. Complexation of iron(III) by natural organic ligands in the Central North Pacific as determined by a new competitive ligand equilibration/adsorptive cathodic stripping voltammetric method. Marine Chemistry 50, 117–138 (1995).
Nolting, R. F., Gerringa, L. J. A., Swagerman, M. J. W., Timmermans, K. R. & de Baar, H. J. W. Fe (III) speciation in the high nutrient, low chlorophyll Pacific region of the Southern Ocean. Marine Chemistry 62, 335–352 (1998).
Gledhill, M. & van den Berg, C. M. G. Determination of complexation of iron(III) with natural organic complexing ligands in seawater using cathodic stripping voltammetry. Marine Chemistry 47, 41–54 (1994).
Gledhill, M., van den Berg, C. M. G., Nolting, R. F. & Timmermans, K. R. Variability in the speciation of iron in the northern North Sea. Marine Chemistry 59, 283–300 (1998).
Green, S. A. & Blough, N. V. Optical absorption and fluorescence properties of chromophoric dissolved organic matter in natural waters. Limnology and Oceanography 39, 1903–1916 (1994).
Coble, P. G. Marine optical biogeochemistry: the chemistry of ocean color. Chemical Reviews 107, 402–418 (2007).
Kowalczuk, P., Tilstone, G. H., Zabłocka, M., Röttgers, R. & Thomas, R. Composition of dissolved organic matter along an Atlantic Meridional Transect from fluorescence spectroscopy and Parallel Factor Analysis. Marine Chemistry 157, 170–184 (2013).
Coble, P. G., Del Castillo, C. E. & Avril, B. Distribution and optical properties of CDOM in the Arabian Sea during the 1995 Southwest Monsoon. Deep Sea Research Part II: Topical Studies in Oceanography 45, 2195–2223 (1998).
Stedmon, C. A. & Markager, S. Resolving the variability in dissolved organic matter fluorescence in a temperate estuary and its catchment using PARAFAC analysis. Limnology and Oceanography 50, 686–697 (2005).
Stedmon, C. A., Markager, S. & Bro, R. Tracing dissolved organic matter in aquatic environments using a new approach to fluorescence spectroscopy. Marine Chemistry 82, 239–254 (2003).
Parlanti, E., Wörz, K., Geoffroy, L. & Lamotte, M. Dissolved organic matter fluorescence spectroscopy as a tool to estimate biological activity in a coastal zone submitted to anthropogenic inputs. Organic Geochemistry 31, 1765–1781 (2000).
Sierra, M. M. D., Giovanela, M., Parlanti, E. & Soriano-Sierra, E. J. Fluorescence fingerprint of fulvic and humic acids from varied origins as viewed by single-scan and excitation/emission matrix techniques. Chemosphere 58, 715–733 (2005).
Acknowledgements
We thank the chief scientists Pascale Lherminier, Marie Robert, Bernard Quéguiner and Stéphane Blain, and the captains, crew and all members of the trace metal sampling teams on each cruise. We thank Jody Spence who runs the ICP lab at UVic, and Stephen Richardson for running the C/FDOM analyses (NC State University) NSF Award OCE 1459406 (CLO). We thank Sébastien Hervé (IUEM) for the schematic, and Andrew Ross, Maria Maldonado, Marie Cheize and Alessandro Tagliabue for their input. This work was supported by the French National Research Agency (ANR-13-BS06-0014, ANR-12-PDOC-0025-01, ANR-2010-BLAN-614Keops2), the French National Centre for Scientific Research (CNRS-LEFE-CYBER), Ifremer, the French CNES, the French Polar Institute IPEV, the “Laboratoire d’Excellence” LabexMER (ANR-10-LABX-19) and co-funded by a grant from the French government under the program “Investissements d’Avenir”, by a grant from the Regional Council of Brittany (SAD programme), and by the EU FP7 Marie Curie actions (PCOFUND- GA-2013-609102), through the PRESTIGE programme coordinated by Campus France. It was also supported for the logistic of the GEOVIDE cruise by DT-INSU and GENAVIR, and from Canada’s Natural Sciences and Engineering Research Council (NSERC) Discovery Grant program. AGG was also funded by a Postdoctoral grant at the Universidad de Las Palmas de Gran Canaria.
Author information
Authors and Affiliations
Contributions
Led design of the study, writing of the manuscript, assembly of the iron and H.S. datasets and H.S. measurements (H.W.); ligand measurements (H.W., A.G.G.), FDOM measurements (C.L.O.) and Pacific iron measurements (D.J.J.). Provided samples and cruise data (G.S., H.P., J.T.C.). Participated in discussion of the hypothesis and underlying concepts (H.W., N.C., C.L.O., E.B., G.S., H.P., C.V.). All authors contributed to the overall discussion of the results and their implications, as well as commenting on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Whitby, H., Planquette, H., Cassar, N. et al. A call for refining the role of humic-like substances in the oceanic iron cycle. Sci Rep 10, 6144 (2020). https://doi.org/10.1038/s41598-020-62266-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-62266-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
