
Abstract
Lethal total body irradiation (TBI) triggers multifactorial health issues in a potentially short time frame. Hence, early signatures of TBI would be of great clinical value. Our study aimed to interrogate microRNA (miRNA) and metabolites, two biomolecules available in blood serum, in order to comprehend the immediate impacts of TBI. Mice were exposed to a lethal dose (9.75 Gy) of Cobalt-60 gamma radiation and euthanized at four time points, namely, days 1, 3, 7 and 9 post-TBI. Serum miRNA libraries were sequenced using the Illumina small RNA sequencing protocol, and metabolites were screened using a mass spectrometer. The degree of early impacts of irradiation was underscored by the large number of miRNAs and metabolites that became significantly expressed during the Early phase (day 0 and 1 post-TBI). Radiation-induced inflammatory markers for bone marrow aplasia and pro-sepsis markers showed early elevation with longitudinal increment. Functional analysis integrating miRNA-protein-metabolites revealed inflammation as the overarching host response to lethal TBI. Early activation of the network linked to the synthesis of reactive oxygen species was associated with the escalated regulation of the fatty acid metabolism network. In conclusion, we assembled a list of time-informed critical markers and mechanisms of significant translational potential in the context of a radiation exposure event.
Similar content being viewed by others

Introduction
In the current geopolitical climate, the threat of radiation exposure either through intentional detonation or accidental exposure represents an ever present concern. To this end, it is imperative that we bolster our understanding of the systems and biological processes that are affected by exposure to ionizing radiation. Collectively the symptoms and health consequences that follow radiation exposure are referred to as acute radiation syndrome (ARS). It has been established that exposure to different levels of radiation can lead to different sets of sub-syndromes (i.e., hematopoietic, gastro-intestinal [GI], and central nervous system [CNS])1. Each of these is associated with the severity of the absorbed dose of radiation; bone marrow is regarded as the most susceptible tissue to radiation exposure, and the hematopoietic sub-syndrome occurs at the lowest doses of radiation. Many medicinal countermeasures are targeted at doses of radiation in the hematopoietic range. At higher doses of radiation, there can be damage to the gastro-intestinal system (GI). At very high doses, organisms can be susceptible to damage in the CNS leading to the rapid onset of symptoms that often result in mortality.
In the mouse, the range for hematopoietic ARS is generally accepted to be from 2 to 10 Gray (Gy)2,3; however, even amongst various strains of mice, the radiation dose for lethality can vary. For instance, in a recent study investigating the medical countermeasure Filgrastim4, four mouse strains were studied, and four different radiation doses for 50% lethality at 30 days (Lethal Dose or LD50/30) were identified. For the inbred strains such as C3H/HeN and C57BL/6, the doses for LD50/30 were identified as 7.56 and 7.74 Gy, respectively. In the B6C3F1 strain, which is a cross between the two inbred strains, the value for LD50/30 was identified as 8.12 Gy, and in the CD2F1 strain, which is a cross between the BALB/c and DBA/2, the LD50/30 value is even higher at 8.65 Gy. Furthermore, the sex of the mouse changes the response to radiation exposure5. Mice experiencing hematopoietic ARS often exhibit decreased numbers of peripheral blood cells, leading to neutropenia and thrombocytopenia, which can result in death often from infection or bleeding.
At higher radiation doses in the mouse (~10 Gy and above), damage resulting from radiation exposure can be observed in the GI system in addition to bone marrow, and weight loss; and diarrhea are typically observed, which can lead to dehydration and mortality6. There is an increasing traction about determining the role of serum miRNAs in responding to radiation, particularly in the context of biodosimetry7,8. A number of reviews highlighted the potential utility of miRNAs as predictors of carcinoma prognosis9,10 and of drug efficacy11,12. The direct relationship of miRNA and radiation exposure was further underscored by a study demonstrating differential expression of miRNAs in C57BL/6 mice exposed to either a sub-lethal dose of 6.5 Gy or a lethal dose of 8 Gy, and in which, the levels of miRNA perturbation reverted back to baseline after administration of amifostine as a medical countermeasure13. In this study, authors identified several differentially expressed miRNAs, such as miR-126-3p, miR-150, miR-342-3p, miR-151-3p, miR-139-3p, and miR-142 and saw overlap with results from previous studies7,14,15. In an earlier report (2016)16, we have shown that radiation-induced miRNAs in the spleen might be involved in radioprotection by a vitamin E analog, gamma-tocotrienol (GT3). Functional analysis indicated that the radiation-induced miRNAs, which were modulated by GT3 were involved in hematopoiesis and lymphogenesis16. The radiation dose and time lag following exposure are two critical factors that essentially define the degree of consequences of radiation. For instance, the hematopoietic symptoms of ARS typically takes a certain period of time to develop in vivo, as the mortality begins weeks after radiation exposure1. By contrast, animals that have been treated with a radiation dose high enough to induce the GI sub-syndrome will typically exhibit symptoms within the first several days, and mortality begins in less than a week1. For the purpose of our present study, we functionally defined the Early period relative to radiation exposure as the first two days post-TBI. Likewise, we defined the Late period as the ninth day after irradiation. The interim period was defined as the Mid period. Bridging the knowledge gap about the temporal impact of radiation exposure17,18; here we present a longitudinal pattern of miRNA shift promoted by lethal dose of radiation.
Another emerging field is the study of metabolomics in biosamples collected from animals that have been exposed to radiation19. Metabolomics is a powerful tool to study post-radiation changes in metabolism and bioenergetics even before the onset of clinical symptoms and can augment triage of an exposed population in real-life scenarios19,20. Past metabolomics analysis investigated mouse21 and rat22 urine, rat plasma23, and mouse livers24,25. In 2013, we reported for the first time radiation-induced metabolite changes in the GI tissues of mice using ultraperformance liquid chromatography (UPLC) coupled with Time-of-flight mass spectrometry (TOF-MS)20. Mice were exposed to TBI doses of 4 and 8 Gy, and blood and jejuna were collected at 1 and 4 days post-irradiation20. Novel metabolites including lipids, glutamate, tryptophan, taurocholate and the Cysteine-Glycine dipeptide were identified20. In addition, human serum samples have been analyzed in individuals following radiotherapy26,27; these studies indicated upregulation of biomarkers corresponding to energy metabolism including increased branched chain amino acid synthesis and protein biosynthesis27. C57BL/6 mouse biosamples including bone marrow, ileum, muscle, lung, serum, and urine have been analyzed within 12 hours of 6 Gy irradiation and demonstrated differences in DNA methylation pattern, energy expenditure and metabolism of amino acid, glutathione, and bile, respectively28. Unlike miRNA profiling, radiation-induced metabolite profiles at various time points relative to radiation exposure warrants further study. In the current study, we aimed to comprehend the longitudinal dynamics of serum miRNA-metabolite landscape perturbed by lethal radiation. For these studies, we used CD2F1 male mice that were exposed to TBI at a GI sub-syndrome-inducing level of 9.75 Gy. These mice were ~12 weeks old at the time of irradiation which is equivalent to the age of a young adult when comparing the mouse lifespan of a mouse to a human29. Serum samples were collected in Early, Mid, and Late time points relative to TBI. Our result identified a set of miRNA and metabolite markers. The networks co-enriched by differentially expressed miRNA and metabolites suggested a surge of inflammatory response to lethal radiation. A number of disease footprints related to hematopoiesis and cardiovascular atrophy emerged as the potential targets for immediate intervention.
Materials and Methods
Handling of mice
Mouse were sourced and housed as described previously30. Briefly, CD2F1 male mice (8–10 weeks old) were purchased from Harlan (Indianapolis, IN) and housed in an air-conditioned facility at the Armed Forces Radiobiology Research Institute (AFRRI) which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). All mice were kept in rooms with a 12 h light/dark cycle at 21 ± 2 °C with 10–15 hourly cycles of fresh air and a relative humidity of 50 ± 10%. Mice were held in quarantine for 2 weeks and were used after microbiology, serology, and histopathology examination of representative samples ensured the absence of Pseudomonas aeruginosa and common murine diseases. Mice were given certified rodent rations (Harlan Teklad Rodent Diet #8604, Harlan Teklad, WI, USA) and acidified water (with HCl, pH 2.5–3.0) ad libitum. Animal procedures were executed following a protocol approved by the AFRRI’s Institutional Animal Care and Use Committee (IACUC). Research was conducted according to the Guide for the Care and Use of Laboratory Animals, prepared by the Institute of Laboratory Animal Resources, the National Research Council, U.S. National Academy of Sciences16.
Irradiation
Mice were irradiated bilaterally in the AFRRI cobalt-60 gamma radiation facility in well ventilated plexiglass boxes (8 mice per box) at a dose rate of ~0.6 Gy/minute (min) to total midline doses of 9.75 Gy, as described below. An alanine/electron spin resonance (ESR) dosimetry system (American Society for Testing and Material Standard E 1607) was used to measure dose rates (to water) in the cores of acrylic mouse phantoms. Phantoms were 3 inches long and 1 inch in diameter and were located in 50% of the compartments of the exposure rack. The ESR signals were measured with a calibration curve based on standard calibration dosimeters provided by the National Institute of Standard and Technology (NIST, Gaithersburg, MD). The accuracy of the calibration curve was verified by inter-comparison with the National Physical Laboratory (NPL) in the United Kingdom. The only corrections applied to the dose rates in phantoms were for the decay of the 60Co source and for a small difference in mass energy-absorption coefficients for water and soft tissue. The radiation field was uniform within ±2%. After irradiation, mice were returned to their original cages with access to food and water ad libitum17,30. All irradiations were performed in the morning to minimize the diurnal effect.
Blood collection
The experimental animals received either 0 or 9.75 Gy radiation (lethal dose) at a dose rate ~0.6 Gy min−1 in the AFRRI 60Co gamma radiation facility. Blood was collected from inferior vena cava under anesthesia on days 0 (2 h post-TBI), 1, 3, 7, and 9 from either 9.75 Gy-irradiated or unirradiated mice followed by euthanasia. Serum was separated and stored at −80 °C until use.
microRNA sequencing and analysis
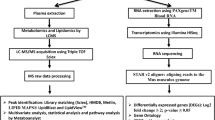
We followed the protocol reported by us elsewhere with necessary modification31. Briefly, 5 µL serum was used to construct sequencing libraries with the Illumina (Illumina, CA, USA) TruSeq Small RNA Sample Prep Kit, following the manufacturer’s guideline. Briefly, 3′ and 5′ adapters were sequentially ligated to small RNA molecules, and the ligated products were reverse transcribed, amplified and subsequently size-selected screened by gel purification and by microfluidics using the Agilent Bioanalyzer High Sensitivity DNA chip. The indexed libraries were pooled in equimolar amounts, clustered and loaded into different lanes of a HiScanSQ Illumina instrument to generate 50 base-pair reads. Image analysis and base calling were performed using Illumina pipeline Version 1.5.15.1 and Illumina CASAVA sequencing analysis software Version 1.7.32.0. The raw sequence reads were processed using a robust miRNA-seq quality control pipeline. First, the samples were evaluated as per quality control assurance using matrices that included acceptable duplication, k-mer or GC content generated using FASTQC. The samples that showed more than 30% disagreement with rest of the samples were excluded henceforth. The low quality reads (Q20 quality score threshold) were filtered out, and adaptor sequences were pruned. The ensuing products were 48%–75% mapped against the mature miRBase miRNA database Version 21 for Mus musculus (mm9), a pool of RNA species was curated, and the effective library sizes were normalized using the trimmed mean of M-values (TMM) normalization method provided by edgeR (Bioconductor.org). In a second phase of filtration, the samples with <1% abundance were also discarded. Meanwhile, we validated the miRBase output by mapping the same filtered sequence reads against the UCSC reference genome for Mus musculus (mm9 build) (University of California Santa Cruz; http://genome.ucsc.edu/).
The short read aligner Bowtie (v 1.0.0) allowed one pair of mismatch. For the baseline, we combined the miRNA readouts obtained from the serum samples collected from two control time points (C1 and C7). Differentially expressed miRNAs were mined by pair-wise comparison between the baseline and individual time points using a moderated t test with the cutoff at p < 0.05.
Protein ELISA assay
Mouse Erythropoietin (EPO) Quantikine ELISA and mouse/Rat Flt3 ligand (Flt3L) quantikine ELISA kits were purchased from R&D Systems Inc. (Minneapolis, MN). Mouse Serum Amyloid A (SAA) quantikine ELISA kits were purchased from Tridelta Development Limited (Maynooth, County Kildare, Ireland). The cytokine detection limits were 18 pg mL−1, >5 pg mL−1 and 30 ng mL−1 for EPO, Flt-3L and SAA ELISAs, respectively. The quantitative levels of EPO, Flt3L, and SAA were evaluated from serum samples collected on days 1, 3, 7 and 9 post-TBI following standard protocols from the vendor. The ELISA result in pg/mL was first computed for individual pre-irradiation baseline and post-irradiation time points, respectively. Fold change was calculated by dividing the irradiation data points by the pre-irradiation baseline data. One-way ANOVA followed by multiple comparison was computed to find the significant changes.
Global metabolite mass spectroscopy assay
The metabolite profiling of serum samples was conducted as per the report published previously32. Here each sample (5 μL) was injected onto a reverse-phase 50 × 2.1 mm Acquity 1.7-μm C18 column (Waters Corp, Milford, MA) using an Acquity UPLC system (Waters) with a gradient mobile phase consisting of 2% acetonitrile in water containing 0.1% formic acid (Solvent A) and 2% water in acetonitrile containing 0.1% formic acid (Solvent B) and resolved for 10 min at a flow rate of 0.5 mL/min. The gradient was consisted of 100% A for 0.5 min with a ramp of curve 6–100% B from 0.5 to 10 min. The column eluent was introduced directly into the mass spectrometer by electrospray. Mass spectrometry was performed on a Quadrupole Time-of-Flight (Q-TOF) Premier mass spectrometer (Waters) operating in either positive-ion or negative-ion electrospray ionization (ESI+/−) mode with a capillary voltage of 3200 V and a sampling cone voltage of 20 V in negative mode and 35 V in positive mode. The desolvation gas flow was set to 800 L/hour, and the temperature was set to 350 °C. The cone gas flow was 25 L/hour, and the source temperature was 120 °C. Accurate mass was maintained by introduction of LockSpray interface of sulfadimethoxine (311.0814 [M + H]+ or 309.0658 [M − H]−) at a concentration of 250 pg/μL in 50% aqueous acetonitrile and a rate of 150 μL/min. Data were acquired in centroid mode from 50 to 850 mass to change ratio (m/z) in mass spectrometry scanning. Centroided and integrated mass spectrometry data obtained in duplicates from the UPLC-TOF mass spectrometer were processed using XCMS (Scripps Institute) to generate a data matrix containing ion intensities, m/z and retention time values. Peaks were machine normalized and mined with ppm error cutoff <1. Baseline was defined as the serum samples collected at two control cohorts (C1 and C7). Significantly different peaks at individual time points were selected using moderated t-test p < 0.05.
Statistical analysis
GeneSpring 14.9 (Agilent Technologies, Inc., Santa Clara, CA) was used to find statistically significant markers and to compute principal component analysis (PCA) and clustering analysis. K-means clustering was performed using Euclidian similarity measures and 50 iterations. Hierarchical clustering was conducted using Similarity Measure: Squared Euclidean similarity measures and Wards linkage rule. Prism 7 (GraphPad, Inc. San Diego, CA) was used for data visualization.
The differentially expressed mass spectroscopy peaks were annotated using the CEU Mass Mediator 3.0 database (www.ceumass.eps.uspceu.es), and the molecules were screened based on the following guidelines: (a) Ppm error less than 1; (b) chemical formula comprised of the adducts +H, −H, +Na, +K, +NH4, −Cl and (c) chemicals listed as endogenous mammalian.
Ingenuity Pathway Analysis (IPA, QIAGEN, Inc., version 01–13) was used for functional analysis and network building. Those miRNAs and metabolites, which were differentially expressed at each phases post-TBI, that is Early-, Mid- and Late-phase post-TBI, were seeded in the IPA analysis portal; and the canonical and non-canonical networks significantly enriched (hypergeometric test, p < 0.05) at each phase were curated. We further screened the list to select those significantly enriched networks, which were enriched by at least 5 molecules that included miRNA and/or metabolites. Finally, we used Venn diagram to screen the networks which were significantly enriched across the time phases post-TBI.
Results
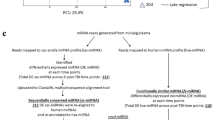
The mice were exposed to 9.75 Gy TBI. Post-irradiation, the mice were longitudinally euthanized at five different time points. One cohort was euthanized two hours after the irradiation, and the time point was defined as day 0 (d0). The post-TBI time points included day one (d1), day three (d3), day seven (d7) and day nine (d9). Sham animals were euthanized at two different time points to take into account the aging factor: day one (C1) and day seven (C7), which were equivalent to d1 and d7 post-TBI. All the cohorts including both irradiated and sham consisted of five mice per group (Fig. 1). At this radiation dose (9.75 Gy) CD2F1 male mice (12–14 weeks old) typically exhibit clinical symptoms from days 9 to 10 post-TBI18. Clinical symptoms include hunched posture, difficulty in breathing and slow movement33. About 90–95% mortality has been observed over 30 days at this dose18,30.

Study design. CD2F1 male mice were exposed to LD90 total body irradiation (TBI). Post-TBI, mice were euthanized at five time points: 6 h (marked as d0), one day (d1), three days (d3), seven days (d7) and nine days (d9). Two control cohorts (C1 and C7) accounted for the baseline. Whole genome miRNA profiling, global metabolomics and targeted metabolomics were conducted at each time point.
miRNA analysis
PCA analysis of serum miRNA of individual mice was plotted in Fig. 2A. The first two principal components explained 31.2% of total variance with 28% attributable to PC1. Baseline controls (C1 and C7) were clustered together with early irradiation time points (d0 and d1). Mice euthanized on d7 post-irradiation were clustered separately, and mice euthanized at d9 were clustered far from the rest the mice across PC1.

The whole genome miRNA profile responding to lethal radiation. (A) Principal component analysis (PCA) of the whole genome miRNA data shows a clear separation between d9 post-TBI and rest of the time points including the controls. A dotted line marks the separation. (B) Venn diagram shows the longitudinal distribution of differentially expressed miRNAs across three time phases post-TBI, namely, Early (d0–d1), Mid (d3–d7) and Late (d9). The largest number of differentially expressed miRNAs (115 miRNAs) are found at the cross section of the three phase. Nevertheless, a considerable number of miRNAs are exclusively regulated during each of the three phases. For instance, there are 72, 61 and 67 miRNAs exclusively regulated in the Early, Mid and Late phases, respectively. (C) Venn diagram shows the longitudinal distribution of significantly enriched networks across three time phases post-TBI, namely, Early (d0–d1), Mid (d3–d7) and Late (d9). Nearly all of these networks are concentrated at the cross section of all three time phases. (D) Top 5 most significantly enriched networks during the Early, Early-Mid and Early-Mid-Late phases are shown in this bar chart. Early and Early-Mid phases enlist only 3 networks, so all of them are included here. Top horizontal axis represents the −log(p value), and the bar charts represents the hypergeometric p-values converted to log10 associated with individual networks. The bottom horizontal axis represents the sample size (N), and the open circles represent the number of miRNAs associated with individual networks.
Based on the time course and the clustering trend in PCA, we grouped the mice in three temporal phases. Early phase included those that were euthanized at d0 and d1 time points. Table 1 lists the top 5 over-expressed and top 5 under-expressed miRNAs and their relevant functions associated with the Early phase post-TBI. Mid phase included d3 and d7 time points. The Late phase had a single entry of d9 time point.
A Venn diagram (Fig. 2B) curated 72 miRNAs, which were exclusively regulated during the Early phase post-TBI. This Early-phase exclusive group included 57 over-expressed and 15 under- expressed miRNAs. In addition, 31 miRNAs were concurrently expressed in the Early and Mid phases. This set included 6 and 10 miRNAs consistently over expressed and under expressed in both time phases; the remaining 15 miRNAs altered their regulations between the Early and Mid phases. There were 115 miRNAs conserved among all three post-TBI phases that included 51 consistently over-expressed and 42 consistently under-expressed miRNAs. Figure S1 showed a hierarchical clustering of these 115 miRNAs conserved across the entire post-TBI phases. Table S1 lists the entire set of differentially expressed miRNAs sorted based on their temporal distribution as noted in the Venn diagram.
Biological networks
For functional analysis, we curated the biological networks differentially enriched by the three sets of miRNA linked to the respective temporal phase post-TBI. The Venn diagram (Fig. 2C) displayed the temporal distribution of those networks (see Table S2 for complete list of networks). The networks linked to cardiomyopathy, cell proliferation, and inflammation were exclusively enriched during the Early phase (Fig. 2D). It is important to note that 109 out of 144 networks (i.e., nearly 75% of all networks enriched by three time phases combined) were significantly enriched during the Early phase. Furthermore, 100 out of the 109 networks (i.e., nearly 91% of all networks enriched by three time phases combined) were co-enriched by all three time phases. Analyzing further, we noted 23 networks significantly enriched by over-expressed miRNAs during the Early phase post-TBI, and 12 of these 23 networks were related to abnormal cell growth. In fact, a substantial number of tumorigenesis and carcinogenesis networks were significantly enriched across all three time phases; Fig. 2D lists those networks that were associated with tumorigenesis and carcinogenesis, and emerged significantly enriched starting from the Early phase post-TBI.
Inflammatory response after radiation injury
To demonstrate the radiation-induced damage in the irradiated animals exposed to 9.75 Gy of TBI, we tested the markers of bone marrow aplasia (EPO and FLT-3L) and sepsis (SAA) in serum at different times post-radiation (Fig. 3). Starting from the d1 post-TBI, FLT-3L levels were significantly elevated compared to the non-irradiated control (p < 0.001), and the trend continued through d9 post-TBI. A linear regression model found a significantly increasing trend in the protein expression dynamics across this time point (slope = 2385 ± 282; F = 71.6; DFn = 1; DFd = 3, p = 0.003). On the other hand, the level of EPO was significantly elevated (p < 0.001) from d7 post-TBI and onwards. Yet again, the linear regression model demonstrated an increasing load of EPO across these time points (slope = 2085 ± 528; F = 15.6; DFn = 1; DFd = 3, p = 0.03). A very high fold change was registered in SAA starting from day 1 (Fold change: 70 ± 35; p < 0.001) and remained highly upregulated through d7 post-TBI.

Expression of FLT-3L, EPO, and SAA from ELISA measurement. The longitudinal expression of FLT-3L, EPO, and SAA is plotted in the bar chart. The fold changes are calculated from the baseline control (C0). ***p < 0.001; N = 10 for each time point.
Metabolomic analysis
Table S3 lists all the peaks identified by the mass spectrometer. Henceforth, we used a subset of these peaks, which were annotated by Human Metabolite Database (HMDB) and differentially expressed from the baseline (see Table S4 for entire metabolite set). Table 2 lists the top 5 over-expressed and top 5 under-expressed metabolites found in the Early phase, and their biofunctions relevant to present study.
A Venn diagram (Fig. 4A) showed that 10 metabolites were exclusively expressed in the Early phase, which included 4 over-expressed and 6 under-expressed metabolites. There were 27 metabolites co-regulated in the Early and Mid-phases, of which 6 and 16 metabolites were consistently over expressed and under expressed, respectively, in the Early and Mid phases post-TBI. The remaining 5 metabolites changed their regulations between the Early and Mid phases. In addition, 40 metabolites (Fig. S2) were co-regulated in all three post-irradiation phases, including 25 consistently over expressed and 11 consistently under expressed metabolites among the Early, Mid and Late phases post-TBI.

The global metabolite profile responding to lethal radiation. (A) Venn diagram shows the longitudinal distribution of differentially expressed metabolites across three time phases post-TBI, namely, Early (d0–d1), Mid (d3–d7) and Late (d9). The largest number of differentially expressed metabolites (40 metabolites) is found at the cross section of the three phases. (B) Venn diagram shows the longitudinal distribution of significantly enriched networks across three time phases post-TBI, namely, Early (d0–d1), Mid (d3–d7) and Late (d9). Unlike the miRNA profile, here most of the networks are linked to the Mid phase post-TBI. (C) The top 5 most significantly enriched networks during the Early, Early-Mid and Early-Mid-Late phases are shown in this bar chart. The Early phase enlists only 2 networks, so all of them are included here. The top horizontal axis represents the −log(p value), and the bars represent the hypergeometric p-values converted to log10 associated with individual networks. The bottom horizontal axis represents the sample size (N), and the open circles represent the number of metabolites associated with individual networks.
Network analysis
The functional analysis was performed for the differentially expressed HMDB-annotated metabolites expressed in the Early, Mid and Late phases post-TBI. The Venn diagram (Fig. 4B) depicted the temporal trend of the networks significantly enriched in all three time phases (Table S4 enlists all networks). Figure 4C shows the most significant networks potentially relevant to early markers of TBI. Unlike the miRNA-enriched networks, a majority of the metabolite-enriched networks (62 out of 70) were discovered to be enriched during the Mid phase post-TBI. Interestingly, seven out of ten networks co-enriched during the Early and Mid phases post-TBI were associated with metabolism. These metabolism-associated networks included oxidation of fatty acids (p-values at Early phase: 1.74 × 10−5, and Mid phase: 9.49 × 10−7), concentration of triglyceride (p-values at Early phase: 3.45 × 10−3, and Mid phase: 3.87 × 10−5), fatty acid metabolism (p-values at Early phase: 2.47 × 10−4, and Mid phase: 2.14 × 10−4), concentration of lipid (p-values at Early phase: 1.22 × 10−3, and Mid phase: 3.04 × 10−4) and concentration of cholesterol (p-values at Early phase: 2.78 × 10−3, and Mid phase: 2.53 × 10−5). The network linked to blood pressure was consistently enriched during the Early (p = 2.70 × 10−3) and Mid phases (p = 2.80 × 10−3) post-TBI. In concert, a network linked to coronary disease was significantly perturbed in the Early phase (p = 0.045).
Finally, 13 networks were consistently perturbed across the 3 time phases post-TBI, of which 8 were linked to reactive oxygen species (ROS) signaling and immune functions. Figure S3 depicts the network linked to ROS synthesis and causally related to metabolic functions, such as glucose and fatty acid metabolism. This non-canonical network was comprised of 3 upregulated and 9 downregulated metabolites.
Integrating the proteins and miRNA, a non-canonical hematopoiesis network was generated using FLT-3L and EPO as the two-hub molecule (Fig. 5). In addition to these two proteins, this network consists of 20 miRNAs differentially expressed at the Early phase. Relevant non-canonical networks enriched by miRNAs included bone marrow cancer (p = 0.0038), hematologic cancer (p = 1.2 × 10−5) and leukopoiesis (p = 0.02).

The non-canonical network linked to hematopoiesis. This network was generated around the two hub proteins FLT-3LG and EPO. Both of these proteins were upregulated during the Early phase post-TBI. FLT-3LG was significantly (p < 0.001) elevated from baseline. EPO was not significantly elevated during the Early phase, but its expression gradually increased to meet the significance level at d7 post-TBI. In this network, these proteins are marked by triangular nodes. The miRNAs enriching hematopoiesis networks are marked by the oval-shaped nodes. Furthermore, we identified five significantly enrihed (p < 0.05) sub-networks under the overarching hematopoiesis network, namely development of hematopoietic progenitor cells, differentiation of erythroid cells, differentiation of erythroid precursor cells, bone marrow cancer and leukemia. The nodes representing these biological functions and dieases are rectangular in shapes. The arrow-headed and blunt-headed edges represent activating and associative relationship between two nodes, respectively. All of the protein and miRNA nodes are colored based on their regulation levels observed during the Early phase post-TBI, and the color code is at right bottom. The rectangular nodes of the subnetworks representing the biological functions and diseases remained uncolored.
Cardiovascular disorder emerged as one of the major disease footprints co-enriched by miRNA and metabolites (Fig. S4). Relevant networks enriched by miRNAs included bone marrow cancer (p = 0.003), dilated cardiomyopathy (p = 5.3 × 10−6) and fibrosis of heart (p = 3.5 × 10−7). In addition, the blood pressure (p = 0.003) and coronary disease networks (p = 0.04) were also co-enriched by the metabolites linked to cardiovascular disorder.
Figure S5A depicts the alterations between miRNA and metabolites at three different time points for immune related networks. We identified 64 miRNAs and 17 metabolites linked to inflammation. Interestingly, a majority of these molecules, namely, 44 miRNAs and 9 metabolites, were upregulated during the Early phase. A hierarchically clustering of the miRNAs and metabolites that are exclusively linked to immune functions is presented in Fig. S5B.
Discussion
Curating microRNAs as radiation markers has been gaining significant traction in recent years13,34,35,36. The small endogenous RNA theoretically modulates up to 30% of the entire human genome and thereby regulates a wide variety of cellular and molecular functions. Furthermore, the miRNA-mediated epigenetic regulations can sustainably influence the bio-functional landscape. miRNA expressions and biogenesis have been posited to maintain a bi-directional relationship to metabolic stimuli, such as hormones and endocrines37. A shock in the metabolic stimuli influences the miRNA expressions; while the altered miRNA profile can disturb the metabolic homeostatis via epigenetic modulations and transcriptional changes37. Therefore, it is clear that metabolites, as the end product of metabolic function, are distinctively connected to miRNA expression. Taken this bi-directional relationship between miRNA-metabolite in consideration, we postulate that a systems integration of serum proteins, miRNAs and metabolites can shed light on the temporal impacts of a lethal dose of radiation on underlying the biofunctions and disease onset.
The clustering trend in the PCA of the global miRNA and metabolite landscape suggested a time lag between the shift in global miRNA expressions and the onset of perturbation in metabolite landscape. The degree of divergence from the baseline miRNA expressions increased gradually with the time lag from TBI. Baseline controls clustered together with Early phase specimens, while Late phase samples (d9 post-TBI) stayed furthest apart from the baseline. In contrast, the metabolite landscape divulged an early separation from the baseline, suggesting an immediate impact of lethal radiation on the metabolic framework.
Taking cues from the sample distribution in the miRNA PCA plot, we segmented the post-TBI time scale into three phases, Early, Mid and Late, respectively. The majority of the miRNA-enriched networks (100 out of 141 networks) emerged significantly perturbed during the Early phase post-TBI and remained significantly enriched across the entire post-TBI time period. Arguably, this trend supports the fact that miRNA mediates an epigenetic mechanism that marks the lasting impacts on biofunctions. Contrastingly, Mid phase post-TBI contained the largest number of metabolite-enriched networks. Unlike the miRNA-enriched networks, only a few of metabolite-enriched networks (18.6% of all 70 networks) remained perturbed through the entire post-TBI time period.
Despite certain differences between miRNA and metabolite-enriched networks, a concerted perturbation of miRNA and metabolite landscape emerged from the systems analysis. miRNA-metabolite integrative analysis suggested an early onset of cardiac atrophy, bone marrow aplasia, and cancer. Tumorigenesis and oncogenesis were linked to 70% of those networks, which were consistently enriched across the entire post-TBI time period. All of the top five highly enriched networks and most of the highly significant miRNAs are linked to various types of carcinoma, a major comorbidity of lethal TBI38.
The hematopoietic system is highly susceptible to radiation, and effective countermeasure development is still elusive despite the decades long investigation39. Radiation triggers apoptosis in the bone marrow cells, a major site of hematopoiesis40. Hence the early enrichment of the bone marrow cancer network was of particular interest. Early accumulation of FLT-3L protein in serum indicated potential perturbation in the network linked to the development of hematopoietic progenitor cells. Overexpressed FLT-3L has been reported in a wide number of hematologic cancers41,42; however, several attempts at inhibiting FLT-3L failed to control the malignancies, which essentially raised the speculation about the presence of additional independent factors involved in modulating the progenitor cell growth43. Our study identified a number of miRNAs with known involvement in regulating the differentiation of hematopoietic cells under lethal radiation. A major subset of these miRNAs are associated with NFkB signal, a known regulator of hematopoietic cell differentiation44. For instance, we reported an early escalation of miR-155–5p, which has been found to be co-expressed with NFkB signals in malignant cells; otherwise miR-155-5p maintains at low-level expression in mature hematopoietic cells45. In concurrence, we reported a set of inhibited miRNAs, such as miR-486-5p, miR-21-5p and miR-351-5p that are known to regulate hematopoiesis via NFkB signal46,47.
As a potential promoter of survival mechanisms, the activation of EPO is typically triggered by hypoxia and low hematocrit48,49. Highlighting the relevance of EPO in responding to radiation, several independent studies reported elevated EPO as the consequence of both lethal and sublethal radiation doses18,50. In concurrence, we found a gradual increment of EPO load that reached significant levels at d7 post-TBI. As an established modulator of cancer51 miR-125b-5p is also an upstream regulator of EPO52; the inhibited signal of miR-125-5p during the Early phase was probably linked to the continued increase of EPO load during the Mid to Late phases post-TBI. Furthermore, activated EPO is an established upstream regulator of miR-486-5p53 and miR-451a54. Together this molecular axis comprised of mir-125b-5p → EPO → miR-486-5p and miR-451a could be of great importance in the context of erythropoiesis caused by lethal radiation dose.
Radiation-induced cardio atrophy, otherwise known as radiation cardiotoxicity, is of noteworthy clinical interest. With the present treatment regimen, cardiotoxicity typically manifests as a delayed effect of radiation55. However, in the present study exposing mice to a lethal dose of radiation, the compromised heart and vascular system emerged as an acute health issue. Our study suggested inhibition of serum fatty acids, which are often identified as the integral part of the protective mechanism against of cardiotoxicity56. In addition, we identified a number of miRNAs that have been linked to cardiomyopathy57,58. For instance, miR-30c-5p, miR-126a-5p, miR-378, miR-16-5p, miR-140-3p, miR-19b-3p, miR-199a-3p and miR-17-5p were linked to the cardiovascular atrophy caused by a lethal dose of radiation. According to the literature, all of these miRNAs have been linked to cardiac diseases caused by non-radiation insults58. A recent study exploring the cardiac atrophy caused by lethal radiation underscored the key role of the TGF-beta signal in heart fibrosis, metabolic disordering, and impaired contractility59. Supporting this evidence we found two TGF-beta-associated molecules, namely, miR-17-5p60 and sildenafil61, differentially expressed during the Early phase post-TBI.
Release of ROS is a typical comorbidity of radiation, a phenomenon that is regularly observed among patients undergoing radiotherapy62. In agreement, we found a significant enrichment of the ROS synthesis network in irradiated mouse plasma. A set of fatty acids including linoleic acid, docosahexaenoic acid, octadecadienoic acid, docosahexaenoic acid, and palmitic acid became inhibited as the immediate response of TBI, which could be attributed to that fact that fatty acid metabolism is a key source of ROS biogenesis63. It also important to note that the release of ROS is a key signature of cardiovascular disorder64 and bone marrow aplasia65.
An early surge of inflammation is the hallmark signature of a host response to radiation66. Our miRNA-metabolite integrative analysis found a consistent perturbation of inflammatory signals across the time line. A number of studies have diligently established miRNAs as a strategic check point of host immunity, in particular the innate immunity67. Via transcriptional alteration of the immunomodulating factors, miRNAs can act as pro-inflammatory or anti-inflammatory agents68. For instance, miR-155 can act in a dual role accomplishing both anti- and pro-inflammatory functions, and our study reported an elevated serum miR-155-5p during the Early phase post-TBI. The potential synergistic role of miR-155-5p and the NFkB network in hematopoiesis69 could be exploited further to develop a next generation therapeutic strategy against radiation. miR-124-3p is another anti-inflammatory candidate of interest that was consistently inhibited across all time phases post-TBI. This miRNA also directly targets NFkB, and its downregulated form is known to elicit an inflammatory response. In addition, we identified several other pro-inflammatory (miR-125-5p and miR-23a-3p)70 and anti-inflammatory (miR-124-3p, miR-145-5p, miR-181a-5p, miR-21-5p and miR-223-3p)68 miRNA signatures of potential translational values. Furthermore, this inflammatory response was likely causally related to the early comorbidities of lethal radiation, namely, bone marrow aplasia71, cardiovascular disease72 and ROS synthesis73, as discussed earlier.
In conclusion, the investigation of the lethal radiation’s temporal influence on serum miRNAs and metabolites curated time critical markers and relevant molecular mechanisms triggered by TBI. At present, we precluded the analysis of the whole genome mRNA profile despite the fact that mRNA readouts could be the logical bridge between miRNA and metabolite readouts. Instead, we focused on the molecules available in the blood serum. We validated the longitudinal loads of three proteins of interest and built non-canonical networks around these proteins to understand the molecular etiology. Inflammation emerged as an overarching host response, which was potentially associated with some of the early disease footprints, such as dysregulated hematopoiesis and cardiovascular disorder. In future studies, we will embark on validating the translational potential of these markers using animal models from a higher phylogenetic order, as well as identifying differences in the sexes.
References
Singh, V. K., Hanlon, B. K., Santiago, P. T. & Seed, T. M. A review of radiation countermeasures focusing on injury-specific medicinals and regulatory approval status: part III. Countermeasures under early stages of development along with ‘standard of care’ medicinal and procedures not requiring regulatory approval for use. International journal of radiation biology 93, 885–906, https://doi.org/10.1080/09553002.2017.1332440 (2017).
Dainiak, N., Waselenko, J. K., Armitage, J. O., MacVittie, T. J. & Farese, A. M. The hematologist and radiation casualties. Hematology Am Soc Hematol Educ Program, 473–496 (2003).
Coleman, C. N. et al. Molecular and cellular biology of moderate-dose (1-10 Gy) radiation and potential mechanisms of radiation protection: report of a workshop at Bethesda, Maryland, December 17–18, 2001. Radiat Res 159, 812–834 (2003).
Satyamitra, M. et al. Impact of Abbreviated Filgrastim Schedule on Survival and Hematopoietic Recovery after Irradiation in Four Mouse Strains with Different Radiosensitivity. Radiation research 187, 659–671, https://doi.org/10.1667/RR14555.1 (2017).
Jones, J. W. et al. Effect of Sex on Biomarker Response in a Mouse Model of the Hematopoietic Acute Radiation Syndrome. Health Phys, https://doi.org/10.1097/HP.0000000000000961 (2019).
Potten, C. S. A comprehensive study of the radiobiological response of the murine (BDF1) small intestine. Int J Radiat Biol 58, 925–973 (1990).
Cui, W., Ma, J., Wang, Y. & Biswal, S. Plasma miRNA as biomarkers for assessment of total-body radiation exposure dosimetry. PloS one 6, e22988, https://doi.org/10.1371/journal.pone.0022988 (2011).
Menon, N. et al. Detection of acute radiation sickness: a feasibility study in non-human primates circulating miRNAs for triage in radiological events. PloS one 11, e0167333 (2016).
Lu, J. et al. MicroRNA expression profiles classify human cancers. Nature 435, 834–838, https://doi.org/10.1038/nature03702 (2005).
Farazi, T. A. et al. Bioinformatic analysis of barcoded cDNA libraries for small RNA profiling by next-generation sequencing. Methods 58, 171–187, https://doi.org/10.1016/j.ymeth.2012.07.020 (2012).
Sebio, A. et al. The LCS6 polymorphism in the binding site of let-7 microRNA to the KRAS 3′-untranslated region: its role in the efficacy of anti-EGFR-based therapy in metastatic colorectal cancer patients. Pharmacogenet Genomics 23, 142–147, https://doi.org/10.1097/FPC.0b013e32835d9b0b (2013).
Wynendaele, J. et al. An illegitimate microRNA target site within the 3′ UTR of MDM4 affects ovarian cancer progression and chemosensitivity. Cancer Res 70, 9641–9649, https://doi.org/10.1158/0008-5472.CAN-10-0527 (2010).
Acharya, S. S. et al. Serum microRNAs are early indicators of survival after radiation-induced hematopoietic injury. Science translational medicine 7, 287ra269, https://doi.org/10.1126/scitranslmed.aaa6593 (2015).
Templin, T., Amundson, S. A., Brenner, D. J. & Smilenov, L. B. Whole mouse blood microRNA as biomarkers for exposure to gamma-rays and (56)Fe ion. Int J Radiat Biol 87, 653–662, https://doi.org/10.3109/09553002.2010.549537 (2011).
Jacob, N. K. et al. Identification of sensitive serum microRNA biomarkers for radiation biodosimetry. PloS one 8, e57603–e57603, https://doi.org/10.1371/journal.pone.0057603 (2013).
Ghosh, S. P. et al. Gamma-Tocotrienol Modulates Radiation-Induced MicroRNA Expression in Mouse Spleen. Radiation research 185, 485–495, https://doi.org/10.1667/RR14248.1 (2016).
Kulkarni, S. et al. Gamma-tocotrienol protects hematopoietic stem and progenitor cells in mice after total-body irradiation. Radiat Res 173, 738–747, https://doi.org/10.1667/rr1824.1 (2010).
Kumar, V. P. et al. PEGylated IL‐11 (BBT‐059): A Novel Radiation Countermeasure for Hematopoietic Acute Radiation Syndrome. Health physics 115, 65–76 (2018).
Menon, S. S. et al. Radiation Metabolomics: Current Status and Future Directions. Front Oncol 6, 20, https://doi.org/10.3389/fonc.2016.00020 (2016).
Ghosh, S. P. et al. Metabolomic changes in gastrointestinal tissues after whole body radiation in a murine model. Mol Biosyst 9, 723–731, https://doi.org/10.1039/c3mb25454b (2013).
Goudarzi, M. et al. The effect of low dose rate on metabolomic response to radiation in mice. Radiation and environmental biophysics 53, 645–657, https://doi.org/10.1007/s00411-014-0558-1 (2014).
Mak, T. D. et al. Exposure to ionizing radiation reveals global dose- and time-dependent changes in the urinary metabolome of rat. Metabolomics: Official journal of the Metabolomic Society 11, 1082–1094, https://doi.org/10.1007/s11306-014-0765-4 (2015).
Wang, C., Yang, J. & Nie, J. Plasma phospholipid metabolic profiling and biomarkers of rats following radiation exposure based on liquid chromatography-mass spectrometry technique. Biomedical chromatography: BMC 23, 1079–1085, https://doi.org/10.1002/bmc.1226 (2009).
Park, H. M. et al. Mass spectrometry-based metabolite profiling in the mouse liver following exposure to ultraviolet B radiation. PloS one 9, e109479, https://doi.org/10.1371/journal.pone.0109479 (2014).
Cheema, A. K. et al. Liver metabolomics reveals increased oxidative stress and fibrogenic potential in gfrp transgenic mice in response to ionizing radiation. J Proteome Res 13, 3065–3074, https://doi.org/10.1021/pr500278t (2014).
Boguszewicz, Ł. et al. 1H NMR based metabolomic approach to monitoring of the head and neck cancer treatment toxicity. Metabolomics: Official journal of the Metabolomic Society 12, 102, https://doi.org/10.1007/s11306-016-1043-4 (2016).
Ros-Mazurczyk, M. et al. Ionizing radiation affects profile of serum metabolites: increased level of 3-hydroxybutyric acid in serum of cancer patients treated with radiotherapy. Acta biochimica Polonica 64, 189–193, https://doi.org/10.18388/abp.2016_1301 (2017).
Golla, S. et al. Metabolomic Analysis of Mice Exposed to Gamma Radiation Reveals a Systemic Understanding of Total-Body Exposure. Radiation research 187, 612–629, https://doi.org/10.1667/RR14592.1 (2017).
Dutta, S. & Sengupta, P. Men and mice: Relating their ages. Life Sciences 152, 244–248, https://doi.org/10.1016/j.lfs.2015.10.025 (2016).
Ghosh, S. P. et al. Gamma-tocotrienol, a tocol antioxidant as a potent radioprotector. Int J Radiat Biol 85, 598–606, https://doi.org/10.1080/09553000902985128 (2009).
Chakraborty, N. et al. Contributions of polyunsaturated fatty acids (PUFA) on cerebral neurobiology: an integrated omics approach with epigenomic focus. The Journal of nutritional biochemistry 42, 84–94 (2017).
Chakraborty, N. et al. Gene-metabolite profile integration to understand the cause of spaceflight induced immunodeficiency. npj Microgravity 4, 4 (2018).
Kulkarni, S., Singh, P. K., Ghosh, S. P., Posarac, A. & Singh, V. K. Granulocyte colony-stimulating factor antibody abrogates radioprotective efficacy of gamma-tocotrienol, a promising radiation countermeasure. Cytokine 62, 278–285, https://doi.org/10.1016/j.cyto.2013.03.009 (2013).
Tomasik, B., Fendler, W. & Chowdhury, D. Serum microRNAs–potent biomarkers for radiation biodosimetry. Oncotarget 9, 14038 (2018).
Singh, V. K. & Pollard, H. B. Ionizing radiation-induced altered microRNA expression as biomarkers for assessing acute radiation injury. Expert Rev Mol Diagn 17, 871–874, https://doi.org/10.1080/14737159.2017.1366316 (2017).
Gao, F. et al. Changes in miRNA in the lung and whole blood after whole thorax irradiation in rats. Scientific reports 7, 44132 (2017).
Dumortier, O., Hinault, C. & Van Obberghen, E. MicroRNAs and metabolism crosstalk in energy homeostasis. Cell Metab 18, 312–324, https://doi.org/10.1016/j.cmet.2013.06.004 (2013).
El Ghissassi, F. et al. A review of human carcinogens—part D: radiation. The lancet oncology 10, 751–752 (2009).
Singh, V. K., Romaine, P. L. & Newman, V. L. Biologics as countermeasures for acute radiation syndrome: where are we now? Expert Opin Biol Ther 15, 465–471, https://doi.org/10.1517/14712598.2015.986453 (2015).
Fliedner, T. M., Graessle, D., Paulsen, C. & Reimers, K. Structure and function of bone marrow hemopoiesis: mechanisms of response to ionizing radiation exposure. Cancer Biotherapy and Radiopharmaceuticals 17, 405–426 (2002).
Gilliland, D. G. & Griffin, J. D. The roles of FLT3 in hematopoiesis and leukemia. Blood 100, 1532–1542, https://doi.org/10.1182/blood-2002-02-0492 (2002).
Tsapogas, P., Mooney, C. J., Brown, G. & Rolink, A. The Cytokine Flt3-Ligand in Normal and Malignant Hematopoiesis. Int. J. Mol. Sci. 18 https://doi.org/10.3390/ijms18061115 (2017).
Kiyoi, H. Flt3 Inhibitors: Recent Advances and Problems for Clinical Application. Nagoya J Med Sci 77, 7–17 (2015).
Gonzalez-Murillo, A. et al. The NFKB Inducing Kinase Modulates Hematopoiesis During Stress. Stem Cells 33, 2825–2837, https://doi.org/10.1002/stem.2066 (2015).
Ranganath, P. MicroRNA-155 and Its Role in Malignant Hematopoiesis. Biomark Insights 10, 95–102, https://doi.org/10.4137/BMI.S27676 (2015).
Havelange, V. & Garzon, R. MicroRNAs: emerging key regulators of hematopoiesis. American journal of hematology 85, 935–942 (2010).
Song, L. et al. miR-486 sustains NF-kappaB activity by disrupting multiple NF-kappaB-negative feedback loops. Cell Res 23, 274–289, https://doi.org/10.1038/cr.2012.174 (2013).
Rosenberger, C. et al. Expression of hypoxia-inducible factors-1alpha and-2alpha in hypoxic and ischemic rat kidneys. FASEB J 16, A887–A888 (2002).
Barshishat-Kupper, M. et al. Captopril modulates hypoxia-inducible factors and erythropoietin responses in a murine model of total body irradiation. Experimental hematology 39, 293–304 (2011).
Peslak, S. A. et al. EPO-mediated expansion of late-stage erythroid progenitors in the bone marrow initiates recovery from sublethal radiation stress. Blood 120, 2501–2511 (2012).
Bousquet, M., Harris, M. H., Zhou, B. & Lodish, H. F. MicroRNA miR-125b causes leukemia. Proceedings of the National Academy of Sciences 107, 21558–21563 (2010).
Ferracin, M. et al. miR-125b targets erythropoietin and its receptor and their expression correlates with metastatic potential and ERBB2/HER2 expression. Molecular cancer 12, 130 (2013).
Wang, L.-S. et al. MicroRNA-486 regulates normal erythropoiesis and enhances growth and modulates drug response in CML progenitors. Blood 125, 1302–1313 (2015).
Alural, B. et al. EPO mediates neurotrophic, neuroprotective, anti-oxidant, and anti-apoptotic effects via downregulation of miR-451 and miR-885-5p in SH-SY5Y neuron-like cells. Frontiers in immunology 5, 475 (2014).
Palta, M., Lee, C.-L., Yusuf, S. W. & Kirsch, D. G. In Cardio-Oncology 161–174 (Springer, 2017).
Bowen, K. J., Harris, W. S. & Kris-Etherton, P. M. Omega-3 fatty acids and cardiovascular disease: are there benefits? Current treatment options in cardiovascular medicine 18, 69 (2016).
Wang, J. & Yang, X. The function of miRNA in cardiac hypertrophy. Cellular and molecular life sciences 69, 3561–3570 (2012).
Thum, T., Catalucci, D. & Bauersachs, J. MicroRNAs: novel regulators in cardiac development and disease. Cardiovascular research 79, 562–570 (2008).
Subramanian, V. et al. Role of TGF beta and PPAR alpha signaling pathways in radiation response of locally exposed heart: integrated global transcriptomics and proteomics analysis. Journal of proteome research 16, 307–318 (2016).
Suzuki, H. MicroRNA control of TGF-β signaling. International journal of molecular sciences 19, 1901 (2018).
Guimarães, D. A. et al. Atorvastatin and sildenafil decrease vascular TGF-β levels and MMP-2 activity and ameliorate arterial remodeling in a model of renovascular hypertension. Redox biology 6, 386–395 (2015).
Renschler, M. F. The emerging role of reactive oxygen species in cancer therapy. European Journal of Cancer 40, 1934–1940 (2004).
Schönfeld, P. & Wojtczak, L. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radical Biology and Medicine 45, 231–241 (2008).
Panth, N., Paudel, K. R. & Parajuli, K. Reactive oxygen species: a key hallmark of cardiovascular disease. Advances in medicine 2016 (2016).
Chatterjee, R. & Law, S. Epigenetic and microenvironmental alterations in bone marrow associated with ROS in experimental aplastic anemia. European journal of cell biology 97, 32–43 (2018).
Multhoff, G. & Radons, J. Radiation, inflammation, and immune responses in cancer. Frontiers in oncology 2, 58 (2012).
Gantier, M. P. New perspectives in MicroRNA regulation of innate immunity. Journal of Interferon & Cytokine Research 30, 283–289 (2010).
Tahamtan, A., Teymoori-Rad, M., Nakstad, B. & Salimi, V. Anti-Inflammatory MicroRNAs and Their Potential for Inflammatory Diseases Treatment. Front Immunol 9, 1377, https://doi.org/10.3389/fimmu.2018.01377 (2018).
Wu, X. Y., Fan, W. D., Fang, R. & Wu, G. F. Regulation of microRNA‐155 in Endothelial Inflammation by Targeting Nuclear Factor (NF)‐κB P65. Journal of cellular biochemistry 115, 1928–1936 (2014).
Rebane, A. & Akdis, C. A. MicroRNAs: Essential players in the regulation of inflammation. Journal of Allergy and Clinical Immunology 132, 15–26 (2013).
Libregts, S. F. & Nolte, M. A. Parallels between immune driven-hematopoiesis and T cell activation: 3 signals that relay inflammatory stress to the bone marrow. Experimental cell research 329, 239–247 (2014).
Berg, A. H. & Scherer, P. E. Adipose tissue, inflammation, and cardiovascular disease. Circulation research 96, 939–949 (2005).
Mittal, M., Siddiqui, M. R., Tran, K., Reddy, S. P. & Malik, A. B. Reactive oxygen species in inflammation and tissue injury. Antioxidants & redox signaling 20, 1126–1167 (2014).
Gao, F. et al. MiR-467a is upregulated in radiation-induced mouse thymic lymphomas and regulates apoptosis by targeting Fas and Bax. International journal of biological sciences 11, 109 (2015).
Wei, H. et al. Benzene-induced aberrant miRNA expression profile in hematopoietic progenitor cells in C57BL/6 mice. International journal of molecular sciences 16, 27058–27071 (2015).
Chen, X., Liu, X., Liu, H., Lu, Y. & Li, Y. Reduced expression of serum miR-204 predicts poor prognosis of gastric cancer. Genet Mol Res 15.2, 15027702 (2016)
Ress, A. L. et al. MiR‐96‐5p influences cellular growth and is associated with poor survival in colorectal cancer patients. Molecular carcinogenesis 54, 1442–1450 (2015).
Zhang, F. et al. Decreased miR-124-3p Expression Prompted Breast Cancer Cell Progression Mainly by Targeting Beclin-1. Clinical laboratory 62, 1139–1145 (2016).
Xie, C.-H. et al. Long non-coding RNA TUG1 contributes to tumorigenesis of human osteosarcoma by sponging miR-9-5p and regulating POU2F1 expression. Tumor Biology 37, 15031–15041 (2016).
Díaz-Martínez, M. et al. miR-204-5p and miR-211-5p contribute to BRAF inhibitor resistance in melanoma. Cancer research 78, 1017–1030 (2018).
Benjamin, D. I. et al. Diacylglycerol metabolism and signaling is a driving force underlying FASN inhibitor sensitivity in cancer cells. ACS chemical biology 10, 1616–1623 (2015).
Timmers, S., Schrauwen, P. & de Vogel, J. Muscular diacylglycerol metabolism and insulin resistance. Physiology & behavior 94, 242–251 (2008).
Werumeus Buning, J. et al. Effects of hydrocortisone on the regulation of blood pressure: results from a randomized controlled trial. The Journal of Clinical Endocrinology & Metabolism 101, 3691–3699 (2016).
Mateos, M. V., Uranga, R. M., Salvador, G. A. & Giusto, N. M. Activation of phosphatidylcholine signalling during oxidative stress in synaptic endings. Neurochemistry international 53, 199–206 (2008).
Jones, P. J. et al. High-oleic canola oil consumption enriches LDL particle cholesteryl oleate content and reduces LDL proteoglycan binding in humans. Atherosclerosis 238, 231–238 (2015).
Zahednezhad, F., Saadat, M., Valizadeh, H., Zakeri-Milani, P. & Baradaran, B. Liposome and immune system interplay: Challenges and potentials. Journal of Controlled Release (2019).
Wang, H. C., Yang, J.-H., Hsieh, S.-C. & Sheen, L.-Y. Allyl sulfides inhibit cell growth of skin cancer cells through induction of DNA damage mediated G2/M arrest and apoptosis. Journal of agricultural and food chemistry 58, 7096–7103 (2010).
Acknowledgements
The authors gratefully acknowledge Sasha Stone, Kefale Wuddie, and Zemenu Aschenake for their technical support in the animal study. Support for the work was provided by AFRRI Intramural funding.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: S.P.G., N.C., R.H. and A.O.O. Performed the experiments: S.B., V.P.K., A.G., R.K., D.H. and G.D. Analyzed the data: N.C., G.P.H.H., R.K., D.H. and G.D. Wrote the paper: N.C., G.P.H.H., S.P.G., A.O.O. and R.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Disclaimer: AFRRI/USUHS. The opinions and assertions expressed herein are those of the authors and do not necessarily reflect the official policy or position of the Uniformed Services University or the Department of Defense. USACEHR. The views, opinions, and/or findings contained in this report are those of the author(s) and should not be construed as official Department of the Army position, policy, or decision, unless so designated by other official documentation. Citations of commercial organizations or trade names in this report do not constitute an official Department of the Army endorsement or approval of the products or services of these organizations. Opinions, interpretations, conclusions, and recommendations are those of the author(s) and are not necessarily endorsed by the US Army. This research complied with the Animal Welfare Act and implementing Animal Welfare Regulations, the Public Health Service Policy on Humane Care and Use of Laboratory Animals, and adhered to the principles noted in The Guide for the Care and Use of Laboratory Animals (NRC, 2011).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chakraborty, N., Gautam, A., Holmes-Hampton, G.P. et al. microRNA and Metabolite Signatures Linked to Early Consequences of Lethal Radiation. Sci Rep 10, 5424 (2020). https://doi.org/10.1038/s41598-020-62255-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-62255-w
This article is cited by
-
microRNA blood signature for localized radiation injury
Scientific Reports (2024)
-
Early to sustained impacts of lethal radiation on circulating miRNAs in a minipig model
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
