
Abstract
Ilmenite, FeTiO3, is a common mineral in nature, existing as an accessory phase in the most basic igneous and metamorphic rocks, for example, it is derived from the upper mantle. Therefore, an understanding of the high-pressure physics of FeTiO3 is of fundamental importance in the study of rock magnetization. Here, we provide experimental evidence of lattice compression of FeTiO3 powder using super-high-energy ball milling, enabling the very high collision energy of 420 times gravitational acceleration. A sample obtained as an ilmenite- hematite 0.5FeTiO3·0.5Fe2O3 solid solution showed a decrease in molar volume of approximately 1.8%. Consequently, the oxidation state in FeTiO3 powder was changed into almost Fe3+Ti3+, corresponding to 87% Fe3+ of the total Fe for FeTiO3, resulting in the emergence of ferromagnetism. This new ferromagnetic behaviour is of crucial importance in the study of rock magnetization which is used to interpret historical fluctuations in geomagnetism. In addition, the super-high-energy ball mill can be used to control a range of charge and spin states in transition metal oxides with high pressure.
Similar content being viewed by others

Introduction
Ilmenite, FeTiO3, is a common mineral in nature, existing as an accessory phase in the most basic igneous and metamorphic rocks. For example, FeTiO3 is derived from the upper mantle down to depths of some 400 km and thus pressures of 12–13 GPa1. Therefore, an understanding of the high-pressure physics of FeTiO3 is of fundamental importance in the study of rock magnetization2,3. FeTiO3 is a typical transition metal oxide where the partially occupied d shells of the cations allow for a range of charge and spin states. Two types of cation charge ordering exist consistent with O2− anions, namely, Fe2+Ti4+ and Fe3+Ti3+, in which Fe has d-electron configurations of d6 and d5, respectively. The charge transfer excitation between these two oxidation states is known and has been observed in a number of Fe- and Ti-bearing minerals4. This results in a rich variation of magnetic, electronics and structural transitions. The Fe3+/Fe2+ ratios in natural FeTiO3 minerals under high pressures, closely representative of the upper mantle pressure condition, have been studied experimentally using a diamond anvil cell in which the rate of Fe3+ formation by charge transfer in FeTiO3 samples showed a rapid increase from ambient up to 2 GPa and saturated at 40% beyond 2–4 GPa to the highest pressure of 14 GPa5.
In a previous study, we reported that the lattice for trigonal FeTiO3 powder can be compressed by the high collision energy of 150 gravity using super-high-energy ball milling6, whereas little lattice compression was found for FeTiO3 using conventional high-energy ball milling7. This lattice compression is to be expected for the charge transfer in FeTiO3 with high pressure. Therefore, we have investigated the oxidation state of Fe cations in a FeTiO3 sample milled at a high-energy collision of 150 G by 57Fe Mössbauer spectroscopy (Fig. S1 and Table S1). Although the raw FeTiO3 powder only showed the oxidation state of Fe2+, i.e., before ball milling the sample, mixed oxygen states of Fe2+ and Fe3+ were confirmed in the FeTiO3 sample after ball milling at 150 G with a Fe3+ rate of 15%. In this study, we mill the trigonal FeTiO3 powder at a higher-energy collision of 420 G to increase the oxidation state of Fe3+ with high pressure. Consequently, the oxidation state in FeTiO3 powder was changed into almost Fe3+Ti3+, resulting in the emergence of ferromagnetism. This magnetic behaviour with high pressure is of crucial importance in the study of rock magnetization which is used to interpret historical fluctuations in geomagnetism8,9. Thus, a super-high-energy ball mill can be used to control a range of charge and spin states in transition metal oxides with high pressure, yielding the emergence of a large spectrum of functionalities such as metal-insulator transitions10, superconductivity11, thermoelectricity12, and multiferroicity13 as well as magnetism.
Results and Discussion
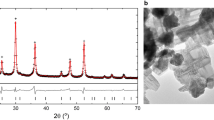
The raw trigonal FeTiO3 powder, labelled as raw FeTiO3, was milled at a very high-energy collision of 420 G (see Methods). The sample was then characterized by measurement of X-ray diffraction (XRD) pattern with synchrotron radiation, as shown in Fig. 1. Notably, the peak position for the as-milled sample was remarkably shifted to a lower d-spacing in comparison with that for the raw powder. This shift in peak position suggests that the trigonal FeTiO3 lattice can be compressed by the collision shock between the balls using super-high-energy ball milling, leading to charge transfer from Fe2+Ti4+ to Fe3+Ti3+ in FeTiO3 with high pressure. Further, the peak position of the as-milled sample was considerably shifted towards a lower d-spacing with increasing collision energy from 150 to 420 G (Fig. S2).

XRD patterns for raw FeTiO3 powder and a sample milled at 420 G. The bar graphs are based on the PDF database of XRD patterns; FeTiO3: 00-029-0733, α-Fe2O3: 00-033-0664, Fe3O4: 00-019-0629, γ-Fe2O3: 00-039-1346, Fe: 00-006-0696.
Although metal iron, Fe, was generated from the surface of the steel balls by the super-high-energy ball milling of 150 G6 (Fig. S2), no peak due to iron was observed in the FeTiO3 sample as-milled at 420 G, as shown in Fig. 1. This absence of iron is considered to be due to the difference in the experimental setup for the ball milling apparatus. The raw FeTiO3 powder was milled at 150 G in a closed type steel vial, i.e., under a reduction atmosphere, and at 420 G in an open type steel vial, i.e., under an oxidation atmosphere. Therefore, the formation of haematite, α-Fe2O3, is presumed to occur via a reaction between oxygen gas in the air and the metal iron generated from the surface of the steel balls by employing an open type steel vial for the ball milling apparatus operated at 420 G. However, it is difficult to identify the formation from α-Fe2O3 by the XRD pattern (Fig. 1) because both FeTiO3 and α-Fe2O3 crystallize in a corundum-derived structure and they usually exist in natural minerals as solid solutions. On the other hand, other iron oxides such as magnetite, Fe3O4, and maghemite, γ-Fe2O3, did not appear as new peaks in the FeTiO3 sample as-milled at 420 G. However, one weak impurity peak marked with a cross appeared with an intensity of less than 5% of the main peak intensity, as shown in Fig. 1. The d value for this peak of 2.036 Å did not agree with the first peak for Fe metal (2.027 Å); this remains an unidentified phase.
To clarify the formation of α-Fe2O3 in the FeTiO3 sample as-milled at 420 G, we have investigated the composition of Fe and Ti for the powder sample by an electron dispersion spectroscopy (EDS). The morphological and compositional features of raw FeTiO3 powder and an as-milled sample are shown in Fig. 2. Notably, the intensity of Fe in the as-milled sample was stronger that in the raw powder, although the Fe and Ti elemental distribution was very homogeneous for the as-milled sample as well as the raw powder, as shown in Fig. 2. The compositional ratio for Fe: Ti was approximately 3: 1 for the as-milled sample, while the ratio was 1: 1 for the raw powder. Similar results were obtained for another as-milled powder sample by additional EDS analyses (Fig. S3). In general, the FeTiO3 shows a solid solution towards α-Fe2O3 and its chemical composition possesses xFeTiO3·(1-x)Fe2O3. Therefore, it is considered that the ilmenite-hematite (IH) solid solution of 0.5FeTiO3·0.5Fe2O3 was formed and the lattice of the IH solid solution powder was compressed by super-high-energy ball milling at 420 G.

Morphological and compositional feature of raw FeTiO3 powder and a sample milled at 420 G. Secondary-electron image and elemental maps for Fe and Ti. (a) Raw powder and its compositional ratio of Fe: Ti = 1.0: 1.0 and (b) as-milled sample and its compositional ratio of Fe: Ti = 3.2: 1.0.
The molar volume for the raw powder and obtained IH solid solution sample determined by least-squares refinement of their XRD patterns was 315.9 Å3 and 302.5 Å3, respectively, with the value for the raw FeTiO3 powder in good agreement with that reported previously14. The decrease in molar volume for the IH solid solution powder as-milled at 420 G was approximately 1.8%, which was estimated from the molar volume of 308 Å3 for 0.5FeTiO3·0.5Fe2O3 bulk14. This percentage of volume decrease due to the very high-energy collision of 420 G corresponds to that of a single crystal of trigonal FeTiO3 under a high pressure condition of approximately 5 GPa at 300 K generated using a lever-type diamond anvil cell15.
Figure 3 shows the 57Fe Mössbauer spectra measured at room temperature, with Table 1 listing the corresponding Mössbauer parameters. The spectrum obtained before ball milling showed a quadrupole doublet with an isomer shift, δ, of 1.08 mm s−1, which is ascribed to Fe2+, with the parameters typical for paramagnetic ilmenite16,17. On the other hand, the spectrum consisted of two doublets and three sextets fit lines (Fig. 3b) after super-high-energy ball milling at 420 G. The sextet due to Fe metal was not found, which is in good agreement with the result from the XRD pattern. Two sextets, red with δ of 0.39 mm s−1 and orange with δ of 0.40 mm s−1 fit lines, are ascribed to Fe3+ in the Fe2O3 component in IH solid solution because their Mössbauer parameters nearly agree with those of α-Fe2O318. This appearance of two sextets in the Fe2O3 component seems to be caused by the R\(\bar{3}\) space group of the IH solid solution14, whereas the α-Fe2O3 has space group R\(\bar{3}\)c. These two sextets for the Fe2O3 component had a total absorption area of 61.2%, which is coherent with the composition of 0.5FeTiO3·0.5Fe2O3 estimated by EDS analysis. Consequently, the residual two doublets and the other sextet are ascribed to the FeTiO3 component in the IH solid solution.

57Fe Mössbauer spectra for raw FeTiO3 powder and a sample milled at 420 G. (a) Raw powder and (b) as-milled sample measured at room temperature. The coloured solid lines are fits to the data (see text and Table 1 for details).
Two doublets, purple with δ of 0.78 mm s−1 and blue with δ of 0.20 mm s−1 fit lines, are ascribed to Fe2+ and Fe3+, with an absorption area of 4.6% and 16.0%, respectively. The sextet, green fit line, with δ of 0.32 mm s−1 is ascribed to Fe3+, with an absorption area of 18.2%. Therefore, the amount of Fe2+ for the FeTiO3 component in IH solid solution is only 13%, which seems to be a mixed oxidation state with Fe3+ because of its δ value of 0.78 mm s−1. It is found that this large oxidation change from Fe2+ to Fe3+, corresponding to 87% Fe3+ of the total Fe for FeTiO3, is caused by the lattice compression of FeTiO3 by the super-high-energy ball milling at 420 G. Such very high-energy collision is effective on the oxidation change in Fe cation because of the slight oxidation rate for Fe3+ of 5–8%7 and 15% (Table S1) for conventional high-energy ball milling and super-high-energy ball milling at 150 G, respectively.
The compression rate for the IH solid solution powder sample obtained by the super-high-energy ball milling at 420 G is approximately 1.8%, which corresponds to that of a single crystal of trigonal FeTiO3 under a high pressure condition of approximately 5 GPa generated using a diamond anvil cell15. The Fe2+ to Fe3+ ratio in natural FeTiO3 minerals under high pressures has been studied using a diamond anvil cell in which the oxidation rate for Fe3+ was approximately 40% at 5 GPa5. It is considered that the higher Fe3+ rate of 87% obtained by the very high-energy collision of 420 G is achieved by a characteristic of the ball milling process in which the collision is continuously repeated during milling. The unit-cell compression in FeTiO3 is quite anisotropic, with the c-axis being more compressible than the a-axis19, so that this anisotropic compression seems to take place preferentially for the FeTiO3 in IH solid solution powder by repeated very high-energy collisions during the super-high-energy ball milling.
Notably, the FeTiO3 component in compressed IH solid solution possessed the sextet, green fit line, as shown in Fig. 3b. This is to be expected for the emergence of ferromagnetism in FeTiO3. Wilson et al. have reported computation of the magnetic property of FeTiO3 from first principles20, and according to them, ferromagnetism is stable in the charge transferred Fe3+Ti3+ state within the Hartree-Fock approximation. Therefore, we have measured the magnetic properties of the powder sample as-milled at 420 G by a superconducting quantum interference device (SQUID) magnetometer. Figure 4a shows magnetization M as a function of the applied magnetic field H at 300 and 100 K. The as-milled sample showed a mixed magnetic behaviour between ferromagnetism and paramagnetism, whereas the raw trigonal FeTiO3 powder showed a paramagnetic state at 300 K. These results are consistent with the Mössbauer spectra (Fig. 3). It is found that the FeTiO3 in compressed IH solid solution was weakly ferromagnetic and that its dependence on temperature was negligible. This magnetic behaviour is coherent with the temperature dependence of magnetization, as shown in Fig. 4b.

Magnetic properties of raw FeTiO3 powder and a sample milled at 420 G. (a) Magnetization measured at 300 and 100 K. (b) Temperature dependence of the magnetization at 60 kOe.
It is known that IH solid solution exhibiting R\(\bar{3}\) symmetry shows large ferrimagnetism in thin films21,22,23 and bulk samples14,24. In this case, the cation charge orderings in FeTiO3 is Fe2+Ti4+ without charge transfer and the slope in magnetization versus temperature is quite steep in comparison with that for the powder sample as-milled at 420 G (Fig. 4b). Therefore, it is considered that the ferromagnetism in compressed IH solid solution is not related to the conventional ferrimagnetism in the IH solid solution and that it emerges from the cation charge orderings of Fe3+Ti3+ by charge transfer in the compressed FeTiO3. This ferromagnetic behaviour is of crucial importance to studies in which rock magnetization under high pressure is used to interpret historical fluctuations in the earth’s magnetic field8,9. It is now an open question as to the reason why the compressed FeTiO3 sample can be quenched by super-high-energy ball milling. It may be due to the charge transfer in FeTiO3 because the irreversible nature of charge transfer in natural FeTiO3 minerals has been shown in experimental study using a diamond anvil cell5.
Conclusion
In conclusion, we milled trigonal FeTiO3 powder using super-high-energy ball milling at 420 G and succeeded in lattice compression of FeTiO3 powder. A sample obtained as an IH solid solution showed a decrease in molar volume of approximately 1.8%. Consequently, charge transfer from Fe2+Ti4+ to Fe3+Ti3+ took place in FeTiO3 with high pressure, resulting in the emergence of ferromagnetism. This finding enables us to infer that such simple and intense collisions induced by super-high-energy ball milling can be used to control the range of charge and spin states in transition metal oxides with high pressure. We believe that property tuning for many functional materials such as graphite25 and graphene26 with high pressure will be realized and that various high pressure materials27 including silica28 and titania29 with α-PbO2 structures will be synthesized based on the super-high-energy ball milling process.
Methods
Materials
The raw material was commercially available FeTiO3 powder (99.9%, Mitsuwa Chemicals Co., Ltd) with a mean particle size of 149 µm.
Super-high-energy ball milling
One gram of FeTiO3 powder was loaded into a 200 cm3 cylindrical steel vial along with 35 g of milling balls. The milling balls were commercial stainless steel balls such as SUS440C, which is a solid solution of iron (Fe, 83 wt.%), chromium (Cr, 16 wt.%), and carbon (C, 1 wt.%) with a diameter of 3 mm. The powder was super-high-energy ball milled using a Thinky Nano Pulverizer (NP-100, Thinky Corporation). The ball milling apparatus operated for 10 min in air atmosphere using an open type steel vial under 420 centrifugal forces, with the ball milling treatment repeated 3 times. It is considered that this short milling time of 30 min is suitable for maintaining the crystallinity of the powder sample. In this experiment, the open type vial was used in order to release the heat in the vial generated by the high collision energy of 420 G.
XRD measurement
Powder x-ray diffraction patterns were measured for the samples filled inside a capillary with an inner diameter of 200 μm using a synchrotron x-ray source (λ = 0.65296 Å) in the BL15XU beam line at SPring-8, Harima, Japan30.
SEM-EDS measurements
A small piece of the produced powder was suspended in ethanol by ultrasonication until a homogeneous suspension was obtained. The suspension was dropped onto an aluminium sample holder, dried, and examined by scanning electron microscopy (SEM) with electron dispersion spectroscopy (EDS) operated at 15 kV (JSM-7600F, JEOL).
Mössbauer measurements
We measured the 57Fe Mössbauer spectra using the constant acceleration method, using a 57Co(Rh) source and an α-Fe a reference. γ-Ray radiation was monitored with a multi-channel analyser (MCA-7700, Seiko EG&G) using 512 channels for each spectrum. The obtained spectra were analysed by Lorentzian fitting using Mösswinn 3.0i XP.
Magnetic measurements
We measured the magnetic properties using a conventional superconducting quantum interference device (SQUID) magnetometer (MPMS-XL, Quantum Design) under a magnetic field of up to 50 kOe in the temperature range from 5 to 300 K for dc-magnetization.
References
Haggerty, S. E. & Sautter, V. Ultradeep (Greater than 300 kilometers), ultramafic upper mantle xenoliths. Science 248, 993–996 (1990).
Lawson, C. A. Antiphase domains and reverse thermoremanent magnetism in ilmenite-hematite minerals. Science 213, 1372–1374 (1981).
Harrison, R. J. et al. Origin of self-reversed thermoremanent magnetization. Phys. Rev. Lett. 95, 268501–1-4 (2005).
Sherman, D. M. Molecular orbital (SCF-Xα-SW) theory of metal-metal charge transfer processes in minerals, I. Application to Fe2+ → Fe3+ charge transfer and “electron delocalization” in mixed-valence iron oxides and silicates. Phys. Chem. Minerals 14, 355–363 (1987).
Seda, T. & Hearne, G. R. Pressure induced Fe2+ + Ti4+ → intervalence Fe3+ + Ti3+ charge transfer and Fe3+/Fe2+ ratio in natural ilmenite (FeTiO3) minerals. J. Phys.: Condens. Matter 16, 2707–2718 (2004).
Hashishin, T. et al. Quenching ilmenite with a high-temperature and high-pressure phase using super-high-energy ball milling. Scientific Reports 4, 4700–1–6 (2014).
Mørup, S., Rasmussen, H. K., Brok, E., Keller, L. & Frandsen, C. Influence of cation disorder on the magnetic properties of ball-milled ilmenite (FeTiO3). Mater. Chem. Phys. 136, 184–189 (2012).
Nagata, T., Akimoto, S. & Uyeda, S. Origin of reverse thermo-remanent magnetism of igneous rocks. Nature 172, 630–631 (1953).
Nagata, T. & Uyeda, S. Production of self-reversal of thermo-remanent magnetism by heat treatment of Ferromagnetic minerals. Nature 177, 179–180 (1956).
Imada, M., Fujimoto, A. & Tokura, Y. Metal-insulator transitions. Rev. Mod. Phys. 70, 1039–1263 (1998).
Bednorz, J. G. & Müller, K. A. Possible high-T c superconductivity in the Ba-La-Cu-O system. Z. Phys.B 64, 189–193 (1986).
Terasaki, I., Sasago, Y. & Uchinokura, K. Large thermoelectric power in NaCo2O4 single crystals. Phys.Rev. B 56, 12685–12687 (1997).
Eerenstein, W., Mathur, N. D. & Scott, J. F. Multiferroic and magnetoelectric materials. Nature 442, 759–765 (2006).
Brown, N. E., Navrotsky, A., Nord, G. L. Jr. & Banerjee, S. K. Hematite-ilmenite (Fe2O3-FeTiO3) solid solutions: Determinations of Fe-Ti order from magnetic properties. Am. Mineral. 78, 941–951 (1993).
Nishio-Hamane, D., Zhang, M., Yagi, T. & Yanming, M. High-pressure and high-temperature phase transitions in FeTiO3 and a new dense FeTi3O7 structure. Am. Mineral. 97, 568–572 (2012).
Frandsen, C., Mørup, S., McEnroe, S. A., Robinson, P. & Langenhorst, F. Magnetic phases in hemo-ilmenite: Insight from low-velocity and high-field Mossbauer spectroscopy. Geophys. Res. Lett. 34, L07306–1-5 (2007).
Frandsen, C., Burton, B. P., Rasmussen, H. K., McEnroe, S. A. & Mørup, S. Magnetic clusters in ilmenite-hematite solid solutions. Phys. Rev. B 81, 224423–1-5 (2010).
García, K. E., Barrero, C. A., Moralesl, A. L & Greneche, J. M. Enhancing the possibilities of the 57Fe Mössbauer spectrometry to study of the inherent properties of rust layers. In Mössbauer spectroscopy (ed. Sharma, V. K. et al.) 415–428 (John Wiley & Sons inc., 2013).
Wechster, B. A. & Prewitt, C. T. Crystal structure of ilmenite (FeTiO3) at high temperature and at high pressure. Am. Mineral. 69, 176–185 (1984).
Wilson, N. C., Muscat, J., Mkhonto, D., Ngoepe, P. E. & Harrison, N. M. Structure and properties of ilmenite from first principle. Phys. Rev. B 71, 075202–1-9 (2005).
Takada, Y., Nakanishi, M., Fujii, T. & Takada, J. Preparation and characterization of (001)- and (110)-oriented 0.6FeTiO3·0.4Fe2O3 films for room temperature magnetic semiconductors. Appl. Phys.Lett. 92, 252102–1-3 (2008).
Hamie, A. et al. Structural, optical, and magnetic properties of the ferromagnetic semiconductor hematite-ilmenite Fe2-xTixO3-δ thin films on SrTiO3(001) prepared by pulsed laser deposition. J. Appl. Phys. 108, 093710–1-5 (2010).
Bocher, L. et al. Direct evidence of Fe2+-Fe3+ charge ordering in the ferrimagnetic hematite-ilmenite Fe1.35Ti0.65O3-δ thin films. Phys. Rev. Lett. 111, 167202–1-6 (2013).
Ishikawa, Y. & Akimoto, S. Magnetic properties of the FeTiO3-Fe2O3 solid solution series. J. Phys. Soc. Jpn. 12, 1083–1098 (1957).
Tan, Z. et al. Interstellar Analogues from Defective Carbon Nanostructures Account for Interstellar Extinction. Astronomical J. 140, 1456–1461 (2010).
Yankowitz, M. et al. Dynamic band-structure tuning of graphene moiré superlattices with pressure. Nature 557, 404–408 (2018).
Zhang, L., Wang, Y., Lv, J. & Ma, Y. Materials discovery at high pressures. Nat. Rev. Mater. 2, 17005–1-16 (2017).
El Goresy, A., Dubrovinsky, L., Sharp, T. G., Saxena, S. K. & Chen, M. A monoclinic post-stishovite polymorph of silica in the Shergotty meteorite. Science 288, 1632–1634 (2000).
El Goresy, A., Chen, M., Dubrovinsky, Gillet, P. & Graup, G. An ultradense polymorph of rutile with seven-coordinated titanium from the Ries crater. Science 293, 1467–1470 (2001).
Nishibori, E. et al. The large Debye-Scherrer camera installed at SPring-8 BL02B2 for charge density studies. Nuc. Instrum. Methods Phys. Res. A 467, 1045–1048 (2001).
Acknowledgements
We thank K. Ichikawa and T. Takatsuka for assistance with the super-high-energy ball milling experiment. The synchrotron radiation experiments were performed on the BL15XU beam line at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (2017A4508). This work was partially supported by a Grant-in-Aid for the Cooperative Research Project of Creation of Life Innovation Materials for Interdisciplinary and International Researcher Development of the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT).
Author information
Authors and Affiliations
Contributions
S.O. designed the research study, performed characterization of the samples, and wrote the paper. T.N. performed the XRD and the magnetic measurements and analyses. K.S. and S.K. performed the 57Fe Mössbauer measurement and analysis. M.S. offered helpful discussion for the super-high-energy ball milling experiment and the characterization of the samples. T.H. performed the super-high-energy ball milling and SEM-EDS measurement and analysis. S.O. was responsible for the project direction.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ohara, S., Naka, T., Sunakawa, K. et al. Emergence of ferromagnetism due to charge transfer in compressed ilmenite powder using super-high-energy ball milling. Sci Rep 10, 5293 (2020). https://doi.org/10.1038/s41598-020-62171-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-62171-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
