
Abstract
Agronomic characteristics and tolerance to biotic and abiotic stresses in hexaploid wheat can be drastically improved through wheat-alien introgression. However, the transcriptional level interactions of introduced alien genes in the wheat genetic background is rarely investigated. In this study, we report the genome-wide impacts of introgressed chromosomes derived from Ae. longissima on gene transcriptions of the wheat landrace Chinese Spring. RNA-seq analyses demonstrated 5.37% and 4.30% of the genes were significantly differentially expressed (DEGs) in CS-Ae. longissima disomic 3Sl#2(3B) substitution line TA3575 and disomic 6Sl#3 addition line TA7548, respectively when compared to CS. In addition, 561 DEGs, including 413 up-regulated and 148 down-regulated or not transcribed genes, were simultaneously impacted by introgressed chromosomes 3Sl#2 and 6Sl#3, which accounts for 41.25% of the DEGs in TA3575 and 38.79% in TA7548. Seventeen DEGs, annotated as R genes, were shared by both introgression lines carrying chromosomes 3Sl#2 and 6Sl#3, which confer resistance to powdery mildew. This study will benefit the understanding of the wheat gene responses as result of alien gene(s) or chromosome intogression and the plant defense response initiated by powdery mildew resistance genes in chromosomes 3Sl#2 and 6Sl#3.
Similar content being viewed by others


Introduction
Allohexaploid wheat (Triticum aestivum L., 2n = 6x = 42, AABBDD) is a species of the genus Triticum in the grass family Poaceae (or Gramineae). Wheat evolved from two independent natural hybridization events followed by chromosome doubling in the new hybrids. The first hybridization occurred between Triticum urartu Tumanian ex Gandilyan (2n = 2x = 14, AA) and Aegilops speltoides Tausch (2n = 2x = 14, SS) about 0.5 million years ago, forming a new tetraploid species T. turgidum L. (2n = 4x = 28, AABB)1,2. The domesticated T. turgidum subsp. dicoccum then hybridized with another diploid species, Ae. tauschii Coss. (2n = 2x = 14, DD) about 7,000 to 12,000 years ago, producing a fertile hexaploid bread wheat (2n = 6x = 42, AABBDD)3,4,5,6,7. Although of relatively recent origin, hexaploid bread wheat is currently the most favored staple food for human beings planted on the most acres and most traded food crop in the world.
Although common wheat is one of the earliest domesticated crop plants, it is still a young species compared with its ancestors or other wild relatives. Gene pools derived from wild relatives, which have a much longer evolutionary history, play an important role in modern wheat improvement. For example, stem rust, caused by Puccinia graminis f. sp. tritici (Pgt), was a devastating disease during the 20th century. However, epidemics of stem rust were well under control in the past several decades largely due to the deployment of highly effective resistance genes, such as Sr31 from Secale cereale L., Sr24 from Thinopyrum elongatum (Host) D. R. Dewey, and Sr36 from T. timopheevii (Zhuk.) Zhuk. in wheat varieties worldwide. However a novel race named Ug99 (TTKSK) emerged in Uganda (1999) having virulence for Sr31 and many other stem rust resistance genes thus Ug99 has become a major threat to wheat production8,9,10,11,12,13. Continuous efforts are needed to identify and deploy new, effective and durable resistance genes to maintain world food security. Of the currently catalogued genes against the major diseases in wheat like leaf rust, stripe rust, and powdery mildew, at least 50% of those genes have been derived from wheat ancestors or other wild relatives including Ae. tauschii, Ae. speltoides, Ae. geniculata Roth, Ae. longissima Schweinf. & Muschl., S. cereale, Dasypyrum villosum, Th. intermedium (Host) Barkworth & D. R. Dewey, Th. elongatum, and Agropyron cristatum (L.) Gaertn14,15.
Despite the importance of the improvement of genetic diversity in common wheat, most alien genes are currently under-utilized due to the limitation of deleterious linkage drag introduced with alien chromosome segments. Linkage drag could be decreased by either shortening the targeted gene segments or possibly deactivating the undesired deleterious genes. For example, the wheat-rye T1BL·1RS translocation has been widely used in wheat cultivars or advanced breeding lines from Europe, China, India, the USA, and other developing countries since the mid-1980s because of remarkable agronomic and disease-resistant characteristics. The short arm of chromosome 1 R (1RS) from S. cereale not only carries resistance genes against fungal pathogens, such as stripe rust resistance gene Yr9, leaf rust resistance gene Lr26, stem rust resistance gene Sr31 and Pm8 against wheat powdery mildew16,17,18,19, but also enhances root biomass, grain yield, protein content20,21,22,23 and tolerance to drought24,25,26. However, chromosome arm 1RS also carries Sec-1 loci encoding γ-secalin and ω-secalin high molecular weight proteins, which decreases the processing and baking quality of wheat containing the T1BL·1RS translocation27,28,29,30.
In addition, linkage drag may also be caused by the inability of introduced alien chromosome segments to adequately compensate for the loss of their homoeologous counterparts or incompatible gene interactions between the alien donor and the recipient wheat line. Parallel with the recent rapid progress of genome sequencing techniques and ever increasing evidence from genome sequence analyses has shown that linkage drag maybe a result of gene expression/suppression caused by gene interactions between the donor and the recipient, post-translational modifications of gene products, and heteromeric complexes formed by the donor and the recipient gene products31,32,33,34,35. However, only a few investigations of chromosome-wide gene expression in alien chromosome addition lines have been reported36,37,38. Rey et al. (2018) reported that the ditelosomic addition of 7HL from Hordeum vulgare L. into the genetic background of CS could lead to 3% differentially expressed genes (DEGs) of wheat and 42% of the 7HL-derived genes in the wheat-barley 7HL addition line38. Nevertheless, the regulatory mechanism underlying the widespread gene transcription of the alien chromosome(s) is still unclear.
Aegilops longissima (2n = 2x = 14, SlSl) is an S-genome diploid species belonging to the section Sitopsis3. This species contains many traits desirable for wheat improvement, such as the powdery mildew resistance gene Pm13 located on chromosome 3Sl, which has been widely used in wheat breeding programs39,40. In addition, we have previously reported that chromosome 6Sl#3 (TA7548) also confers resistance to wheat powdery mildew41. Several QTLs conferring resistance to wheat eyespot are located on chromosomes 1Sl, 3Sl, 5Sl and 7Sl42,43. Additionally, chromosome 1Sl has been reported to carry genes enhancing bread-making quality44,45. A few wheat-Ae. longissima amphiploids and sets of addition and substitution lines have been developed and several translocation lines, such as T3BL·3BS-3Sl and T3DL·3DS-3Sl translocations carrying Pm13, have been deployed in wheat breeding programs worldwide39,40,44,46,47,48,49.
In this study, we report the genome-wide impact on gene transcription in a recipient wheat landrace CS caused by the introgression of pairs of the alien chromosomes 3Sl#2 and 6Sl#3 (Ae. longissima), each of which harbors resistance genes to powdery mildew41, based on comparative transcriptome analyses of CS with a CS-Ae. longissima disomic 3Sl#2(3B) substitution line (TA3575) and a disomic 6Sl#3 addition line (TA7548).
Results
Karyotype analysis of TA3575
Karyotype analysis of TA3575 was identified by integration analyses of PCR amplification using ten 3B-specific and 12 3Sl-specific markers (Supplementary Table S1) and in situ hybridization of root-tip cell mitotic metaphase chromosomes. The PCR results showed that no 3B-specific bands were amplified using 3B-specific primer pairs in both TA3575 and the N3B-T3D control, where a pair of chromosome 3B was replaced by two pairs of 3D. Conversely the 3Sl#2-specific amplifications resulted only from TA3575, indicating the absence of 3B chromosomes and the presence of alien chromosome 3Sl#2 in TA3575. The in situ hybridization results confirmed that in TA3575, 20 pairs of wheat chromosomes plus a pair of 3Sl#2 chromosomes from Ae. longissima replaced the pair of 3B chromosomes (Fig. 1, Supplementary Fig. S1). Thus, TA3575 was redesignated as a CS-Ae. longissima disomic substitution (DS) 3Sl#2(3B) line, rather than the disomic addition (DA) 3Sl#2 line previously identified by the Wheat Genetic Resource Center (WGRC), Kansas State University, Manhattan, KS.

Karyotype analysis of TA3575 using molecular markers and in situ hybridization. (a) Electrophoresis patterns of molecular markers of wheat chromosome 3B-specific. N3B-T3D represents Nullisomic 3B-Tetrasomic 3D. (b). Electrophoresis patterns of molecular markers of 3Sl#2 from Ae. longissima. Arrows show the specific amplification bands of chromosome 3B (a) and 3Sl#2 (b). (c) GISH patterns of TA3575, chromosomes 3Sl#2 were painted in green and wheat chromosomes in blue. (d) FISH patterns of TA3575. Signals from oligos pAs1-3, pAs1-4, pAs1-6, AFA-3, AFA-4 and (AAC)10 displayed in red, those from oligos pSc119.2-1 and (GAA)10 in green, and chromosomes of wheat were stained with DAPI displaying in blue. Arrows indicated chromosome 3Sl#2.
RNA-seq quantity analysis, sequence assemble
A total of 77,962,967, 77,492,142 and 76,266,826 clean reads (>100 bp) were obtained from RNA sequencing for CS, TA3575 and TA7548, respectively. Sequencing quality scores Q30, which inferred a base call accuracy of 99.9%, were greater than 93.06% for all samples, which signifies that the RNA-seq quality was appropriate for subsequent sequence assembly and analysis.
Clean reads of all the samples were further assembled into a total of 158,953 unigenes with a median length of 1,247.95 bp using the short-read assembly software, Trinity. The contig N50 value was 1,725 bp in length. We mapped 91.29% clean reads back to the reference assembly.
Gene expression analysis
Expression levels of the 158,953 genes were normalized based on fragments per kilobase per million mapped reads (FPKM) values of the sequences for further analysis of differential genes. A total of 69,808 genes were transcribed by CS at an FPKM ≥ 0.5 in two independent biological replicates, of which 37,058 (53.10%) were assigned to the wheat genome (expect < =1e-10&qcov > =75%) (Supplementary Tables S2, S3). Pairwise comparison of expression of 37,058 mapped-genes in CS with the CS-Ae. longissima DS 3Sl#2(3B) line TA3575 and CS-Ae. longissima DA 6Sl#3 line TA7548 yielded 1,991 (5.37%) and 1,592 (4.30%) genes, respectively, which were significantly differentially transcribed with a cut-off of |log2(fold change)| ≥ 1 and P < 0.05. These differential genes are described as differentially expressed genes (DEGs) for convenience. Expression pattern analyses revealed that the majority of the DEGs were non-expressed genes (NEGs) or down-regulated expressed genes (DRGs) in both TA3575 (1,253/1,991, 62.93%) and TA7548 (922/1,592, 57.91%). Up-regulated expressed genes (URGs) accounted for 37.07% in TA3575 and 42.09% in TA7548 (Table 1, Supplementary Tables S2–S4, Supplementary Fig. S2).
Distribution of DEGs in TA3575
Analyzing the chromosome distribution of 37,058 mapped-genes in CS demonstrated that these genes were unevenly located across all 21 wheat chromosomes (CS RefSeq v1.0, IWGSC 2018). Of the 21 chromosome pairs, chromosome 2B possessed a maximum number of transcribed genes (2,066/37,058, 5.58%), followed by 5B (2,035, 5.49%), 2D (1,937, 5.23%) and 6B (1,931, 5.21%), whereas the minimum number of genes were mapped on chromosome 6A (138, 3.72%) (Supplementary Table S2).
Similarly, the 1,991 DEGs in TA3575 also covered all seven wheat homoeologous groups and but were unevenly distributed (Table 1) like in CS. The most DEGs were located on group 3 of CS, where the transcription of 781 (14.50%) out of 5,387 genes were significantly affected by the introgression of 3Sl#2 replacing chromosome 3B. Of those 781 DEGs, 511 (65.43%) and 129 (16.52%) genes were non-transcribed and down-regulated, respectively, whereas the remaining 141 genes (18.05%) were up-regulated. In contrast, the transcription of genes in group 7 was least affected by introgression of 3Sl#2, and only 1.83% (98/5,369) of the genes were differentially transcribed (Table 1).
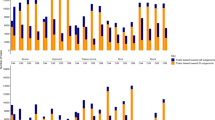
Further analysis of DEG distribution on wheat chromosomes showed that most DEGs mapped to wheat chromosome 3B; 577 (31.43%) out of the 1,839 genes mapped on chromosome 3B in CS were differentially transcribed in TA3575, with 33.43% on the short and 30.18% on the long arm of chromosome 3B (Fig. 2). Analysis of the expression pattern of these 577 DEGs showed that the majority (461, 79.90%) were non-transcribed and 100 genes (17.33%) were down-regulated. Only 16 (2.77%) DEGs were up-regulated, indicating that at least 34.57% (461/1,839) of genes on the missing chromosome 3B were not genetically compensated by the introgressed 3Sl#2 chromosome from Ae. longissima, regardless of gene expression level differences. The DEGs from others chromosome-arms averaged 4.01% (1,413/35,219), ranging from 1.47% (7/475) for the short arm of 5 A (5AS) to 7.23% (86/1,190) for the long arm (5AL) and 7.55% (52/689) for the short arm of chromosome 3D (Fig. 2). Major transcription pattern changes were different for different wheat chromosomes. Besides chromosome 3B, 80% of the DEGs for chromosome 4BS were non-transcribed or down-regulated genes (Fig. 2), whereas, for chromosome 6DS, at least 75% of the DEGs were up-regulated in the presence of chromosome 3Sl#2 in TA3575 (Fig. 2).

Chromosome distribution of DEGs in TA3575. (a) Distribution of DEGs on different chromosome arms. (b) Ratios of different DEGs in different chromosomes.
Validation of differentially transcribed genes in TA3575
We selected nine genes mapped to chromosome 3B and three genes from other chromosomes to validate gene transcription changes in TA3575 using 18S rRNA as a control (Supplementary Table S1). PCR amplification using genomic DNA (gDNA) from control CS, N3B-T3D, and TA3575 as templates showed that all nine 3B genes were present in CS and absent in N3B-T3D, as expected. However, in TA3575, six of the nine 3B-specific genes, including CL22200Contig1, CL27257Contig1, CL24323Contig1, CL29051Contig1, CL53819Contig1 and CL51762Contig1, were amplified and the remaining three (CL40247Contig1, CL35206Contig1 and CL23073Contig1) were absent (Fig. 3, Supplementary Fig. S3). These results indicate that the six 3B-specific sequences were also present on introgressed Ae. longissima chromosome 3Sl#2, whereas the other three were absent on chromosome 3Sl#2. Using the non-3B-specific primer pairs (CL53958Contig1, CL78654Contig1, and CL23519Contig1), gDNA of CS, N3B-T3D, and TA3575 produced the expected PCR amplification (Fig. 3, Supplementary Fig. S3).

Validation of differentially transcribed genes in TA3575 by PCR amplification of gDNA and semi-quantitative RT-PCR. 1. gDNA of CS; 2. gDNA of N3B-T3D; 3. gDNA of TA3575; 4. cDNA of CS; 5. cDNA of TA3575.
Based on semi-quantitative RT-PCR, which was performed using nine 3B-specific primer pairs, four (CL24323Contig1, CL40247Contig1, CL35206Contig1, and CL23073Contig1) were not expressed, two (CL22200Contig1 and CL27257Contig1) were down-regulated and transcribed, and three, including CL29051Contig1, CL53819Contig1, and CL51762Contig1, were up-regulated and expressed in TA3575 as compared to CS (Fig. 3, Supplementary Fig. S3). On the other hand, genes not located on chromosome 3B, CL53958Contig1 and CL78654Contig1 were down-regulated and expressed, and CL23519Contig1 was up-regulated in TA3575, compared with CS. The results of both PCR amplifications using gDNA and semi-quantitative RT-PCR using RNA as templates were consistent with the RNA-seq data analyses and suggested that RNA-seq analyses in this study were valid and reproducible.
Distribution of DEGs in TA7548
A total of 1,592 (4.3%) DEGs, including 396 NEGs (24.87%), 526 DRGs (33.04%) and 670 URGs (42.09%), were identified based on a pairwise comparison of expressed genes in CS-Ae. longissima DA 6Sl#3 line TA7548 and CS on comparison of 37,058 mapped genes (Supplementary Tables S2, S3). These 1,592 DEGs were also asymmetrically distributed on different chromosomes in TA7548. The least affected genes were those located on the short arm of 5B; only nine out of 506 genes (1.78%) showed a change in transcription in TA7548 compared to that in CS. On the contrary, chromosome 6A of wheat contained the most DEGs (190/1,385, 7.50%), which included 54 (54/190, 28.42%) NEGs, 119 (119/190, 62.63%) DRGs and 17 (17/190, 8.95%) URGs (Fig. 4, Supplementary Table S2). On an average, about 90% of the DEGs were down-regulated or non-transcribed on both the short and the long arms of chromosome 6A, followed by chromosome 4BS, where more than 80% of DEGs were negatively regulated. In contrast, close to 70% of DEGs on the short arm of chromosome 4D were positively impacted by chromosome 6Sl#3 in TA7548 (Fig. 4, Supplementary Table S2).

Chromosome distribution of DEGs in TA7548. (a) Distribution of DEGs on different chromosome arms. (b) Ratios of different DEGs on chromosome arms.
Unlike in TA3575, where the highest ratio of non-transcribed to transcribed genes, were those genes that were located to the missing chromosome 3B. The percentage of DRGs mapped to chromosome 6A in TA 7548 was significantly higher than that for any other chromosomes. The DEGs included 13.76% on the short arm and 13.65% on the long arm of chromosome 6A (Fig. 4). In addition, down-regulated genes (DRGs) were a major DEG type on both arms of chromosome 6A (Fig. 4), indicating that genes on both arms were negatively affected to the some extent by introgressed chromosome 6Sl#3 in TA7548.
Transcription-affected genes shared by both introgressed 3Sl#2 and 6Sl#3 in CS
Based on a pairwise comparison of DEGs between TA3575 and TA7548, 561 DEGs were simultaneously impacted by introgression of both chromosomes 3Sl#2 and 6Sl#3, which accounted for 41.25% and 38.79% of the total, excluding those on chromosome 3B in TA3575 (1,360) and 6A in TA7548 (1,446). Of the 561 shared DEGs, the overwhelming majority of genes (99.29%, 557/561) were consistently transcribed in both TA3575 and TA7548, with 84 (14.97%) not expressed, 60 (10.70%) down-regulated, and the majority of 413 up-regulated, accounting for 73.62% shared genes (Supplementary Table S3). Only four DEGs (0.71%, 4/561), including CL120979Contig1, CL14430Contig1, CL60355Contig1 and CL77981Contig1, were transcribed with different expression patterns between TA3575 and TA7548 (Supplementary Table S3). It is worth noting that the ratio of shared URGs to total shared DEGs (413/561, 73.62%) was much higher than that of URGs/DEGs in both TA3575 (722/1414, 51.06%) and TA7548 (665/1499, 44.36%).
The 561 DEGs shared by chromosomes 3Sl#2 and 6Sl#3 from Ae. longissima were annotated by blastx alignment against the GO database. The results indicated that most of the enriched, shared DEGs were categorized biologically as negative regulators of programmed cell death, followed by those involved in protein phosphorylation, defense response to fungus and response to chitin. Of those categorized as cellular components, cytoplasmic membrane-bounded vesicle and an integral component of the plasma membrane were the top terms. The molecular function category included protein serine/threonine kinase activity and transferase activity (Fig. 5, Supplementary Table S5).

Go enrichment of DEGs shared by both 3Sl#2 in TA3575 and 6Sl#3 in TA7548. P-value = number of genes/the total genes annotated to a particular GO term in the whole genome. Only 40 pathways with the highest Go enrichment confidence P-value were represented. The number on the right of each lane in the figure represents the number of genes in the pathway.
Impact of introgressed chromosomes 3Sl#2 and 6Sl#3 on putative disease resistance genes of CS
Both chromosomes 3Sl#2 and 6Sl#3 from Ae. longissima carry resistance gene(s) to powdery mildew of wheat41. The impact of 3Sl#2 in TA3575 and 6Sl#3 in TA7548 on the transcription of CS-derived putative plant disease resistance (R) genes was analyzed in this study. A sequence homology search of DEGs in the PRGdb discovered 34 (1.71%) R genes in seven classes from 1,991 DRGs in TA3575, with the maximum number of R genes in the RLP class (16, 47.06%), followed by those in classes NL (8, 23.53%), CNL and T (3, 8.82% each), and N, TNL and other (1, 2.94% each) (Table 2, Supplementary Table S6). Of these 34 putative R genes, 26 (76.47%) were up-regulated and eight (23.53%) down-regulated by chromosome 3Sl#2. There were also two up-regulated R genes (RLP class) (CL77976Contig1 and CL29051Contig1) mapped on chromosome 3B (in CS) in TA3575 where chromosome 3B was missing, indicating that these two R genes are derived from 3Sl#2 rather than CS wheat chromosome 3B (Table 2, Supplementary Table S6).
In TA7548, 34 (34/1,592, 2.14%) DEGs were annotated as R genes, which were grouped in six classes, including CNL (6), NL (6), other (1), RLP (16), T (3) and TNL (2) R genes. Of these R genes, five in class CNL and 12 in class RLP were down-regulated; the 17 other R genes were up-regulated in TA7548 (Table 2). Three of the 34 R genes (CL33063Contig1, CL80133Contig1, and CL69182Contig1) were located on chromosome 6A, the CL33063Contig1 was up-regulated, whereas, the remaining two down-regulated in TA7548 that carries chromosome 6Sl#3 (Supplementary Table S6).
A pairwise comparison of effected R genes between TA3575 and TA7548 further revealed that 17 R genes were impacted together by the introgression of both chromosomes 3Sl#2 and 6Sl#3. These 17 shared R genes were grouped into six classes, including one R gene for each class CNL, TNL and others, four in NL, seven in RLP and three in class T. Twelve (70.59%) R genes were transcribed with up-regulation, whereas the remaining five (29.41%), which all belong to class RLP, were down-regulated in both TA3575 and TA7548 (Supplementary Table S6). Transcription patterns of these 17 shared R genes were similar in both TA3575 and TA7548.
Identification of a CS-Ae. longissima DS 6Sl#3(6A) line from TA7548
As shown in TA3575, if a pair of wheat chromosomes is missing, non-transcribed genes account for the majority of DEGs located on the substituted chromosomes. In TA3575, where a pair of 3B chromosomes is replaced by a pair of 3Sl#2 chromosomes, 461 out of 577 DEGs located to 3B were NEGs, accounting for as high as 79.90%. However, in TA7548 an unusually high ratio of DRGs (119/190, 62.63%) rather than NEGs (54/190, 28.42%) of chromosome 6A-specific DEGs led to the suspicion that chromosomes 6A might be missing in some TA7548 individuals (progenies) used for RNA-seq in this study.
Molecular markers analysis of seven, randomly selected individual plants from TA7548 using five chromosome 6A-specific SSR and five chromosome 6Sl-specific markers showed that all these chromosome 6A-specific SSR markers were present in five of the seven plants (5/7, 71.43%); two individuals (2/7, 28.57%) presented 6Sl-specific markers but lacked 6A-specific markers (Fig. 6a,b, Supplementary Fig. S4). Subsequent genomic in situ hybridization (GISH) analysis of these individual plants indicated that the two individuals missing 6A-specific markers had 42 chromosomes, 40 wheat chromosomes plus a 6Sl chromosome pair. Further fluorescent in situ hybridization (FISH) analysis verified that a pair of 6A chromosomes was replaced by a 6Sl#3 chromosome pair in these two plants, which resulted in a disomic 6Sl#3(6A) substitutions (Fig. 6c,d). The remaining five plants were confirmed to be disomic 6Sl#3 addition lines, which had 42 wheat chromosomes plus a pair of 6Sl chromosomes (Fig. 6e,f).

Identification of disomic 6Sl#3(6A) substitution line in a TA7548 population. (a) PCR patterns of 6A-specific molecular markers of individuals in the TA7548 population. Arrows show the specific bands of chromosome 6A that are missing in plants 3 and 8. 1. N6A-T6B; 2–8. numbers of the tested plants. (b) PCR patterns of molecular markers of 6Sl#3 from Ae. longissima. Arrows show the specific amplification bands of chromosome 6Sl#3. (c,d) GISH and FISH patterns of a spontaneous disomic 6Sl#3(6A) substitution line. Chromosomes 6Sl#3 were painted green, and wheat chromosomes by blue in GISH photo. Arrows indicate chromosome 6Sl#3 pairs. (e,f) GISH and FISH patterns of disomic 6Sl#3(6A) addition line. Arrows show a pair of chromosome 6Sl#3 painted green in GISH photo.
Therefore, the TA7548 population used in this study was composed of about 71.43% DA 6Sl#3 and 28.57% DS 6Sl#3(6A) plants. This novel CS-Ae. longissima DS 6Sl#3(6A) line, spontaneously formed during the long process of reproduction of the original DA 6Sl#3 plants, was named TA7548-6A. The substitution of chromosome 6A by chromosome 6Sl#3 in some individuals of TA7548 led to the unusually high ratio of DRGs of chromosome 6A in TA7548 used for RNA-seq in this study.
Discussion
Linkage drag, caused by alien chromosome segments introduced into wheat genetic backgrounds, has led to unfavorable agronomic and end-use quality traits and, thus, limited the utilization of alien genes in wheat improvement programs. The linkage drag caused by deleterious genes associated with targeted genes had been widely demonstrated21,50,51,52. However, linkage drag that may be attributed to the inability of alien genes to replace missing wheat genes, or the interactions between the introduced alien genes and recipient wheat genes, has, until now, been relatively under-researched38,53. In this study, we verified the genome-wide impact on wheat gene expression by the presence of introduced alien chromosomes. Transcription of 1,413 (4.01%) of 35,219 genes mapped to non-3B chromosomes in CS were significantly changed by the substitution of a pair of chromosome 3B by chromosome 3Sl#2 derived from Ae. longissima in TA3575. Whereas 1,402 (3.93%) of 35,673 non-6A genes in CS were affected by introgression of chromosome 6Sl#3 in TA7548. The ratios of affected genes in this study were higher than the 3% (960/35,301) reported by Rey et al. (2018), investigating the effect of introgression of barley telosomic 7HL in CS38. Further analyses of DEGs on different wheat chromosomes demonstrated that the impact of introgressed chromosomes 3Sl#2 and 6Sl#3 was obviously different for wheat chromosomes. The minimum ratio of DEGs was associated with chromosome 5AS in TA3575 (1.47%) and chromosome 5BS in TA7548 (1.78%); whereas the highest ratios were on chromosomes 3DS in TA3575 (7.55%) and 6DL in TA7548 (6.35%). Those negatively impacted genes, including non-transcribed and down-regulated, accounted for 48.90% (691/1,413) in TA3575, and 53.42% (749/1,402) in TA7548. Whether these impacts may lead to linkage drag of agronomic traits, or the effects can be decreased in different recipient wheat backgrounds, are currently under investigation. The present study will benefit the understanding of gene interactions between recipient wheat and the alien donor species.
Of those DEGs caused by introgressed chromosomes 3Sl#2 and 6Sl#3 from Ae. longissima, 17 genes in six R gene classes were annotated as putative R genes from 561 DEGs shared by introgressed 3Sl#2 in TA3575 and 6Sl#3 in TA7548. Transcription analysis of these 17 R genes, based on transcriptome analyses, showed that 12 shared R genes (70.59%) were significantly up-regulated and transcribed in both TA3575 and TA7548, whereas only five genes (29.41%) in class RLP were down-regulated. Because both chromosomes 3Sl#2 and 6Sl#3 confer resistance to powdery mildew of wheat41, whether the transcription changes of these shared R genes were involved in the pathogen-defense processes deserved to be further investigated. Molecular marker could be developed from these R gene for mapping of the two powdery mildew resistance genes introgressed from Ae. longissima.
RNA-sequencing techniques have provided a very useful means for examining chromosome structural changes38,54,55. In this study, the discovery of an abnormally high percentage of DRGs on both the short and the long arm of wheat chromosome 6A, based on RNA-seq analyses, implied that chromosome variation of chromosome 6A might exist in the CS-Ae. longissima DA 6Sl#3 line TA7548. We further verified that 28.57% of the plants in the TA7548 population were, in fact, spontaneously formed DS 6Sl#3(6A) by integration analyses of RNA-seq data, molecular markers and in situ hybridization. This novel DS 6Sl#3(6A) line (TA7548-6A) will be useful for further transfer of 6Sl#3 resistance genes to powdery mildew into wheat by inducing wheat-Ae. longissima 6Sl-6A translocations. In a similar way, we selected a spontaneous Robertsonian translocation T4Sl#2·4BL carrying the powdery mildew resistance gene Pm6656. In summary, integrated use of genome sequencing, molecular markers, and classic cytogenetic techniques can speed up the introgression of alien targeted genes, thus promoting the utilization of wild relatives of wheat in breeding programs.
Conclusion
In the current study, transcriptome analyses were performed using two CS-Ae. longissima introgression lines carrying powdery mildew resistance gene(s) and their genetic background line wheat CS. The results showed that introgression of chromosomes 3Sl#2 and 6Sl#3 derived from Ae. longissima had genome-wide impact on gene transcription in wheat. A total of 5.37% and 4.30% genes (or 4.01% of non-3B-specific genes and 3.93% of non-6A-specific genes) were differentially transcribed due to the introduction of chromosomes 3Sl#2 in TA3575 and 6Sl#3 in TA7548, respectively. Furthermore, 17 putative R genes of wheat were significantly impacted together in both TA3575 and TA7548, which carry chromosomes 3Sl#2 and 6Sl#3. The majority of these shared R genes (70.59%) were significantly up-regulated by introgression of chromosomes 3Sl#2 and 6Sl#3. Whether these shared putative R genes were involved in the defensive processes initiated by powdery mildew resistance genes located on chromosomes 3Sl#2 and 6Sl#3 needs to be further investigated.
Materials and methods
Plant materials
Wheat landrace Chinese Spring (CS); nullisomic 3B-tetrasomic 3D (N3B-T3D, TA3272), where a pair of chromosome 3B is replaced by two pairs of 3D; N6A-T6B (TA3152) where 6A is substituted by two pairs of 6B; a CS-Ae. longissima disomic 3Sl#2(3B) substitution line TA3575, where a pair of 3Sl#2 chromosomes replaces the 3B chromosome pair in CS; and a CS-Ae. longissima disomic 6Sl#3 addition line TA7548, where a pair of 6Sl#3 chromosomes are added to the chromosome complement of CS; were used in this study. Both chromosomes 3Sl#2 and 6Sl#3 confer resistance to powdery mildew41. The number following the chromosome designation is used to distinguish between the same Ae. longissima chromosome derived from different Ae. longissima accessions57. All plant materials were kindly provided by the Wheat Genetic Resources Center (WGRC) at Kansas State University, USA, propagated and stored at the experimental station of Henan Agricultural University.
Plant growth and tissue collection
Seeds of the two wheat-Ae. longissima introgression lines (TA3575 and TA7548) and CS were sterilized with 5% sodium hypochlorite solution for 10 min at room temperature and then planted in 8-cm diameter pots filled with an autoclaved vermiculite potting medium. The seedlings were grown in an illuminated incubator at 18–20 °C, 18 h light and 6 h dark, and 75% relative humidity. Pooled, equal-length segments from the first leaf of 10 individuals for each line were then immediately frozen in liquid nitrogen for subsequent RNA isolation. Two independent, biological replicates were collected for subsequent cDNA library construction and RNA sequencing.
Construction of cDNA libraries for Illumina sequencing
Total RNA was extracted from leaf samples of three plants using an mirVana miRNA Isolation Kit (Cat. No. AM1561, Ambion, Thermo Fisher Scientific Inc., Waltham, MA, USA) following the manufacturer’s protocol. RNA integrity was evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The samples with an RNA Integrity Number (RIN) ≥ 7 were subjected to subsequent analysis. The libraries were constructed using TruSeq Stranded mRNA LTSample Prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. These libraries were sequenced on the Illumina sequencing platform (HiSeqTM 2500 or Illumina HiSeq X Ten) by OE Biotech (Shanghai), China, and 125 bp/150 bp paired-end reads (raw reads) were generated.
RNA-seq data analysis
Paired-end reads were pretreated using the NGS QC Toolkit software to remove sequences containing adapters or poly-N above 5%, and low-quality reads to produce valid data (clean data) for downstream sequence assembly58. Valid ratio, Q30 and GC content of each sample were calculated after pretreatment.
De novo assembly of clean data was applied using Trinity (version: trinityrnaseq_r20131110)59, followed by the removal of abundant sequences using TGICL60. Annotation and function of these genes were assigned with a cut off e-value <1e−5 based on blastx alignment against protein sequences in public databases NR (NCBI non-reduction protein sequences, ftp://ftp.ncbi.nih.gov/blast/db), GO (Gene Ontology, http://www.geneontology.org) and PRGdb (Plant Resistance Gene Database)61.
For DEGs analysis, the read counts of genes were first normalized as FPKM. Then, the expression significance of genes of CS vs. TA3575 and CS vs. TA7548 were calculated using DESeq (http://bioconductor.org/packages/release/bioc/html/DESeq.html) with a threshold FDR ≤ 0.05 & |log2(fold change)| ≥ 1. Genes with an FPKM ≥ 0.5 were considered expressed, whereas genes with FPKM = 0 were not transcribed. Genes of CS were pair-wise compared with those of TA3575 and TA7548 to identify genes present in CS and absent in TA3575 and TA7548. All the genes were assigned to chromosome arms based on blastn alignment against wheat reference genomic sequences (CS RefSeq v1.0)62 with a cutoff of expect < =1e-10&qcov > =75% at UGRI BLAST (https://urgi.versailles.inra.fr/blast/blast.php).
Molecular marker analysis
Genomic DNA (gDNA) was extracted from young leaf segments with a DNeasy Plant Mini Kit (Qiagen, Cat No. 69104) following the manufacturer’s directions. Total RNA was isolated using an RNA Prep Pure Plant Kit (Tiangen Biotech Co. Ltd., Code No. DP432), and cDNA synthesized using a PrimeScript™ Double Strand cDNA Synthesis Kit (Takara, Code No. 6111A).
In total, 44 molecular markers were used in the study (Supplementary Table S1). Of these, 10 3B-specific SSR and 12 3Sl#2-specific EST markers were used for the karyotype analysis of TA357563,64. Another nine PCR primer pairs of 3B-specific and three non-3B chromosomes were designed based on transcription sequences of CS and used for validation of gene expression changes in TA3575. The remaining five 6A-specific SSR and five 6Sl#3-specific markers were used for molecular marker analyses of TA754863,65,66,67. PCR reaction mixture preparation and amplification by “F50SSR” (for SSR markers) or “Touch-down 63” (for the remaining markers) followed Liu et al.14. The PCR products were digested with restriction enzymes followed by Liu et al.14. The PCR products or restricted fragments were resolved in 2.5% agarose gels and visualized by ethidium bromide staining under UV light.
Chromosome preparation and in situ hybridization
Root tip collection, nitrous oxide treatment, fixation, and slide preparations were according to Liu et al.14. GISH using fluorescein-12-dUTP labeled genomic DNA of Ae. longissima as the probe, was followed the procedure of Liu et al.14. The ratio of genomic Ae. longissima DNA and CS blocking DNA was 1:120. FISH using FAM (6-carboxyfuorescein) and TAMRA (6-carboxytetramethylrhodamine) modified oligonucleotide probes followed Huang et al.68. After hybridization and slide washing, 25–30 µl of Vectashield mounting medium containing 1 µg/ml DAPI (Vector Laboratories Inc, Burlingame, CA, USA) was added to each slide and then covered with a 24 × 30 cm glass coverslip. Observation of fluorescent images was on a Zeiss Axio Scope A1 fluorescence microscope (Germany) and captured with an AxioCam MRc5 CCD camera. Images were further processed with Adobe Photoshop CS3 (Version 10.0.1) (Adobe Systems Inc., San Jose, CA, USA).
References
Ling, H. Q. et al. Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 496, 81–90 (2010).
Akhunov, E. D., Akhunova, A. R. & Dvořák, J. BAC libraries of Triticum urartu, Aegilops speltoides and Ae. tauschii, the diploid ancestors of polyploid wheat. Theor. Appl. Genet. 111, 1617–1622 (2005).
Feldman, M., Lupton, F. & Miller, T. E. Wheats, in Evolution of Crop Plants, Ed. 2nd, edited by J. Smartt, NW Simmonds. Longman Scientific, London, pp 184–192 (1995).
Huang, S. X. et al. Genes encoding plastid acetyl-CoA carboxylase and 3-phosphoglycerate kinase of the Triticum/Aegilops complex and the evolutionary history of polyploid wheat. Proc. Natl. Acad. Sci. USA 99, 8133–8138 (2002).
Brenchley, R. et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491, 705–710 (2012).
Liu, F. et al. Molecular evolution of Wcor15 gene enhanced our understanding of the origin of A, B and D genomes in Triticum aestivum. Sci. Rep. 6, 31706 (2016).
El Baidouri, M. et al. Reconciling the evolutionary origin of bread wheat (Triticum aestivum). New Phytol. 213, 1477–1486 (2017).
Pretorius, Z. A., Singh, R. P., Wagoire, W. W. & Payne, T. S. Detection of virulence to wheat stem rust resistance gene Sr31 in Puccinia graminis f. sp. tritici in Uganda. Plant Dis. 84, 203–203 (2000).
Singh, R. P. et al. Will stem rust destroy the world’s wheat crop? Adv. Agron. 98, 271–309 (2008).
Jin, Y. & Singh, R. P. Resistance in US wheat to recent eastern African isolates of Puccinia graminis f. sp. tritici with virulence to resistance gene. Sr31. Plant Dis. 90, 476–480 (2006).
Jin, Y. et al. Detection of virulence to resistance gene Sr24 within race TTKS of Puccinia graminis f. sp. tritici. Plant Dis. 92, 923–926 (2008).
Jin, Y. et al. Detection of virulence to resistance gene Sr36 within the TTKS race lineage of Puccinia graminis f. sp. tritici. Plant Dis. 93, 367–370 (2009).
Liu, W. X. et al. Development and characterization of wheat-Ae. searsii Robertsonian translocations and a recombinant chromosome conferring resistance to stem rust. Theor. Appl. Genet. 122, 1537–1545 (2011).
Liu, W. X. et al. Homoeologous recombination-based transfer and molecular cytogenetic mapping of powdery mildew-resistant gene Pm57 from Aegilops searsii into wheat. Theor. Appl. Genet. 130, 841–848 (2017).
McIntosh, R. A. et al. Сatalogue of gene symbols for wheat: 2017 Supplement, https://shigen. nig. ac. jp/wheat/komugi/genes/macgene/supplement2017 (2017).
Singh, N. K., Shepherd, K. W. & McIntosh, R. A. Linkage mapping of genes for resistance to leaf, stem and stripe rusts and ω-secalins on the short arm of rye chromosome 1R. Theor. Appl. Genet. 80, 609–616 (1990).
Friebe, B., Jiang, J., Raupp, W. J., McIntosh, R. A. & Gill, B. S. Characterization of wheat-alien translocations conferring resistance to diseases and pests: current status. Euphytica 91, 59–87 (1996).
Mohler, V., Hsam, S., Zeller, F. & Wenzel, G. An STS marker distinguishing the rye-derived powdery mildew resistance alleles at the Pm8/Pm17 locus of common wheat. Plant Breed. 120, 448–450 (2001).
Crespo-Herrera, L. A., Garkava-Gustavsson, L. & Åhman, I. A systematic review of rye (Secale cereale L.) as a source of resistance to pathogens and pests in wheat (Triticum aestivum L.). Hereditas 154, 14 (2017).
Schlegel, R. & Meinel, A. A quantitative trait locus (QTL) on chromosome arm 1RS of rye and its effect on yield performance of hexaploid wheat. Cereal Res. Commun. 22, 7–13 (1994).
Kumlay, A. M. et al. Understanding the effect of rye chromatin in bread wheat. Crop Sci. 43, 1643–1651 (2003).
Waines, J. G. & Ehdaie, B. Domestication and crop physiology: roots of green-revolution wheat. Ann. Bot. 100, 991–998 (2007).
Lelley, T., Eder, C. & Grausgruber, H. Influence of 1BL.1RS wheat-rye chromosome translocation on genotype by environment interaction. J. Cereal Sci. 39, 313–320 (2004).
Ehdaie, B., Whitkus, R. W. & Waines, J. G. Root biomass, water-use efficiency, and performance of wheat-rye translocations of chromosomes 1 and 2 in spring bread wheat’Pavon’. Crop Sci. 43, 710–717 (2003).
Hoffmann, B. Alteration of drought tolerance of winter wheat caused by translocation of rye chromosome segment 1RS. Cereal Res. Commun. 36, 269–278 (2008).
Karki, D., Wyant, W., Berzonsky, W. A. & Glover, K. D. Investigating physiological and morphological mechanisms of drought tolerance in wheat (Triticum aestivum L.) lines with 1RS translocation. Am J Plant Sci 5, 1936–1944 (2014).
Zhou, Y., He, Z. H., Liu, J. J. & Liu, L. Distribution of 1BL/1RS translocation in Chinese winter wheat and its effect on noodle quality. In Proceedings of 10th International Wheat Genetics Symposium. (Eds. Pogna, N. E., Romano, M & Galterio, G.). Paestum, Volume 3, pp, 1419–1421 (2003).
Payne, P. I. Genetics of wheat storage proteins and the effect of allelic variation on bread-making quality. Annu. Rev. Plant. Physiol. 38, 141–153 (1987).
Burnett, C. J., Lorenz, K. J. & Carver, B. F. Effects of the 1B/1R translocation in wheat on composition and properties of grain and flour. Euphytica 86, 159–166 (1995).
Clarke, B. C., Mukai, Y. & Appels, R. The Sec-1 locus on the short arm of chromosome 1R of rye (Secale cereale). Chromosoma 105, 269–275 (1996).
Kerber, E. R. & Aung, T. Leaf rust resistance gene Lr34 associated with nonsuppression of stem rust resistance in the wheat cultivar Canthatch. Phytopathology 89, 518–521 (1999).
Chen, P. D. et al. Development and characterization of wheat-Leymus racemosus translocation lines with resistance to Fusarium Head Blight. Theor. Appl. Genet. 111, 941–948 (2005).
McIntosh, R. A. et al. Rye-derived powdery mildew resistance gene Pm8 in wheat is suppressed by the Pm3 locus. Theor. Appl. Genet. 123, 359–367 (2011).
Stirnweis, D. et al. Suppression among alleles encoding nucleotide-binding-leucine-rich repeat resistance proteins interferes with resistance in F1 hybrid and allele-pyramided wheat plants. Plant J. 79, 893–903 (2014).
Hurni, S. et al. The powdery mildew resistance gene Pm8 derived from rye is suppressed by its wheat ortholog Pm3. Plant J. 79, 904–913 (2014).
Cho, S., Garvin, D. F. & Muehlbauer, G. J. Transcriptome analysis and physical mapping of barley genes in wheat-barley chromosome addition lines. Genetics 172, 1277–1285 (2006).
Wilson, M. D. et al. Species-specific transcription in mice carrying human chromosome 21. Science 322, 434–438 (2008).
Rey, E. et al. Transcriptome reprogramming due to the introduction of a barley telosome into bread wheat affects more barley genes than wheat. Plant Biotechnol. J. 16, 1767–1777 (2018).
Ceoloni, C., Signore, G., Ercoll, L. & Donini, P. Locating the alien chromatin segment in common wheat-Aegilops longissima mildew resistant transfers. Hereditas 116, 239–245 (1992).
He, Z. H., Xia, X. C., Chen, X. M. & Zhang, Q. S. Progress of wheat breeding in China and the future perspective. Acta Agron. Sin. 37, 202–215 (2011).
Xia, Q., Mai, Y. N., Dong, Z. J. & Liu, W. X. Identification of powdery mildew resistance resources from wheat-wild relative disomic addition lines and development of molecular markers of alien chromosome-specialty. J. Henan Agric. Sci. 47, 64–69 (2018).
Sheng, H., See, D. R. & Murray, T. D. Mapping QTL for resistance to eyespot of wheat in Aegilops longissima. Theor. Appl. Genet. 125, 355–366 (2012).
Huang, S., Steffenson, B. F., Sela, H. & Stinebaugh, K. Resistance of Aegilops longissima to the rusts of wheat. Plant Dis. 102, 1124–1135 (2018).
Wang, S. et al. Molecular mechanisms of HMW glutenin subunits from 1Sl genome of Aegilops longissima positively affecting wheat breadmaking quality. PLoS One 8, e58947 (2013).
Garg, M., Kumar, R., Singh, R. & Tsujimoto, H. Development of an Aegilops longissima substitution line with improved bread-making quality. J. Cereal Sci. 60, 389–396 (2014).
Netzle, S. & Zeller, F. J. Cytogenetic relationship of Aegilops longissima chromosomes with common wheat chromosomes. Plant Syst. Evol. 145, 1–13 (1984).
Friebe, B., Tuleen, N., Jiang, J. & Gill, B. S. Standard karyotype of Triticum longissimum and its cytogenetic relationship with T. aestivum. Genome 36, 731–742 (1993).
Rawat, N. et al. Evaluation and utilization of Aegilops and wild Triticum species for enhancing iron and zinc content in wheat. Genet Resour Crop Evol 56, 53 (2009).
Neelam, K. et al. Development and molecular characterization of wheat-Aegilops longissima derivatives with high grain micronutrients. Aust. J. Crop Sci. 7, 508–514 (2013).
Zeven, A. C., Knott, D. R. & Johnson, R. Investigation of linkage drags in near isogenic lines of wheat by testing for seedling reaction to races of stem rust, leaf rust and yellow rust. Euphytica 32, 319–327 (1983).
Olson, E. L. et al. Development of wheat lines having a small introgressed segment carrying stem rust resistance gene. Sr22. Crop Sci. 50, 1823–1830 (2010).
Niu, Z. et al. Development and characterization of wheat lines carrying stem rust resistance gene Sr43 derived from Thinopyrum ponticum. Theor. Appl. Genet. 127, 969–980 (2014).
Dong, Z. et al. Transcriptional and epigenetic adaptation of maize chromosomes in Oat-Maize addition lines. Nucleic Acids Res. 46, 5012–5028 (2018).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Van Dijk, E. L., Auger, H., Jaszczyszyn, Y. & Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 30, 418–426 (2014).
Li, H. H. et al. A spontaneous wheat-Aegilops longissima translocation carrying Pm66 confers resistance to powdery mildew. Theor. Appl. Genet., https://doi.org/10.1007/s00122-020-03538-8 (2020).
Raupp, J. W. Suggested guidelines for the nomenclature and abbreviation of the genetic stocks of wheat, Triticum aestivum L. em Thell., and its relatives. Wheat Inf Serv., Volume 81, pp, 50–55 (1995).
Patel, R. K. & Jain, M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PloS One 7, e30619 (2012).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Pertea, G. et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19, 651–652 (2003).
Osuna-Cruz, C. M. et al. PRGdb 3.0: a comprehensive platform for prediction and analysis of plant disease resistance genes. Nucleic Acids Res. 46, D1197–D1201 (2017).
Appels, R. et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361, eaar7191 (2018).
Somers, D. J., Isaac, P. & Edwards, K. A high-density wheat microsatellite consensus map for bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 109, 1105–1114 (2014).
Wang, K. et al. Development of a set of PCR markers specific to Aegilops longissima chromosome arms and application in breeding a translocation line. Theor. Appl. Genet. 131, 13–25 (2018).
Sourdille, P. et al. Wheat genoplante SSR mapping data release: a new set of markers and comprehensive genetic and physical mapping data, https://wheat.pw.usda.gov/ggpages/SSRclub/GeneticPhysical (2004).
Lowe, I. et al. Mapping and validation of QTL which confer partial resistance to broadly virulent post-2000 North American races of stripe rust in hexaploid wheat. Theor. Appl. Genet. 123, 143–157 (2011).
Röder, M. S. et al. A microsatellite map of wheat. Genetics 149, 2007–2023 (1998).
Huang, X. et al. Structural chromosome rearrangements and polymorphisms identified in Chinese wheat cultivars by high-resolution multiplex oligonucleotide FISH. Theor. Appl. Genet. 131, 1967–1986 (2018).
Acknowledgements
This project was financially supported by National Natural Science Foundation of China (No. 31571658 and 31971887), The State Key Laboratory of Crop Genetics and Germplasm Enhancement at Nanjing Agricultural University, China (No. ZW2011002), Henan Province Special Major Science and Technology Project (No. 161100110400) and National Key Laboratory of Wheat and Maize Crop Science at Henan Agricultural University, China (No. 39990022). We would like to thank Dr. Sunish Sehgal (Department of Agronomy, Horticulture and Plant Science, South Dakota State University) for critical reading and amendment of the manuscript.
Author information
Authors and Affiliations
Contributions
W.L. and H.L. conceptualized the experiments and designed the methodology. Z.D., C.M., X.T. and Y.L. performed the isolation of DNA and RNA samples, molecular marker analysis and gene expression validation by semi-quantitative RT-PCR. C.Z. and B.F. analyzed the RNA-seq data. H.L. and G.W. performed in situ hybridization. W.L., B.F. and H.L. wrote and modified the manuscript. All authors commented and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dong, Z., Ma, C., Tian, X. et al. Genome-wide impacts of alien chromatin introgression on wheat gene transcriptions. Sci Rep 10, 4801 (2020). https://doi.org/10.1038/s41598-020-61888-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61888-1
This article is cited by
-
Wheat powdery mildew resistance gene Pm13 encodes a mixed lineage kinase domain-like protein
Nature Communications (2024)
-
Global transcriptome analysis of allopolyploidization reveals large-scale repression of the D-subgenome in synthetic hexaploid wheat
Communications Biology (2023)
-
Introgression of the bread wheat D genome encoded Lr34/Yr18/Sr57/Pm38/Ltn1 adult plant resistance gene into Triticum turgidum (durum wheat)
Theoretical and Applied Genetics (2023)
-
Genomic sequencing of Thinopyrum elongatum chromosome arm 7EL, carrying fusarium head blight resistance, and characterization of its impact on the transcriptome of the introgressed line CS-7EL
BMC Genomics (2022)
-
Rye B chromosomes differently influence the expression of A chromosome–encoded genes depending on the host species
Chromosome Research (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
