
Abstract
With the advent of whole genome-sequencing (WGS) studies, family-based designs enable sex-specific analysis approaches that can be applied to only affected individuals; tests using family-based designs are attractive because they are completely robust against the effects of population substructure. These advantages make family-based association tests (FBATs) that use siblings as well as parents especially suited for the analysis of late-onset diseases such as Alzheimer’s Disease (AD). However, the application of FBATs to assess sex-specific effects can require additional filtering steps, as sensitivity to sequencing errors is amplified in this type of analysis. Here, we illustrate the implementation of robust analysis approaches and additional filtering steps that can minimize the chances of false positive-findings due to sex-specific sequencing errors. We apply this approach to two family-based AD datasets and identify four novel loci (GRID1, RIOK3, MCPH1, ZBTB7C) showing sex-specific association with AD risk. Following stringent quality control filtering, the strongest candidate is ZBTB7C (Pinter = 1.83 × 10−7), in which the minor allele of rs1944572 confers increased risk for AD in females and protection in males. ZBTB7C encodes the Zinc Finger and BTB Domain Containing 7C, a transcriptional repressor of membrane metalloproteases (MMP). Members of this MMP family were implicated in AD neuropathology.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is the most common form of dementia worldwide, with a substantial burden for not only patients, but their families, society and the healthcare system. The impact of the disease is expected to increase further by 2050, with a projected 13.9 million Americans to develop AD or related dementias1. Like most complex diseases, AD is caused by a mixture of genetic and environmental factors. Early-onset familial AD (monogenic) is caused by rare fully penetrant mutations in APP, PSEN1, PSEN2 genes2. The more prevalent form, late-onset (sporadic) AD, is caused by a complex polygenic architecture, including large-effect variants in the APOE gene3. Environmental and lifestyle factors also affect the prevalence of the disease, however this domain is less well elucidated to date. Although one of the strongest predictors for AD is age, there are several other risk factors, including race4, high blood pressure5, brain trauma6 and sex7,8,9,10.
Not only are women at twofold greater risk than men, the progression of the disease and neurodegeneration is more rapid among women versus men11,12. In contrast, men with AD have higher mortality, as compared to women12,13. Interactions between sex and APOE ε4 have been previously reported. For instance, Altmann et al. showed that women have greater AD risk in the presence of APOE ε4, and this APOE-related risk in women may be associated with tau pathology14. Another study showed opposite directions on cognition among male APOE ε4 carriers versus female APOE ε4 carriers during intranasal insulin treatment15. A few studies have assessed other genes or performed a systematic gene-by-sex genetic analysis. For example, female- or male-specific effects have been reported in ACE16, BDNF17 and RELN18 genes. Large-scale meta-analyses of genotyped data have largely focused on the AD affection status itself, rather than on sex-specific AD effects19,20,21. This may be attributed to the fact that understanding and modelling gene-by-environment interactions still remain major challenges in the field, due to lack of power given current analytic methods.
FBATs have been recognized to be robust to population structure and to have the advantage of flexible model building based on solely Mendelian transmissions22. This feature of FBATs becomes particularly important when statistical inference is made for an environment interaction effect in the context of WGS-based data, where most of the variants are rare. Adjustment approaches that are based on common variant data might not capture the population structure of the rare variant information. Furthermore, FBATs require only minimal assumptions in terms of modelling the phenotypes22. The correct specification of the phenotypic model increases the power of the FBAT, but a misspecification does not affect the validity of its test results, i.e. type-1 error. Several extensions of FBATs have been proposed for gene-environment interaction analyses23,24,25,26,27,28. While some of these do not scale well with WGS data and require several statistical assumptions, others are better suited for such analysis settings.
However, in the context of rare-variant data, family-based studies face one major hurdle: they are sensitive to genotyping/sequencing errors. In the context of sex-specific analyses, this issue is further aggravated as many genetic regions show sequence homology with the X-chromosome29. This can lead to differential genotyping error rates for females and males due to different X chromosome dosage. Ignoring the impact of such sex-specific genotyping/sequencing errors can lead to substantially inflated type-1 errors29.
Here, we sought to model sex-specific genetic effects within the traditional FBAT framework to analyze two WGS-based AD family datasets for sex-specific genetic associations. We discuss several approaches to test for locus-by-sex interactions in family-based designs where the analysis is restricted to affected individuals only. It is important to note that implementation of an affected-only approach in a population-based design is not straightforward, as the inclusion of covariates to adjust for population substructure is non-trivial30.
While affected-only analysis approaches can be implemented in the FBAT-framework, e.g. in application to AD-WGS datasets, they can be sensitive to sex-specific genotyping errors that are not filtered out by standard QC-pipelines. To minimize such effects, we also discuss the implementation of additional QC filters in this setting.
Our analysis identified four novel putative AD-associated loci, for which corresponding p-values of the sex-specific FBATs achieve the level of 2e-07. Most notably, our analyses nominate ZBTB7C to represent a sex-specific AD gene. ZBTB7C encodes the Zinc Finger and BTB Domain Containing 7C, a transcriptional repressor of membrane metalloproteases (MMP). Members of this MMP family have been implicated in AD neuropathology in previous work31.
Results
We used a combined dataset from two WGS family-based cohorts: The NIMH Alzheimer’s disease genetics initiative study (NIMH)32 and the family component of the NIA ADSP sample(NIA)33. To reduce the number of misclassified unaffected individuals, we performed a case-only analysis and focused on strong sex-specific effects. Our combined dataset contained 18,413,698 variants, after performing regular quality control and filtering by genotyping rate (Methods), in 2,247 individuals from 605 families (Table 1).
Sex-specific only and joint FBAT analysis
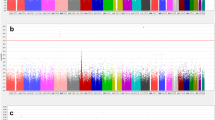
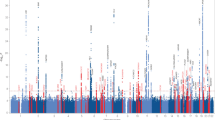
In our primary analysis, we tested for a joint signal of main genetic effect for AD affection status and sex-specific interaction effect. Many of the identified genome-wide significant variants were located in the APOE gene cluster, clearly driven by the main, i.e. not sex-specific, effect (Fig. 1 and Supplementary Fig. 1, Methods). To disentangle the main effects from the sex-specific effects, we performed a sex-specific AD analysis (Fig. 2 and Supplementary Fig. 2, Methods). To this end, we excluded 2,864,446 variants, some of which showed genome-wide significant association, because they were located in loci, which were found to have a pseudogene on either X or Y chromosome (for example RFTN1) and/or did not pass quality control in TOPMED (Methods, Supplementary Figs. 2 and 3).

Manhattan plot for the joint FBAT-GEE analysis. The dashed line corresponds to the significance level p < 5e-08. Highlighted are genes, which correspond to loci with p < 5e-06.

Manhattan plot for the FBAT sex-specific only analysis. The dashed line corresponds to the significance level p < 5e-08. Highlighted are genes, which correspond to loci with p < 5e-06.
After this additional QC filtering, described in the previous paragraph and Methods, and after excluding the markers eliciting significant associations in the APOE region we found 16 variants with pjoint <= 5-06 (Supplementary Table 1). Among these, rs1008912 (GRID1; Glutamate Ionotropic Receptor Delta Type Subunit 1, chromosome 10) had pjoint = 2.01e-10 and pinter = 3.97e-11 with an over-transmission of the minor allele to affected males. Additionally, rs181239893 (RIOK3; RIO Kinase 3, chromosome 18) had pjoint = 8.78e-07 and pinter = 1.42e-07 with an over-transmission of the minor allele in males and rs13259125 (MCPH1, Microcephalin 1, chromosome 8) had pjoint = 9.84e-07 and pinter = 1.5e-07 with an over-transmission of the minor allele in affected males. Finally, rs1944572 (ZBTB7C; Zinc Finger And BTB Domain Containing 7C, chromosome 18) had pjoint = 1.09e-06 and pinter = 1.83e-07 with an over-transmission of the minor allele to affected females. (Table 2).
We next sought to eliminate the possibility that the association signals around our top regions were affected by mismapped reads from the X or Y chromosome. We found that variants in GRID1 and RIOK3 are located in repeat regions within retrotransposons. In addition, a BLAST analysis of these loci identified many matching sequences among the whole genome. Thus, lack of associated variants in LD (Supplementary Figs. 4 and 5) and the fact that those two loci are located in repeat regions suggest that they could represent read errors or mismapped reads. In contrast, ZBTB7C and MCPH1 were mapped to only one region based on the BLAST analysis. Furthermore, there were additional variants in linkage disequilibrium with rs1944572 and rs13259125 that lent further statistical support for the sex-specific association with AD (Supplementary Figs. 6 and 7). Since our combined family-based dataset consisted to a large extent of sibships without observed parents or with only one observed parent, standard Mendel QC assessment was based only on two families, where both parents were observed. We next searched for additional Mendelian inconsistencies among our top associations in nuclear families with at least one parental genotype observed (Methods). Based on 133 such nuclear families, we didn’t observe any Mendelian errors for all 4 loci.
Next, we performed a case-only FBAT association analysis in males and females separately (Methods, Supplementary Table 1). All four variants exhibited opposite effects in males and females. Specifically, the minor allele of rs1008912 (GRID1) was genome-wide significant for protection in females (p = 1.83e-10) and significant for risk in males (p = 3.75e-07). Rs181239893 (RIOK3) and rs13259125 (MCPH1) showed the same pattern: protection in females and risk in males (Supplementary Table 1). However, the minor allele of rs1944572 (ZBTB7C) exhibited an opposite direction: risk in females (p = 1.28e-06) and protection in males (p = 8e-05). Together, these data illustrate AD-associated variants with opposite effects between males and females.
Finally, we performed a robust gene-environment FBAT interaction test by Hoffman et al.23 for the top selected variants, in which the environmental variable was selected to be sex. The results (Table 3) of this test confirmed the sex-specific signals and the validity of our approach. The p-values of this approach are slightly less significant than for the original analysis, as the approach by Hoffmann et al. has additional robustness features.
Sex-specific effects in known AD genes
Next, we sought to identify sex-specific effects in known AD susceptibility loci and gained access to summary statistics from two large AD GWAS with approximately 455,000 individuals and approximately 64,000 individuals20,21. In Supplementary Tables 2 and 3 we list the reported genome-wide significant variants and our corresponding sex-specific results for these variants in our WGS dataset. Three loci, BIN1 (rs4663105 in Jansen et al.; rs6733839 in Kunkle et al.) with elevated risk in females, KAT8 (rs59735493 in Jansen et al.) with elevated risk in males and FERMT2 (rs17125924 in Kunkle et al.) with elevated risk in males, showed nominally significant (pinter < 0.05) sex-specific AD association. We further report, based on our WGS dataset, all nominally significant variants (pinter <= 0.05) for sex-specific AD effects 500 kb up- and downstream of the reported variants from those two studies (Supplementary Table 4). The top loci included CASS4 (pinter = 2.2e-05) with elevated risk for males and protection for females, and KLK5 (pinter = 9.6e-05) with elevated risk for females and protection for males.
Discussion
Genetic associations may have sex differences for complex human diseases. We performed a whole genome sequencing study of sex-specific effects in AD. To our knowledge, this is the first large family-based WGS study for sex-specific AD effects to date. We identified four loci that exhibited sex-specific association with AD. However, at the quality control filtering stage, we found two of the four loci are located within transposable elements of the human genome. Transposable elements make up almost half of the whole genome and they have recently been shown to be important for disease heritability34. But the lack of other variants in linkage disequilibrium with those two loci showing association with AD, raises questions about their validity as novel sex-specific AD loci and suggests that GRID1 and RIOK3 are likely artifacts, emphasizing the importance of standardized additional critical quality assessment of sequencing data when performing WGS sex-specific or other stratified analyses. Meanwhile, for the third and fourth loci at MCPH1 and ZBTB7C, BLAST analysis revealed only one match on the correct chromosome, and additional variants in linkage disequilibrium with the ZBTB7C SNP, rs1944572, and the MCPH1 SNP, rs13259125, also exhibited sex-specific association with AD. These data support ZBTB7C and MCPH1 as a novel AD genes in which the minor allele of rs1944572 conferred risk for AD in females and protection in males and rs13259125 conferred risk for AD in males and protection in females.
Although many consortia have combined their efforts in collecting WGS data, currently, few WGS datasets are publicly available. For our top signals, we sought replication in the ADNI cohort, for which WGS data is available on 494 affected individuals. A proper assessment using the same methodology was impossible in this dataset, due to different study design, different available loci for analysis and small sample size, deeming the study not reliable for replication. But we note, that, for ZBTB7C we observed similar minor allele frequency (MAF) differences between affected males and females as in our family-based cohorts, although the sample size was too small to obtain significance. We also note that for MCPH1 we observed a higher MAF frequency in females, than in males, as opposed to our study, which is not supportive of our finding.
MCPH1, Microcephalin 1, encodes a response protein for DNA damage, which is also implicated in chromosome condensation. The encoded protein may play a role in regulating the development of cerebral cortex in the fetal brain and neurogenesis35,36,37. Common variants in the MCPH1 locus were shown to be associated with brain structure measures, such as brain volume and cortical area38. However, the same study could not replicate these findings for MCPH1 in the ADNI dataset. Another study examined AD association with four microcephaly genes, including MCPH139 and did not find convincing evidence, that MCPH1 is associated with AD.
Our most robust finding was for sex-specific association with ZBTB7C, demonstrating a protective effect in males (and risk in females). This gene encodes Zinc Finger and BTB Domain Containing 7C, a transcription factor, which is expressed in the brain (GTEX) and is a known repressor of membrane metalloproteases (MMP 8,10,13,16)40. Recently, a study by Blue et al. identified variants associated with age at onset of AD in ZBTB4, another gene from the same gene family, located on chromosome 1741. In addition, knock out of ZBTB7C in mice led to decreased glucose blood levels42. Moreover, this same study showed that Zbtb7c deacetylated forkhead box O1 (Foxo1), leading to increased Foxo1 binding and transcriptional activation of genes involved with glucogenesis. Interestingly, the FOXO families of transcription factors have previously been implicated in AD pathogenesis by influencing neuronal survival43 and Aβ-induced neuroinflammation44. ZBTB7C has also been suggested as a susceptibility gene for ischemic stroke through modulation of neuronal apoptosis45. Finally, a paternally inherited translocation of ZBTB7C has been associated with non-syndromal mental retardation in male twins46. In a search for other variants in linkage disequilibrium with the sex-specific, AD-associated SNP in ZBTB7C, rs1944572, one rare (minor allele frequency = 0.008 in gnomAD) missense variant, rs61729532 (Proline250Serine) was detected, which was in strong linkage disequilibrium with rs1944572 (D’ = 1), and yielded a pinter = 0.028 for sex-specific association with AD with an elevated risk for females and protection for males. Future studies assessing how this missense mutation affects ZBTB7C function, particularly in female versus males brain tissue, would be warranted.
Our study has several limitations. First, as noted in the methods, the test we use is particularly powerful in scenarios where the sex-specific effects are in opposite directions, but might miss same direction sex-specific effects of different magnitude. However, it is worth noting that separate tests in males and females are not sufficient to prove interaction, even if both are statistically significant and effect estimates point in opposite directions. Larger sample sizes and more sensitive approaches are needed to detect same direction sex-specific effects. Collection of large datasets with suitable families requires more time and resources as compared to population-based or case-control cohorts. Next, our study was dominated by samples of European ancestry. Racial and ethnic differences in AD prevalence and genetics are recognized in the literature1,47,48,49. Although FBAT is robust to confounding due to population structure, an expanded set of underrepresented populations is necessary to identify AD sex-specific effects unique to other populations.
In summary, we have developed and employed an FBAT-approach using an affected-only analysis to detect sex-specific AD effects. During the analysis, we encountered the need for additional sex-specific filtering steps that have not previously been considered for the association analysis of a WGS scan to reduce the number of false positive findings. Using our analysis approach, we initially identified four loci showing sex-specific association with AD risk. However, after additional quality control filtering, the only candidate remaining was a variant in ZBTB7C, rs1944572, which conferred increased risk for AD in females and protection from AD in males and showed similar MAF differences between affected males and females in an independent cohort. Recently, methods have started to emerge, that address sex chromosome related mismapping in WGS data, but these approaches are still early in development29. Based on our experience, we recommend careful re-mapping of all autosomal variants to search for potential homology on the sex-chromosomes which may drive spurious associations. To the best of our knowledge, our study represents the first large family-based WGS analysis for sex-specific associations with AD. Similar analyses of additional AD WGS samples are needed to confirm our putative association with ZBTB7C, to identify additional novel sex-specific AD loci, and to better understand sex-specific genetic features and potential pathways for AD development.
Methods
Cohort description and sequencing
Briefly, sequencing in NIMH was performed by Illumina HiSeq 2000. Alignment to the human reference genome (GRh37) was done with bwa-mem50 (v0.7.7, default parameters). Variants were jointly called for each family using FreeBayes51 (v0.9.9.2-18) and GATK52 (v3.0) best practices method as part of the bcbio-nextgen workflow53 before being squared-off with bcbio.recall across the whole cohort to distinguish reference calls from no variant calls. Library and read quality were assessed using FastQC54 (v0.10.1) and Qualimap55 (v0.7.1). Variant calls in vcf format for the families from the NIA ADSP cohort were obtained from the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS) under accession number: NG00067 and the database of Genotypes and Phenotypes (dbGaP) under accession number: phs000572v8p4. Both cohorts: NIMH32 and the family component of the NIA ADSP sample33 consisted of multiplex AD families with affected and unaffected siblings (Table 1). A subject was considered to be affected, if he/she was included in one of the following categories: “Definite AD”, “Probable AD” or “Possible AD”. Unaffected subjects had either no dementia, suspected dementia (34 subjects) or non-AD dementia (4 subjects). It is important to note that NIA ADSP families by design did not include individuals with two APOE-ε4 alleles. After standard quality control both cohorts were merged together as described in the next section.
The Alzheimer’s Disease Neuroimaging Initiative (ADNI) was used to lookup minor allele frequencies in males and females for the top candidate findings. Data was obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer’s disease (AD).
Regular quality control
We used PLINK56,57 v1.9 to calculate most of the quality metrics. Initially, we had 1432 individuals (affected/unaffected siblings) from the NIMH cohort and 873 individuals from the NIA. Nineteen individuals in NIA were removed because they were marked as either replicates, duplicates or had a bad GWAS concordance. Three individuals in NIMH were removed because they were outliers based on genotyping rate and inbreeding coefficient. Based on estimated identity by descent (IBD) sharing coefficients we identified 12 duplicate pairs and 24 individuals with wrong family assignments in NIMH. After filtering the analyzed dataset was composed of two WGS familial AD cohorts with 1,393 individuals (NIMH; 446 families) and 854 individuals (NIA; 159 families), which were merged together. For variant quality control we performed the following: we have used only variants which passed all quality control filters in the vcf file (marked by “PASS”), excluded multiallelic variants, monomorphic variants, singletons (i.e. variants with only one alternative allele across the dataset), indels and variants which had one missing allele among 2 alleles in an individual. The remaining variants were filtered based on Mendel errors and genotyping rate (95%).
Additional quality control
We performed additional quality control after our main analysis to eliminate additional sequencing and calling errors. We excluded variants, which were not called in TOPMed58,59 – a large WGS database with >100,000 individuals sequenced jointly. We also screened a pseudogene database60 for genes, which had known pseudogenes on either X or Y chromosome. This allowed us to eliminate possible mapping errors.
For the four novel candidate SNPs, we performed an additional, non-standard Mendel error check. We utilized the genetic data for 133 nuclear families where one parental genotype is available and screened for Mendelian inconsistencies (discordant homozygous genotypes between parent and offspring). No Mendelian inconsistency was found.
In order to eliminate possible mismapping issues we performed a local alignment (BLAST) for the four candidate loci using the BLAT tool from Ensembl Genome Browser61. As the query we used a flanking sequence centered around the variant of interest. The query length was 201 basepairs, 100 basepairs from each side of the selected variant.
Sex-specific FBAT analysis
The NIMH and NIA samples are studies of extended families of multiple generations with affected and unaffected siblings. While the genotypes of all offsprings regardless their phenotypes are observed, most of the parents are missing. As the ascertainment condition for both samples was that at least one of the offspring has late onset AD, we decided to perform a case-only analysis, minimizing the number of misclassified unaffected individuals who might have developed the disease later in life. In order to maximize the power of the FBAT-statistic, we set the phenotype of the unaffected offspring as missing, so that their genotype information can still be used in the construction of the sufficient statistics for each family.
We used the FBAT software62 and the following framework to perform a sex-specific family-based association study for Alzheimer’s disease. Briefly, The FBAT score statistic can be described as:
where n is the number of families, nj is the number of offsprings in the family i, Xij is the genotype, Si – family sufficient statistic for parental genotypes, Tij – the coded trait of offspring. Usually Tij is the phenotypic residual and corresponds to Tij = Yij − μ, where Yij is the phenotype of the offspring and µ is the offset parameter, which we set to 0.15 to approximately correspond to the population prevalence of Alzheimer’s disease. Since we use only affected offsprings in our analysis, we modify Tij to incorporate sex-specific effects into the test statistic and define it as following: Tij = (Yij − μ)(Zij − 0.5), where (Yij − μ) is constant, Zij is 0, if the offspring is male and 1 if it is female. Our modified FBAT score statistics is:
It is important to note that this coding of the sex-specific effect will be especially efficient if the genetic effect direction is different between sexes.
In order to test the joint effect we used FBAT-GEE63 on two phenotypes: T1 = Y − μ and T2 = (Y − μ)(Z − 0.5), which were constructed as described above.
Additionally, we used a robust gene by environment test, developed by Hoffmann et al.23. For this we used the function “fbatge” from the “fbati” package. We used sex as the environmental variable and utilized the hybrid test strategy, which is more efficient for sibships without parental genotypes.
R64 v3.5.0 and Locuszoom65 were used to generate all plots.
Signal direction assessment
In order to identify the AD risk direction for our sex-specific analysis, we performed a case-only family-based analysis for AD affection status with FBAT in males and females separately. We kept all the genotypes to calculate sufficient statistics for each family. By setting the phenotype of one of the sexes to missing, we were able to observe the effect direction for the tested allele in males and females.
Assessment of known AD susceptibility regions
We used summary statistics from two recent large genome-wide association studies of AD20,21. We sought to identify whether any of the reported genome-wide significant loci from those studies showed a sex-specific effect in our analysis. Additionally, we extended our lookup to all variants, located 500 kb up- and downstream of the reported variants in the papers.
Ethical statement
This research project is approved by the Institutional Review Board (IRB) (2015P000111) at Massachusetts General Hospital. Informed consent was obtained from all subjects. All methods were carried out in accordance with relevant guidelines and regulations.
Data availability
The NIMH dataset analysed during the current study is available from the corresponding author on reasonable request. The family component of the NIA ADSP WGS dataset is available from DSS NIAGADS under accession number: NG00067. The ADNI WGS dataset is available at http://adni.loni.usc.edu/.
References
Matthews, K. A. et al. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015–2060) in adults aged ≥65 years. Alzheimer’s Dement. 15, 17–24 (2019).
Tanzi, R. E. & Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 120, 545–555 (2005).
Bertram, L. & Tanzi, R. E. Alzheimer disease risk genes: 29 and counting. Nat. Rev. Neurol., https://doi.org/10.1038/s41582-019-0158-4 (2019).
Howell, J. C. et al. Race modifies the relationship between cognition and Alzheimer’s disease cerebrospinal fluid biomarkers. Alzheimer’s Res. Ther. 9, 1–10 (2017).
Skoog, I. & Gustafson, D. Update on hypertension and Alzheimer’s disease. Neurol. Res. 28, 605–11 (2006).
Perry, D. C. et al. Association of traumatic brain injury with subsequent neurological and psychiatric disease: a meta-analysis. J. Neurosurg. 124, 511–526 (2015).
Kim, S. et al. Gender differences in risk factors for transition from mild cognitive impairment to Alzheimer’s disease: A CREDOS study. Compr. Psychiatry 62, 114–122 (2015).
Podcasy, J. L. & Epperson, C. N. Considering sex and gender in Alzheimer disease and other dementias. Dialogues Clin. Neurosci. 18, 437–446 (2016).
Ferretti, M. T. et al. Sex differences in Alzheimer disease — The gateway to precision medicine. Nat. Rev. Neurol. 14, 457–469 (2018).
Nebel, R. A. et al. Understanding the impact of sex and gender in Alzheimer’s disease: A call to action. Alzheimer’s Dement. 14, 1171–1183 (2018).
Laws, K. R., Irvine, K. & Gale, T. M. Sex differences in cognitive impairment in Alzheimer’s disease. World J. Psychiatry 6, 54 (2016).
Sinforiani, E. et al. Impact of gender differences on the outcome of alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 30, 147–154 (2010).
Todd, S., Barr, S., Roberts, M. & Passmore, A. P. Survival in dementia and predictors of mortality: A review. Int. J. Geriatr. Psychiatry 28, 1109–1124 (2013).
Altmann, A., Tian, L., Henderson, V. W. & Greicius, M. D. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann. Neurol. 75, 563–573 (2014).
Claxton, A. et al. Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or alzheimer’s disease. J. Alzheimer’s Dis. 35, 789–797 (2013).
Crawford, F. et al. Gender-specific association of the angiotensin converting enzyme gene with Alzheimer’s disease. Neurosci. Lett. 280, 215–219 (2000).
Li, G. D. et al. Female-specific effect of the BDNF gene on Alzheimer’s disease. Neurobiol. Aging 53, 192.e11–192.e19 (2017).
Fehér, Á., Juhász, A., Pákáski, M., Kálmán, J. & Janka, Z. Genetic analysis of the RELN gene: Gender specific association with Alzheimer’s disease. Psychiatry Res. 230, 716–718 (2015).
Marioni, R. E. et al. GWAS on family history of Alzheimer’ s disease. Transl. Psychiatry 1–26, https://doi.org/10.1101/246223 (2018).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413 (2019).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019 513 51, 414 (2019).
Laird, N. M. & Lange, C. Family-based designs in the age of large-scale gene-association studies. Nat. Rev. Genet. 7, 385–94 (2006).
Hoffmann, T. J. et al. Combining disease models to test for gene-environment interaction in nuclear families. Biometrics 67, 1260–70 (2011).
Hoffmann, T. J., Lange, C., Vansteelandt, S. & Laird, N. M. Gene-environment interaction tests for dichotomous traits in trios and sibships. Genet. Epidemiol. 33, 691–699 (2009).
Lake, S. L. & Laird, N. M. Tests of gene-environment interaction for case-parent triads with general environmental exposures. Ann. Hum. Genet. 68, 55–64 (2004).
Moerkerke, B., Vansteelandt, S. & Lange, C. A doubly robust test for gene-environment interaction in family-based studies of affected offspring. Biostatistics 11, 213–225 (2010).
Vansteelandt, S. et al. Testing and Estimating Gene – Environment Interactions in Family-Based Association Studies. 458–467, https://doi.org/10.1111/j.1541-0420.2007.00925.x (2008).
Lange, C., DeMeo, D., Silverman, E. K., Weiss, S. T. & Laird, N. M. PBAT: tools for family-based association studies. Am. J. Hum. Genet. 74, 367–9 (2004).
Webster, T. H. et al. Identifying, understanding, and correcting technical biases on the sex chromosomes in next-generation sequencing data. GigaScience, 8(7), 1–11, https://doi.org/10.1093/gigascience/giz074 (2019).
Khoury, M. J. & Flanders, W. D. Nontraditional epidemiologic approaches in the analysis of gene-environment interaction: case-control studies with no controls! Am. J. Epidemiol. 144, 207–13 (1996).
Rosenberg, G. A. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 8, 205–216 (2009).
Blacker, D. et al. ApoE-4 and age at onset of Alzheimer’s disease: the NIMH genetics initiative. Neurology 48, 139–147 (1997).
Beecham, G. W. et al. The Alzheimer’s Disease Sequencing Project: Study design and sample selection. Neurol. Genet. 3, e194 (2017).
Hormozdiari, F. I. F. et al. Functional disease architectures reveal unique biological role of transposable elements. bioRxiv 482281, https://doi.org/10.1101/482281 (2018).
Jackson, A. P. et al. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am. J. Hum. Genet. 71, 136–142 (2002).
Trimborn, M. et al. Mutations in microcephalin cause aberrant regulation of chromosome condensation. Am. J. Hum. Genet. 75, 261–266 (2004).
Xu, X., Lee, J. & Stern, D. F. Microcephalin is a DNA damage response protein involved in regulation of CHK1 and BRCA1. J. Biol. Chem. 279, 34091–34094 (2004).
Rimol, L. M. et al. Sex-dependent association of common variants of microcephaly genes with brain structure. Proc. Natl. Acad. Sci. USA 107, 384–388 (2010).
Erten-Lyons, D. et al. Microcephaly genes and risk of late-onset Alzheimer disease. Alzheimer Dis. Assoc. Disord. 25, 276–282 (2011).
Jeon, B. N. et al. Zbtb7c is a molecular ‘off’ and ‘on’ switch of Mmp gene transcription. Biochim. Biophys. Acta - Gene Regul. Mech. 1859, 1429–1439 (2016).
Blue, E. E. et al. Variants regulating ZBTB4 are associated with age-at-onset of Alzheimer’s disease. Genes, Brain Behav. 17, 1–9 (2018).
Choi, W. I. et al. Zbtb7c is a critical gluconeogenic transcription factor that induces glucose-6-phosphatase and phosphoenylpyruvate carboxykinase 1 genes expression during mice fasting. Biochim. Biophys. Acta - Gene Regul. Mech. 1862, 643–656 (2019).
Maiese, K. Forkhead transcription factors: new considerations for alzheimer’s disease and dementia. J. Transl. Sci. 2, 241–247 (2016).
Fernandez, A. M. et al. Blockade of the Interaction of Calcineurin with FOXO in Astrocytes Protects Against Amyloid-β-Induced Neuronal Death. J. Alzheimer’s Dis. 52, 1471–1478 (2016).
Du, R. et al. Integrative mouse and human studies implicate ANGPT1 and ZBTB7C as susceptibility genes to ischemic injury. Stroke 46, 3514–3522 (2015).
Gilling, M. et al. Biparental inheritance of chromosomal abnormalities in male twins with non-syndromic mental retardation. Eur. J. Med. Genet. 54, e383–e388 (2011).
Rajabli, F. et al. Ancestral origin of ApoE ε4 Alzheimer disease risk in Puerto Rican and African American populations. PLOS Genet. 14, e1007791 (2018).
Jun, G. R. et al. Transethnic genome-wide scan identifies novel Alzheimer’s disease loci. Alzheimer’s Dement. 13, 727–738 (2017).
Hohman, T. J. et al. Global and local ancestry in African-Americans: Implications for Alzheimer’s disease risk. Alzheimer’s Dement. 12, 233–243 (2016).
Burrows-Wheeler Aligner. Available at: http://bio-bwa.sourceforge.net.
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. 1–9 (2012).
GATK. Available at: https://software.broadinstitute.org/gatk/.
bcbio-nextgen workflow. Available at: https://github.com/bcbio/bcbio-nextgen.
FastQC. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Okonechnikov, K., Conesa, A. & García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294 (2016).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 1–16 (2015).
Purcell, S. & Chang, C. PLINK v1.9. Available at: https://www.cog-genomics.org/plink/1.9/.
TOPMed. Available at: https://www.nhlbiwgs.org.
BRAVO browser. Available at: https://www.bravo.sph.umich.edu.
Human Pseudogene Annotation. Available at: http://pseudogene.org/human.
Cunningham, F. et al. Ensembl 2019. Nucleic Acids Res. 47, D745–D751 (2019).
Laird, N., Horvath, S. & Xu, X. Implementing a unified approach to family-based tests of association. Genet. Epidemiol. 19 (2000).
Lange, C., Weiss, S. T. & Laird, N. A. N. M. A multivariate family-based association test using generalized estimating equations: FBAT-GEE. 195–206 (2003).
Team, R. C. R: A Language and Environment for Statistical Computing. Available at: https://www.r-project.org.
Pruim, R. J. et al. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics 27, 2336–2337 (2011).
Acknowledgements
This work was supported by Cure Alzheimer’s Fund. D.P. was supported by the Women’s Alzheimer’s Movement research grant. D.L.D. was supported by PO1 HL 132825, PO1 HL 114501, PO1 HL 105339. R.K., B.A.C. and O.H. at the Harvard Chan Bioinformatics Core was supported in part by the Harvard NeuroDiscovery Center. The computations in this paper were run in part on the Odyssey cluster supported by the FAS Division of Science, Research Computing Group at Harvard University with support from John Morrissey and in part on compute provided by Dell HPC Research Computing Solutions with support by Glen Otero. The funding body has no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript. Please refer to the Supplementary Note for full acknowledgements.
Author information
Authors and Affiliations
Contributions
D.P., C.L. and R.E.T. contributed to the study concept and design, data analysis, statistical support and manuscript writing. J.H. contributed to study concept and design, data analysis and statistical support. N.L. contributed to the study concept and design, statistical support and manuscript writing. D.L.D. contributed to the study concept and design and manuscript writing. L.B. contributed to manuscript writing. R.K., B.A.C., O.H., K.M. and W.H. contributed to data analysis. All authors contributed to the critical review and editing of the manuscript and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Prokopenko, D., Hecker, J., Kirchner, R. et al. Identification of Novel Alzheimer’s Disease Loci Using Sex-Specific Family-Based Association Analysis of Whole-Genome Sequence Data. Sci Rep 10, 5029 (2020). https://doi.org/10.1038/s41598-020-61883-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61883-6
This article is cited by
-
Integrating Multi-omics Data for Alzheimer’s Disease to Explore Its Biomarkers Via the Hypergraph-Regularized Joint Deep Semi-Non-Negative Matrix Factorization Algorithm
Journal of Molecular Neuroscience (2024)
-
Functional variants identify sex-specific genes and pathways in Alzheimer’s Disease
Nature Communications (2023)
-
PILRA polymorphism modifies the effect of APOE4 and GM17 on Alzheimer’s disease risk
Scientific Reports (2022)
-
Epigenome-wide DNA methylation analysis of whole blood cells derived from patients with GAD and OCD in the Chinese Han population
Translational Psychiatry (2022)
-
Region-based analysis of rare genomic variants in whole-genome sequencing datasets reveal two novel Alzheimer’s disease-associated genes: DTNB and DLG2
Molecular Psychiatry (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
