
Abstract
Autism spectrum disorder (ASD) is a complex disorder with an unclear aetiology and an estimated global prevalence of 1%. However, studies of ASD in the Vietnamese population are limited. Here, we first conducted whole exome sequencing (WES) of 100 children with ASD and their unaffected parents. Our stringent analysis pipeline was able to detect 18 unique variants (8 de novo and 10 ×-linked, all validated), including 12 newly discovered variants. Interestingly, a notable number of X-linked variants were detected (56%), and all of them were found in affected males but not in affected females. We uncovered 17 genes from our ASD cohort in which CHD8, DYRK1A, GRIN2B, SCN2A, OFD1 and MDB5 have been previously identified as ASD risk genes, suggesting the universal aetiology of ASD for these genes. In addition, we identified six genes that have not been previously reported in any autism database: CHM, ENPP1, IGF1, LAS1L, SYP and TBX22. Gene ontology and phenotype-genotype analysis suggested that variants in IGF1, SYP and LAS1L could plausibly confer risk for ASD. Taken together, this study adds to the genetic heterogeneity of ASD and is the first report elucidating the genetic landscape of ASD in Vietnamese children.
Similar content being viewed by others


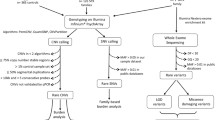
Introduction
Autism spectrum disorder (ASD) is a group of neurodevelopmental disorders that includes autism, Asperger’s syndrome and pervasive developmental disorder not otherwise specified and is characterized by restricted repetitive behaviour, delays in language development and lack of reciprocal social communication and interaction1. The common psychiatric and cognitive comorbidities with ASD include intellectual disability (ID)2,3,4, sleep deprivation5,6 and epilepsy (EP)3,7.
Recent studies have gradually emphasized the role of genetics in ASD, with up to 25% of ASD cases having genetically identifiable causes8. Males have a 4-fold higher incidence of ASD than females9, suggesting the involvement of genetics in the aetiology of ASD. Previous studies have shown that 5% to 15% of patients with ASD have inherited copy number variations (CNVs) or de novo CNVs in some affected genes with synaptic function10. Genome-wide association studies have indicated that common variations are risk factors for ASD11,12. Meanwhile, de novo CNV and loss of function (LoF) mutations merely explain 2.6% of the variance in liability12. Other studies have revealed that non-coding de novo mutations in the promoter regions also affect ASD13,14. Recent genomic studies with large samples have explored new genes linked to ASD15,16. To date, over 100 genes have been identified as strongly linked to the risk of ASD17,18 and enriched in the following three main pathways: chromatin remodelling, transcription and splicing, and synaptic function18,19,20. In fact, ASD is highly genetically heterogeneous due to multiple familial inheritance patterns and the occurrence of a large number of de novo variations. It is estimated that up to 1000 genes are potentially implicated ASD21,22, making it one of the most complex disorders. Genetic factors are known to play an important role in ASD aetiology, but that their elucidation is a work in progress23,24,25.
The prevalence of ASD has been recently reported as high as 16.8 per 1000 or 1/59 children in the US9. Unlike that in Western populations, ASD is not well studied in Asia, where the prevalence of ASD varies from studies and populations, ranging from 0.09–1.07% in south Asia26, 0.1018% in China27 and 2.64% in Korea4. Another study estimated that the prevalence of ASD in Asia from 1980 to recent present is 14.8/10,000 children28. A comprehensive study on 17,277 children aged 18 to 30 months in Vietnam revealed that the prevalence was 0.752%, and the number continues to increase29. It is estimated that ASD affects from 1% to 2% of the global population4,30,31. The reason for the varied prevalence of ASD across populations remains unclear, although sociodemographic factors such as socioeconomic status, ethnicity and parental education level have been reported to influence the diagnosis of ASD, resulting in disparities in ASD prevalence across populations32,33,34,35,36. With an increasing number of new variants/genes associated with ASD and different prevalence rates of ASD, we wanted to help shed light on whether genetic variation linked to ASD varies between populations. Thus, we conducted whole exome sequencing (WES) using a trio-based approach to investigate the genetic pattern of Vietnamese children with ASD.
Results
Clinical characteristics
We initially recruited 105 children who were definitively diagnosed with ASD and their unaffected parents. However, five families dropped out during recruitment. Finally, 100 trios were selected to participate in this study. In general, observations across populations, including Vietnamese populations, indicate that the ASD-affected male/female (M/F) ratio is approximately 4-fold29,37,38. The ratio of M/F with ASD in our study was slightly higher than this value (M/F: 4.9-fold). By the enrolment time, the proband’s age ranged from 3 to 17 years old (y/o). The average age was 6.9 y/o (Supplementary Table S1). The CARS scores ranged from 35 to 55.5, with an average score of 46.8 points. The DSM-V results showed that most probands were ranked at level 3 or required very substantial support, accounting for 68% of the cases. Denver II, comprising five domains including “Personal-social”, “Fine motor”, “Gross motor”, “Understanding” and “Language”, indicated that all probands had a lower developmental level compared to their chronological age. Overall, our cohort included ASD patients from mild to severe levels (Supplementary Table S1).
In addition to common autistic symptoms such as repetitive behaviours, speech delay, jargon, hyperactivity, toe walking and spinning, a quarter of the probands exhibited comorbid conditions such as ID (13 probands), EP (two probands), seizure (seven probands), ADHD (three probands) and cerebral palsy (CP) (one proband). Two cases showed congenital foot defects (ASD025 and ASD082) (Supplementary Table S1). Notably, six subjects had siblings with ASD (ASD004, ASD027) or with other neurological conditions, such as speech delay, CP and ID (ASD062, ASD065, ASD072 and ASD075). Of these, proband ASD004 and his male sibling were twins conceived through in vitro fertilization (IVF), while proband ASD027 and his sibling were monozygotic twins (Supplementary Table S1). Twenty-three probands showed language regression during their early life. Six probands often experienced gastrointestinal (GI) problems, mostly chronic constipation and diarrhoea. We were able to perform brain fluorodeoxyglucose (FDG)-PET/CT indicating hypometabolism of 29 probands and brain MRI of 40 cases (Supplementary Table S1). Brain MRI in 15 probands showed abnormal signal changes, while the rest showed either no abnormality detected or unavailable information.
Screening of Rett and Fragile-X syndrome
Rett syndrome (MIM#312750) is a severe neurodevelopmental disorder that almost exclusively affects females. The disease is caused by heterozygous mutations in the X-linked MECP2 gene, which encodes methyl-CpG binding protein 2. We screened all 17 ASD females for mutations in exons 2, 3 and 4 of the MECP2 gene but did not find any potential causal variants. In other aspect, Fragile-X syndrome (MIM#30064) is caused by an expansion repeat of CGG in the 5′ UTR of the FMR1 gene. This syndrome is a common comorbid condition with ASD as well as ID39,40,41. Therefore, we next screened for this syndrome in all 100 children with ASD. Surprisingly, no subjects in our study had changes in the FMR1 gene. The patients with no mutations found in these two screening tests and their parents were then selected for the WES experiment.
Variant identification
Paired-end sequencing resulted in 36.4 billion reads from 100 trios with an average Q30 score of 95.2. The average sequencing depth in the target region of all samples was 83×, ranging from 52 to 146.7× (Supplementary Table S2). All samples passed the quality control recommended by the manufacturer (Illumina). Although all the parents had self-claimed that they were biological parents to their children, our relatedness analysis revealed unclear parental relations in four cases (ASD080, ASD091, ASD100 and ASD101). In addition, the mother’s sample from case ASD034 was inadvertently sequenced twice (Supplementary Table S3). Therefore, we excluded these cases from the downstream analyses. Reads with low depth, variants with MAF > 0.1%, synonymous variants and variants predicted as benign, tolerated or neutral by PolyPhen-2, SIFT and MutationTaster were removed accordingly. We finally detected and Sanger validated a total of 18 unique variants in 17 genes from 16 unrelated probands. X-linked variants predominated, accounting for 56% of the total, followed by de novo variants (44%) (Fig. 1a, Table 1, Supplementary Fig. S1).

Types of validated variants and inheritance modes. (A) The distribution of inheritance modes; (B) The distribution of variant types. The values indicate the number of variants, and their percentages are presented in parentheses.
We observed that only six variants were recorded in dbSNP (Build 152), and the remaining variants were newly uncovered (Table 1). Missense variants were the major events, accounting for 61% of the total detected variants, followed by frameshift (17%), in-frame deletion (11%) and stop gained variants (11%) (Fig. 1b). Interestingly, we found two different variants in the DMD gene from two unrelated probands (ASD057 and ASD059) (Tables 1, 2). We used SnpEff, SIFT, PolyPhen-2, MutationTaster and CADD to predict the deleteriousness of the nucleotide changes. SnpEff predicted that five different variants detected from four individuals had a high impact (Table 2), and all of them were LoF variants (MDB5:c.14_15delAA, DYRK1A:c.601 C > T, GRIN2B:c.2208dupG, AGTR2:c.757 C > T and SCN2A:c.232delC), while the rest had a moderate impact. Two variants, DYRK1A: c.601 C > T (proband ASD038) and AGTR2:c.757 C > T (proband ASD046), had the highest CADD scores, suggesting these variants were most deleterious (Table 2). Two missense variants, CHM:c.866 T > C (proband ASD086) and SLC16A7:c.260 C > T (proband ASD097), were predicted to be benign by PolyPhen-2 but were damaging by MutationTaster and SIFT. The remaining variants were predicted to be damaging or disease causing by all employed in silico tools (Table 2).
Phenotype-genotype analysis and biological function
In total, we obtained 17 different genes, which showed genetic predispositions from 16 unrelated probands. We used the three most popular autism databases, SFARI (https://gene.sfari.org/), AutDB42 and the syndromic category in AutismKB43, to investigate the association of the detected genes with ASD. We found that 11/17 genes were previously reported in SFARI and AutDB (Table 3). Meanwhile, only four genes, AGTR2, ATRX, DMD and MBD5, were categorized as syndromic ASD genes in AutismKB. We next compared our genes to gene sets of ASD or other neurological disorders and found that CHD8, DYRK1A, GRIN2B, MBD5 and SCN2A overlapped with the ASD gene sets (FDR ≤ 0.1) reported in previous large-scale ASD studies44,45. CHD8, DYRK1A, GRIN2B and SCN2A were found in a gene set containing 94 genes enriched in developmental disorders derived from the DECIPHER project46. Three genes (DYRK1A, GRIN2B and SCN2A) were found in the EP gene set47. Hence, these genes detected in our ASD cohort are highly implicated in ASD or other neurological disorders.
Interestingly, our data showed six ASD candidate genes, CHM, ENPP1, IGF1, LAS1L, SYP and TBX22, which have not been previously reported in the three aforementioned autism databases (Table 3). Human Phenotype Oncology (HPO) analysis showed that all of these candidate genes have been reported to be linked to human diseases (Table 3). Mutations in the IGF148,49, LAS1L50,51 and SYP52 genes have been found in patients with CP and ID. In our study, a de novo missense variant was detected in the IGF1 gene from a female proband (ASD006) whose language and intellectual status were regressed after 18 m/o. Her language and personal-social status were strongly delayed (Supplementary Table S1). Brain PET-CT showed a decrease in FDG uptake in the temporal lobe, bilateral hippocampus, prefrontal region and parietal lobe (Supplementary Tables S1,S4). IGF1 mutations have been previously found in individuals with hyperactivity and short intension48,49. Treatment with IGF1 or expression of SHANK3, which is associated with idiopathic ASD, may restore synaptic deficits in neurons from Phelan-McDermid syndrome53,54. We thus conferred that IGF1 deficiency may reduce the synaptic transmission of neurons.
We also found two X-linked variants in the LAS1L and SYP genes from a male proband (ASD076). SYP encodes synaptophysin, which is involved in the regulation of synaptic plasticity55. GO analysis also showed that this gene is involved in synaptic membrane activity (Fig. 2, Supplementary Table S5). Moreover, several SYP mutations with evidence of segregation have been reported in patients with X-linked nonsyndromic mental retardation52. Therefore, SYP is a plausible gene for ASD. In addition, LAS1L encodes the ribosomal biogenesis protein LAS1L, which is required for cell proliferation, ribosome biosynthesis of the 60 S ribosomal subunit and the maturation of 28 S rRNA56,57. Missense mutations in the LAS1L gene have been detected in individuals with Wilson-Turner X-linked mental retardation syndrome50 and in a proband with congenital lethal motor neuron disease51. Our proband showed language regression after 12 m/o. At 5 y/o, he remained nonverbal and progressively developed more severe symptoms (Supplementary Table S4). Brain PET-CT images showed severe hypometabolism in the hippocampus region and in the frontal lobe (Supplementary Table S1). Interestingly, we observed that two probands (ASD006, ASD076) who carried IGF1, SYP and LAS1L variants showed language regression and brain hypometabolism (Supplementary Tables S1,S4).

Gene ontology analysis. Candidate genes and the gene sets are listed in the vertical line and in the upper horizontal line, respectively.
Gene ontology (GO) analysis of the 17 detected genes against 9996 gene sets derived from the Molecular Signatures Database (MSigDB) with a false discovery rate (FDR) <0.05 revealed that AGTR2, PLXNA3, ATRX, GRIN2B and CHD8 overlapped in the GO categories head development and central nervous system development (Fig. 2, Supplementary Table S5). Four genes, GRIN2B, SCN2A, DMD and SYP, were found in the GO category synaptic membrane. We also observed that ENPP1, IGF1, AGTR2, PLXNA3, DYRK1A and CHD8 were involved in the GO category negative regulation of response to stimulus. These results suggested that these particular biological processes might be related to ASD or other neurological conditions.
Discussion
The rapidly declining cost of next-generation sequencing in recent years has resulted in many genetic studies of neurodegenerative disorders, including ASD58,59,60. As a result, an increasing number of new genes and variants have been found to be enriched in ASD individuals, revealing the highly heterogeneous nature of ASD. In addition, cutting-edge molecular biology or modelling of ASD allows us to better define and understand the aetiology of ASD and biological pathways of associated genes61. In this study, we performed whole exome sequencing of 100 Vietnamese children with ASD and their unaffected parents together with analysis of biological processes to explore the genetic landscape of ASD in Vietnamese children.
Gender bias between males and females in ASD is generally acknowledged, where males have an approximately 4-fold higher incidence of ASD than females. Several hypotheses on male bias, such as the nonrecognition of females with ASD62,63, differences in brain structure and genetic load between females and males64 and female protective effect against autistic behaviour65, have been proposed. It is hypothesized that females carry a higher genetic load and are thus less vulnerable to ASD from genetic causes than males66. Meanwhile, de novo variations are strongly associated with ASD, suggesting that females have a greater resistance to de novo variations than males67,68. Our findings with a predominance of males over females (4.9-fold) are consistent with those from previous studies65,69,70,71. Moreover, the results of all X-linked variants detected in affected males further supported the recent hypothesis of the female protective effect against ASD72.
Regarding the number of de novo variants detected, a previous study using whole genome sequencing from the 1902 quartet family with ASD reported 67.1 de novo variants per child’s genome (61 SNV and 5.6 indel DNV per child)13. Another study using WES73 detected 0.9 de novo variants per child’s exome (869 SNVs and 27 indel de novo variants in a total cohort of 990 ASD samples). In our study, before variant filtering by stringent conditions, we detected 1.3 de novo variants per child’s exome, which was compatible with the results of previous studies. After filtering, the de novo variant rate in our study was 0.08%, including three missense and five LoF variants (three frameshift, one stop gained and one in-frame deletion) (Table 1).
We observed that 11 genes from our cohort have been reported in the autism databases (Table 3). Among these genes, CHD8, DYRK1A, GRIN2B, SCN2A, and MDB5 were highly recognized as ASD risk genes with FDR <0.1, as previously reported17,18,44,45,74,75. In addition, OFD1 is involved in the Wnt pathway and is highly suggested as an ASD risk gene76. Most of these genes also overlapped in the gene sets of developmental disorders46 or EP47. Thus, these genes were highly universal for ASD regardless of population.
Given that ASD is highly genetically heterogeneous, there are several key biological processes involved in the development of ASD, including neurogenesis, synaptic plasticity, synaptogenesis and neurite growth8,20,77. Among the six newly uncovered genes in this study, we observed that IGF1 and SYP encode proteins involved in brain development and synaptic activities, respectively. In addition, a dozen ASD-linked genes are involved in several biological processes, such as ribosomal maturation and mRNA regulation, that are linked to synaptic function and chromosome condensation18,20,78. Defects in genes encoding ribosomal proteins have resulted in downregulation of these genes in children with ASD and healthy woman with autistic children79. Therefore, defects of LAS1L, which plays a role in ribosome biogenesis, likely lead to a disruption of ribosomal maturation.
We found 18 unique variants in 17 genes from 16 unrelated probands. Biological analysis showed that many of these genes were involved in neuronal activities and formation. Together with previously ASD-linked genes, other newly detected genes in this study, including IGF1, LAS1L and SYP, were found to be plausible ASD-related genes due to their involvement in some risk-associated biological processes and their associations with other neurological conditions. In general, ASD is a complex disorder showing a broad range of phenotypes, where 35% of ASD cases have ID, 5–15% have EP and 50% have language developmental delay22. Therefore, phenotype-genotype analysis accompanied by biological process/pathway analysis is critical for the determination of their associations with ASD. However, functional studies on these genes in ASD development should be performed.
Conclusions
Consistent with previous studies, this study found a predominance of ASD-affected males over affected females (4.9-fold). Rett and Fragile-X syndrome are the most common comorbidities with ASD, but there was no individual in our cohort with these syndromes. With a stringent pipeline, we finally identified 18 unique variants that occurred in 17 genes, of which 12 variants were reported for the first time. All X-linked variants were detected in male probands but not in affected females. This finding is consistent with a contribution of X-linked recessive variants to ASD. We found 11 genes formally associated with ASD, some of which have been previously identified as ASD risk genes (CHD8, DYRK1A, GRIN2B, SCN2A, OFD1 and MDB5), indicating the genetic universal aetiology of ASD. Interestingly, this study uncovered variants in six new candidate genes (CHM, ENPP1, IGF1, LAS1L, SYP and TBX22) enriched in our ASD cohort. Analyses of phenotype-genotype and GO showed that many of our detected genes were associated with several neurological conditions and were involved in some neuronal biological processes, such as synaptic processes, regulation of transport and ribosome maturation. We conferred that genetic predispositions in these genes might be causative factors. This study is the first to elucidate the common and unique genetic landscape of ASD in Vietnamese children.
Methods
Ethics statement
The study protocol was approved by the Ethical Committee of Vinmec International Hospital in accordance with the Declaration of Helsinki. Before the enrolments, written informed consent forms including the use of peripheral blood and clinical data for research use and publication were obtained from the parents.
Study cohort and sample collection
Children with suspected ASD were independently examined by trained physicians and psychologists at Vinmec Times City International Hospital, Hanoi, Vietnam, using the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-V)1, the Autism Diagnostic Observation Schedule (ADOS)80, and Childhood Autism Rating Scale (CARS)81. DSM-V classified the severity of ASD into three levels: Level 1 (requiring support), Level 2 (requiring substantial support) and Level 3 (requiring very substantial support). To examine the development of the probands, the Denver Developmental Screening test II82 (Denver II) was used. Development of the proband (in months) was examined based on five domains: personal-social, fine motor, gross motor, understanding and language. The developmental quotient (DQ) was calculated by dividing the average developmental age from five domains by the chronological age of the proband and multiplying by 100. Definitively diagnosed children with ASD and their unaffected parents were recruited to participate in the study. Approximately 3–4 mL of peripheral blood from children with ASD and their unaffected parents was collected and kept in EDTA tubes. Genomic DNA was extracted by using a QIAamp Blood Kit (Qiagen, Germany) and stored at −80 °C before use.
Screening of Fragile-X and RETT syndrome
Fragile-X and Rett syndrome are the most common causes of inherited mental retardation and are reportedly linked to ASD. Therefore, this study aimed to exclude patients with either Fragile-X or Rett syndrome. An AmplideX FMR1 PCR kit (Asuragen, TX, USA) was used for screening Fragile-X syndrome. Genotypes were determined by examining the size of the trinucleotide repeat segment and the methylation status of the FMR1 gene. Since Rett syndrome is predominantly found in females due to the mutations in the MECP2 gene, all females diagnosed with ASD were selected for screening for mutations in exons 2, 3 and 4 of the MECP2 gene83. A detailed method and primer list for MECP2 mutation screening can be found in our previous study84. Direct sequencing was performed on an ABI 3500 DX system as indicated hereinafter.
Whole exome sequencing
Probands without mutations detected in the MECP2 and FMR1 genes and their biological self-claimed parents were selected for the WES experiment. A DNA library was constructed by using a Nextera Rapid Capture Exome Kit (Illumina, USA) capturing over 98% of the exonic contents. The library concentration was quantified by a Qubit dsDNA Broad Range Assay Kit (Invitrogen, USA). Library size was measured by a Lab Chip 3 K Hisense Kit (Perkin Elmer, USA). Paired-end exome sequencing with a read length of 75 × 2 bp was performed on a HiSeq. 4000 (Illumina, USA).
Data analysis
Adapters were removed prior to downstream analysis. BWA version 0.7.15 was used for alignment against the human reference genome version GRCh3785. Short, index and mark duplicates were assessed by using Samtool version 1.386 and Picard version 2.7.2 (http://broadinstitute.github.io/picard/). GATK toolkit version 3.687 and Platypus version 0.8.188 were used to call variants (single nucleotide variants and indels with less than 50 bp). Variants were considered highly reliable if they were called by both GATK and Platypus with a Phred-score of equal or greater than 30. The minor allele frequency (MAF) was attained from the 1000 Human Genome Project89 and gnomAD database (https://gnomad.broadinstitute.org/). Biological relatedness was analysed by using Peddy software version v0.4.390. Trio samples with non-biological relatedness were excluded from the downstream analyses.
Variant classification
Variants were annotated by SnpEff programme version 4.3g91 and fulfilled the following criteria: (i) variant with MAF <0.1%92 against gnomAD, gnomAD East Asia, and 1000 Human Genomes Project; (ii) variant passed the GATK standard filters; (iii) nonsynonymous variants including missense variants with a prediction of damaging impact and LoF variants (nonsense, frameshift and splicing variants); and (iv) variant occurred in genes recorded in the DECIPHER and/or SFARI databases. Putative de novo variants were considered if independent reads in all family members were ≥20 and if both parents were homozygous for the reference and the offspring was heterozygous; an X-linked variant was considered if it occurred on the X chromosome.
In silico prediction, gene function and biological process
PolyPhen-2 version 2.293, SIFT version 4.094 and MutationTaster95 were employed to predict the damaging impact of the missense variants. Combined Annotation-Dependent Depletion (CADD) GRCh37-v.14 was used to score the deleteriousness of variants with a single nucleotide change96. Gene function, protein class and biological process were determined by the PANTHER classification system v.1497. Validated gene sets were computed with the Molecular Signatures Database (MSigDB) v6.298,99 to further explore the gene ontology of the ASD candidate genes. Human Phenotype Ontology (HPO-Web version 1.5.0)100 was used to find the association of candidate genes with human diseases.
Variant validation
After the stringent filtering strategy, the identified variants were validated by Sanger sequencing. Proper primers were designed for each variant by using Primer3Plus software101. Fresh DNA was newly extracted from stored blood samples by using the same DNA extraction protocol. Target fragments were amplified by GoTaq DNA polymerase (Promega, USA). The amplicons were sequenced on an ABI 3500 DX system using a BigDye Terminator v3.1 (Thermo Fisher Scientific, USA). Both forward and reverse read alignment were used to determine the sequence.
Data availability
Sequencing data generated and analysed in this study are included in this article and its Supplementary Information.
References
Association, A. P. Diagnostic and Statistical Manual of Mental Disorders. 5 edn, Vol. 2017 (American Psychiatric Association, 2013).
Perou, R. et al. Mental health surveillance among children–United States, 2005-2011. MMWR Suppl. 62, 1–35 (2013).
Amiet, C. et al. Epilepsy in autism is associated with intellectual disability and gender: evidence from a meta-analysis. Biol. Psychiatry 64, 577–582, https://doi.org/10.1016/j.biopsych.2008.04.030 (2008).
Kim, Y. S. et al. Prevalence of autism spectrum disorders in a total population sample. Am. J. Psychiatry 168, 904–912, https://doi.org/10.1176/appi.ajp.2011.10101532 (2011).
Liu, X., Hubbard, J. A., Fabes, R. A. & Adam, J. B. Sleep disturbances and correlates of children with autism spectrum disorders. Child. Psychiatry Hum. Dev. 37, 179–191, https://doi.org/10.1007/s10578-006-0028-3 (2006).
Krakowiak, P., Goodlin-Jones, B., Hertz-Picciotto, I., Croen, L. A. & Hansen, R. L. Sleep problems in children with autism spectrum disorders, developmental delays, and typical development: a population-based study. J. Sleep. Res. 17, 197–206, https://doi.org/10.1111/j.1365-2869.2008.00650.x (2008).
Besag, F. M. Epilepsy in patients with autism: links, risks and treatment challenges. Neuropsychiatr. Dis. Treat. 14, 1–10, https://doi.org/10.2147/NDT.S120509 (2017).
Huguet, G., Ey, E. & Bourgeron, T. The genetic landscapes of autism spectrum disorders. Annu. Rev. Genomics Hum. Genet. 14, 191–213, https://doi.org/10.1146/annurev-genom-091212-153431 (2013).
Baio, J. et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill. Summ. 67, 1–23, https://doi.org/10.15585/mmwr.ss6706a1 (2018).
Devlin, B. & Scherer, S. W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 22, 229–237, https://doi.org/10.1016/j.gde.2012.03.002 (2012).
Klei, L. et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism 3, 9, https://doi.org/10.1186/2040-2392-3-9 (2012).
Gaugler, T. et al. Most genetic risk for autism resides with common variation. Nat. Genet. 46, 881–885, https://doi.org/10.1038/ng.3039 (2014).
An, J.-Y. et al. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Sci. 362, eaat6576, https://doi.org/10.1126/science.aat6576 (2018).
Turner, T. N. et al. Genomic Patterns of De Novo Mutation in Simplex Autism. Cell 171, 710–722 e712, https://doi.org/10.1016/j.cell.2017.08.047 (2017).
O’Roak, B. J. et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Sci. 338, 1619, https://doi.org/10.1126/science.1227764 (2012).
Yuen, R. K. C. et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. https://doi.org/10.1038/nm.3792 (2015).
Betancur, C. Etiological heterogeneity in autism spectrum disorders: More than 100 genetic and genomic disorders and still counting. Brain Res. 1380, 42–77, https://doi.org/10.1016/j.brainres.2010.11.078 (2011).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nat. 515, 209–215, https://doi.org/10.1038/nature13772 (2014).
Park, H. R. et al. A Short Review on the Current Understanding of Autism Spectrum Disorders. Exp. Neurobiol. 25, 1–13, https://doi.org/10.5607/en.2016.25.1.1 (2016).
Gilbert, J. & Man, H. Y. Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front. Cell Neurosci. 11, 359, https://doi.org/10.3389/fncel.2017.00359 (2017).
Ramaswami, G. & Geschwind, D. H. Genetics of autism spectrum disorder. Handb. Clin. Neurol. 147, 321–329, https://doi.org/10.1016/b978-0-444-63233-3.00021-x (2018).
Geschwind, D. H. & State, M. W. Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol. 14, 1109–1120, https://doi.org/10.1016/s1474-4422(15)00044-7 (2015).
Rylaarsdam, L. & Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell Neurosci. 13, 385, https://doi.org/10.3389/fncel.2019.00385 (2019).
Robert, C. et al. Role of Genetics in the Etiology of Autistic Spectrum Disorder: Towards a Hierarchical Diagnostic Strategy. Int J Mol Sci 18, https://doi.org/10.3390/ijms18030618 (2017).
Vorstman, J. A. S. et al. Autism genetics: opportunities and challenges for clinical translation. Nat. Rev. Genet. 18, 362–376, https://doi.org/10.1038/nrg.2017.4 (2017).
Hossain, M. D. et al. Autism Spectrum disorders (ASD) in South Asia: a systematic review. BMC Psychiatry 17, 281, https://doi.org/10.1186/s12888-017-1440-x (2017).
Wang, F. et al. The prevalence of autism spectrum disorders in China: a comprehensive meta-analysis. Int. J. Biol. Sci. 14, 717–725, https://doi.org/10.7150/ijbs.24063 (2018).
Sun, X. & Allison, C. A review of the prevalence of Autism Spectrum Disorder in Asia. Res. Autism Spectr. Disord. 4, 156–167, https://doi.org/10.1016/j.rasd.2009.10.003 (2010).
Hoang, V. M. et al. Prevalence of autism spectrum disorders and their relation to selected socio-demographic factors among children aged 18–30 months in northern Vietnam, 2017. Int. J. Ment. Health Syst. 13, 29, https://doi.org/10.1186/s13033-019-0285-8 (2019).
Mpaka, D. M. et al. Prevalence and comorbidities of autism among children referred to the outpatient clinics for neurodevelopmental disorders. Pan Afr Med J 25, https://doi.org/10.11604/pamj.2016.25.82.4151 (2016).
Brugha, T. S. et al. Epidemiology of autism spectrum disorders in adults in the community in England. Arch. Gen. Psychiatry 68, 459–465, https://doi.org/10.1001/archgenpsychiatry.2011.38 (2011).
Zaroff, C. M. & Uhm, S. Y. Prevalence of autism spectrum disorders and influence of country of measurement and ethnicity. Soc. Psychiatry Psychiatr. Epidemiol. 47, 395–398, https://doi.org/10.1007/s00127-011-0350-3 (2012).
Thomas, P. et al. The association of autism diagnosis with socioeconomic status. Autism 16, 201–213, https://doi.org/10.1177/1362361311413397 (2012).
Russell, G., Steer, C. & Golding, J. Social and demographic factors that influence the diagnosis of autistic spectrum disorders. Soc. Psychiatry Psychiatr. Epidemiol. 46, 1283–1293, https://doi.org/10.1007/s00127-010-0294-z (2011).
Liu, K.-Y., King, M. & Bearman, P. S. Social influence and the autism epidemic. AJS; Am. J. Sociol. 115, 1387–1434, https://doi.org/10.1086/651448 (2010).
Mazurek, M. O. et al. Age at first autism spectrum disorder diagnosis: the role of birth cohort, demographic factors, and clinical features. J. Dev. Behav. Pediatr. 35, 561–569, https://doi.org/10.1097/dbp.0000000000000097 (2014).
Loomes, R., Hull, L. & Mandy, W. P. L. What Is the Male-to-Female Ratio in Autism Spectrum Disorder? A Systematic Review and Meta-Analysis. J. Am. Acad. Child. Adolesc. Psychiatry 56, 466–474, https://doi.org/10.1016/j.jaac.2017.03.013 (2017).
Werling, D. M. & Geschwind, D. H. Sex differences in autism spectrum disorders. Curr. Opin. Neurol. 26, 146–153, https://doi.org/10.1097/WCO.0b013e32835ee548 (2013).
Niu, M. et al. Autism Symptoms in Fragile X Syndrome. J. Child. Neurol. 32, 903–909, https://doi.org/10.1177/0883073817712875 (2017).
Devitt, N. M., Gallagher, L. & Reilly, R. B. Autism Spectrum Disorder (ASD) and Fragile X Syndrome (FXS): Two Overlapping Disorders Reviewed through Electroencephalography-What Can be Interpreted from the Available Information? Brain Sci. 5, 92–117, https://doi.org/10.3390/brainsci5020092 (2015).
Crawford, D. C., Acuna, J. M. & Sherman, S. L. FMR1 and the fragile X syndrome: human genome epidemiology review. Genet Med 3, 359–371, 10.109700125817-200109000-00006 (2001).
Basu, S. N., Kollu, R. & Banerjee-Basu, S. AutDB: a gene reference resource for autism research. Nucleic Acids Res. 37, D832–D836, https://doi.org/10.1093/nar/gkn835 (2009).
Yang, C. et al. AutismKB 2.0: a knowledgebase for the genetic evidence of autism spectrum disorder. Database 2018, https://doi.org/10.1093/database/bay106 (2018).
Sanders, S. J. et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233, https://doi.org/10.1016/j.neuron.2015.09.016 (2015).
Satterstrom, F. K. et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. bioRxiv. https://www.biorxiv.org/content/ https://doi.org/10.1101/484113v3 (2019).
Deciphering Developmental Disorders, S. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438, https://doi.org/10.1038/nature21062 (2017).
Heyne, H. O. et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat. Genet. 50, 1048–1053, https://doi.org/10.1038/s41588-018-0143-7 (2018).
Woods, K. A., Camacho-Hubner, C., Savage, M. O. & Clark, A. J. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N. Engl. J. Med. 335, 1363–1367, https://doi.org/10.1056/nejm199610313351805 (1996).
Walenkamp, M. J. et al. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J. Clin. Endocrinol. Metab. 90, 2855–2864, https://doi.org/10.1210/jc.2004-1254 (2005).
Hu, H. et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol. Psychiatry 21, 133–148, https://doi.org/10.1038/mp.2014.193 (2016).
Butterfield, R. J. et al. Congenital lethal motor neuron disease with a novel defect in ribosome biogenesis. Neurol. 82, 1322–1330, https://doi.org/10.1212/wnl.0000000000000305 (2014).
Tarpey, P. S. et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 41, 535–543, https://doi.org/10.1038/ng.367 (2009).
Pinto, D. et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nat. 466, 368–372, https://doi.org/10.1038/nature09146 (2010).
Shcheglovitov, A. et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nat. 503, 267–271, https://doi.org/10.1038/nature12618 (2013).
Kwon, S. E. & Chapman, E. R. Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron 70, 847–854, https://doi.org/10.1016/j.neuron.2011.04.001 (2011).
Castle, C. D., Cassimere, E. K., Lee, J. & Denicourt, C. Las1L is a nucleolar protein required for cell proliferation and ribosome biogenesis. Mol. Cell Biol. 30, 4404–4414, https://doi.org/10.1128/MCB.00358-10 (2010).
Castle, C. D., Cassimere, E. K. & Denicourt, C. LAS1L interacts with the mammalian Rix1 complex to regulate ribosome biogenesis. Mol. Biol. Cell 23, 716–728, https://doi.org/10.1091/mbc.E11-06-0530 (2012).
Keogh, M. J. & Chinnery, P. F. Next generation sequencing for neurological diseases: new hope or new hype? Clin. Neurol. Neurosurg. 115, 948–953, https://doi.org/10.1016/j.clineuro.2012.09.030 (2013).
Goodwin, S., McPherson, J. D. & McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333, https://doi.org/10.1038/nrg.2016.49 (2016).
Sener, E. F., Canatan, H. & Ozkul, Y. Recent Advances in Autism Spectrum Disorders: Applications of Whole Exome Sequencing Technology. Psychiatry Investig. 13, 255–264, https://doi.org/10.4306/pi.2016.13.3.255 (2016).
Guerreiro, R., Bras, J., Hardy, J. & Singleton, A. Next generation sequencing techniques in neurological diseases: redefining clinical and molecular associations. Hum. Mol. Genet. 23, R47–53, https://doi.org/10.1093/hmg/ddu203 (2014).
Milner, V., McIntosh, H., Colvert, E. & Happe, F. A Qualitative Exploration of the Female Experience of Autism Spectrum Disorder (ASD). J Autism Dev Disord, https://doi.org/10.1007/s10803-019-03906-4 (2019).
Bargiela, S., Steward, R. & Mandy, W. The Experiences of Late-diagnosed Women with Autism Spectrum Conditions: An Investigation of the Female Autism Phenotype. J. Autism Dev. Disord. 46, 3281–3294, https://doi.org/10.1007/s10803-016-2872-8 (2016).
Baron-Cohen, S. et al. Why are autism spectrum conditions more prevalent in males? PLoS Biol. 9, e1001081, https://doi.org/10.1371/journal.pbio.1001081 (2011).
Robinson, E. B., Lichtenstein, P., Anckarsäter, H., Happé, F. & Ronald, A. Examining and interpreting the female protective effect against autistic behavior. Proc. Natl Acad. Sci. USA 110, 5258–5262, https://doi.org/10.1073/pnas.1211070110 (2013).
Levy, D. et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70, 886–897, https://doi.org/10.1016/j.neuron.2011.05.015 (2011).
Sebat, J. et al. Strong association of de novo copy number mutations with autism. Sci. 316, 445–449, https://doi.org/10.1126/science.1138659 (2007).
Sanders, S. J. et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nat. 485, 237–241, https://doi.org/10.1038/nature10945 (2012).
Szatmari, P. et al. Sex differences in repetitive stereotyped behaviors in autism: implications for genetic liability. Am. J. Med. Genet. B Neuropsychiatr. Genet 159B, 5–12, https://doi.org/10.1002/ajmg.b.31238 (2012).
Taylor, M. J. et al. Is There a Female Protective Effect Against Attention-Deficit/Hyperactivity Disorder? Evidence From Two Representative Twin Samples. J. Am. Acad. Child. Adolesc. Psychiatry 55, 504–512 e502, https://doi.org/10.1016/j.jaac.2016.04.004 (2016).
Jacquemont, S. et al. A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am. J. Hum. Genet. 94, 415–425, https://doi.org/10.1016/j.ajhg.2014.02.001 (2014).
Skuse, D. H. Imprinting, the X-chromosome, and the male brain: explaining sex differences in the liability to autism. Pediatr. Res. 47, 9–16 (2000).
Werling, D. M. et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet. 50, 727–736, https://doi.org/10.1038/s41588-018-0107-y (2018).
Neale, B. M. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nat. 485, 242–245, https://doi.org/10.1038/nature11011 (2012).
O’Roak, B. J. et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589, https://doi.org/10.1038/ng.835 (2011).
Li, J. et al. Targeted sequencing and functional analysis reveal brain-size-related genes and their networks in autism spectrum disorders. Mol. Psychiatry 22, 1282–1290, https://doi.org/10.1038/mp.2017.140 (2017).
Subramanian, M., Timmerman, C. K., Schwartz, J. L., Pham, D. L. & Meffert, M. K. Characterizing autism spectrum disorders by key biochemical pathways. Front. Neurosci 9, https://doi.org/10.3389/fnins.2015.00313 (2015).
Darnell, J. C. et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261, https://doi.org/10.1016/j.cell.2011.06.013 (2011).
Kuwano, Y. et al. Autism-associated gene expression in peripheral leucocytes commonly observed between subjects with autism and healthy women having autistic children. PLoS One 6, e24723, https://doi.org/10.1371/journal.pone.0024723 (2011).
Lord, C. et al. Autism diagnostic observation schedule: a standardized observation of communicative and social behavior. J. Autism Dev. Disord. 19, 185–212, https://doi.org/10.1007/bf02211841 (1989).
Schopler, E., Reichler, R. J., DeVellis, R. F. & Daly, K. Toward objective classification of childhood autism: Childhood Autism Rating Scale (CARS). J. Autism Dev. Disord. 10, 91–103, https://doi.org/10.1007/bf02408436 (1980).
Frankenburg, W. K., Dodds, J., Archer, P., Shapiro, H. & Bresnick, B. The Denver II: a major revision and restandardization of the Denver Developmental Screening Test. Pediatrics 89, 91–97 (1992).
Saunders, C. J., Minassian, B. E., Chow, E. W. C., Zhao, W. & Vincent, J. B. Novel exon 1 mutations in MECP2 implicate isoform MeCP2_e1 in classical Rett syndrome. Am. J. Med. Genet. A 149A, 1019–1023, https://doi.org/10.1002/ajmg.a.32776 (2009).
Le Thi Thanh, H. et al. Spectrum of MECP2 mutations in Vietnamese patients with RETT syndrome. BMC Med. Genet. 19, 137, https://doi.org/10.1186/s12881-018-0658-x (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma. 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics, https://doi.org/10.1093/bioinformatics/btp352 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303, https://doi.org/10.1101/gr.107524.110 (2010).
Rimmer, A. et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 46, 912, https://doi.org/10.1038/ng.3036 (2014).
Auton, A. et al. A global reference for human genetic variation. Nat. 526, 68–74, https://doi.org/10.1038/nature15393 (2015).
Pedersen, B. S. & Quinlan, A. R. Who’s Who? Detecting and Resolving Sample Anomalies in Human DNA Sequencing Studies with Peddy. Am. J. Hum. Genet. 100, 406–413, https://doi.org/10.1016/j.ajhg.2017.01.017 (2017).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 6, 80–92, https://doi.org/10.4161/fly.19695 (2012).
Kosmicki, J. A. et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet. 49, 504–510, https://doi.org/10.1038/ng.3789 (2017).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249, https://doi.org/10.1038/nmeth0410-248 (2010).
Vaser, R., Adusumalli, S., Leng, S. N., Sikic, M. & Ng, P. C. SIFT missense predictions for genomes. Nat. Protoc. 11, 1–9, https://doi.org/10.1038/nprot.2015.123 (2016).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361, https://doi.org/10.1038/nmeth.2890 (2014).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894, https://doi.org/10.1093/nar/gky1016 (2019).
Mi, H. et al. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 14, 703–721, https://doi.org/10.1038/s41596-019-0128-8 (2019).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinforma. 27, 1739–1740, https://doi.org/10.1093/bioinformatics/btr260 (2011).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425, https://doi.org/10.1016/j.cels.2015.12.004 (2015).
Köhler, S. et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 47, D1018–D1027, https://doi.org/10.1093/nar/gky1105 (2019).
Untergasser, A. et al. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 35, W71–74, https://doi.org/10.1093/nar/gkm306 (2007).
Acknowledgements
We thank the physicians, particularly Ms. Dang Thi Thanh Tung of the High Technology Unit for Cerebral Palsy and Autism, Vinmec Times City International Hospital, for their patient recruitment and assessment. This work was financially supported by the Vinmec Health Care system with grant No. ISC17.03.
Author information
Authors and Affiliations
Contributions
Conception of the study: L.T.N., K.T.T. and V.S.L. Patient recruitment and assessment: H.T.P.B., D.H.D., L.T.H. and H.T.T.L. Whole exome sequencing: K.T.T. and D.M.V. Genetic screening and Sanger sequencing validation: H.T.T.L., L.T.H., H.T.N., L.T.M.D. and T.H.N. Bioinformatics analysis: V.S.L. and K.T.T. Data interpretation: K.T.T. Manuscript drafting: K.T.T., V.S.L., L.T.N., A.M., H.T.N., L.T.M.D. and T.H.N.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tran, K.T., Le, V.S., Bui, H.T.P. et al. Genetic landscape of autism spectrum disorder in Vietnamese children. Sci Rep 10, 5034 (2020). https://doi.org/10.1038/s41598-020-61695-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61695-8
This article is cited by
-
Pathogenic/likely pathogenic mutations identified in Vietnamese children diagnosed with autism spectrum disorder using high-resolution SNP genotyping platform
Scientific Reports (2024)
-
Exploring key genes and pathways associated with sex differences in autism spectrum disorder: integrated bioinformatic analysis
Mammalian Genome (2024)
-
Assessment of machine learning strategies for simplified detection of autism spectrum disorder based on the gut microbiome composition
Neural Computing and Applications (2024)
-
The phenotypic spectrum and genotype-phenotype correlations in 106 patients with variants in major autism gene CHD8
Translational Psychiatry (2022)
-
Novel findings from family-based exome sequencing for children with biliary atresia
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
