
Abstract
Vascular bio-scaffolds produced from decellularized tissue offer a promising material for treatment of several types of cardiovascular diseases. These materials have the potential to maintain the functional properties of the extracellular matrix (ECM), and allow for growth and remodeling in vivo. The most commonly used methods for decellularization are based on chemicals and enzymes combinations, which often damage the ECM and cause cytotoxic effects in vivo. Mild methods involving pressurized CO2-ethanol (EtOH)-based fluids, in a supercritical or near supercritical state, have been studied for decellularization of cardiovascular tissue, but results are controversial. Moreover, data are lacking on the amount and type of lipids remaining in the tissue. Here we show that pressurized CO2-EtOH-H2O fluids (average molar composition, ΧCO2 0.91) yielded close to complete removal of lipids from porcine pulmonary arteries, including a notably decrease of pro-inflammatory fatty acids. Pressurized CO2-limonene fluids (ΧCO2 0.88) and neat supercritical CO2 (scCO2) achieved the removal of 90% of triacylglycerides. Moreover, treatment of tissue with pressurized CO2-limonene followed by enzyme treatment, resulted in efficient DNA removal. The structure of elastic fibers was preserved after pressurized treatment, regardless solvent composition. In conclusion, pressurized CO2-ethanol fluids offer an efficient tool for delipidation in bio-scaffold production, while pressurized CO2-limonene fluids facilitate subsequent enzymatic removal of DNA.
Similar content being viewed by others

Introduction
Cardiovascular diseases (CVDs) are responsible for 17.9 million deaths per year in the world (31% of total deaths)1. In 2015, the global prevalence of arterial hypertension (AHT), the most prevalent risk factor for CVD development, was estimated to be around 30–45% of the adult population, increasing up to 60% in people above 60 years of age2. Moreover, the prevalence of AHT is estimated to increase by 15–20% in 20252. Pulmonary arterial hypertension (PAH), a sub-form of AHT, is characterized by breakdown of elastic fibers and alterations in the cross-linking of collagen, resulting in remodeling the extracellular matrix (ECM) in pulmonary arteries3,4. Hypertrophic remodeling of the media and endothelial cell dysfunction result in a high vascular resistance and thrombosis4,5, potentially leading to right ventricular failure and death in severely affected patients.
Organ or tissue transplantation is the last option proposed for such CVDs-affected patients with a poor prognosis. However, the lack of compatible organs and tissues constitutes a major limitation. Even though the global rate of transplantation increased by 7.25% between 2015 and 2016, reaching a rate of 15.5 organs transplanted per hour6, less than 10% of the transplant needs are covered. Consequently, patients often have to wait long time for transplantation, resulting in worsening of their medical condition. Furthermore, those that are offered a transplantation require life-long immune therapy to reduce the risk for organ/tissue rejection7. Large efforts have been invested in approaches such as own-tissue regeneration, synthetic scaffold construction or bio-scaffold production to increase the availability of tissues for transplantation purposes8.
The potential of decellularized blood vessels as a source of vascular grafts have long been recognized, however ideal decellularization strategies are still sought after9. Decellularization of native tissue should reduce immunogenicity while preserving structure and biomechanical properties. Decellularized tissues are composed of natural ECM components and can be modified by repopulating cells allowing for remodeling, repair and growth in vivo. The material produced by decellularization of native tissues, termed bio-scaffold or bio-extracellular matrix (bio-ECM), are mainly composed of long-chain structural components, e.g. elastin and collagen10. These structures must be free from cellular components, but still maintain their native architectural and mechanical characteristics11. Lipid residues may hamper bio-ECM production as: (i) lipid bilayers constitute a barrier that hinders extraction of protein and genetic material; (ii) lipids, such as phosphatidylinositols, which are anchored to membrane proteins are involved in the development of immune responses12,13; (iii) some lipids hamper recellularization by impairing cell attachment14; and iv) certain lipids may act as a source of pro-inflammatory fatty acids, such as arachidonic acid15. Moreover, residual antigens (dsDNA) in the bio-ECM should be kept at a minimum to avoid activation of the immune system, which may lead to tissue rejection16,17. For a successful bio-ECM production, dsDNA and lipids (delipidation) need to be removed, while preserving ECM functionality. Ideally, the decellularization method should allow some bioactive molecules, e.g. growth factors and cell adhesion molecules, such as glycosaminoglycans, to remain in the ECM11.
Currently, the dominating techniques used for tissue decellularization utilize surfactant-based methodologies. Ionic, non-ionic or zwitterionic detergents, i.e. sodium dodecyl sulfate (SDS), Triton X-100 and CHAPS, respectively, results in efficient decellularization of the tissue, including removal of lipids11,18,19,20. Some decellularization protocols also include nucleases, which degrades genetic material in the tissue18,21. However, detergents often damage the ECM by disrupting the protein tertiary and quaternary structures19,22. Studies have revealed SDS to efficiently remove glycosaminoglycans, thereby causing collagen damage11,22. Detergents have also been shown to reduce levels of beneficial growth factors in some tissue23. Moreover, residual surfactants are cytotoxic24, yielding detrimental effects on the subsequent recellularization.
As an alternative to detergents, supercritical carbon dioxide (scCO2) has been proposed as a safer, non-toxic and non-residual technology to obtain bio-ECMs from multiple tissues, including porcine heart valve, equine tendon, porcine esophagus, bovine cartilage, human skin, porcine aorta, and human amniotic membrane17,21,25,26,27,28,29,30. The vast majority of these studies have revealed no or a very moderate influence of pressurized carbon dioxide on ECM structure and mechanical properties. However, while delipidation is crucial for the usefulness of the bio-scaffold, studies on ECM lipid composition before and after the treatment are lacking. Despite claims about the capacity for scCO2-cosolvents to remove lipids from biological tissue, only two of the studies mentioned above show results regarding lipid analysis. Sawada et al. (2008) showed a decrease in the content of total phospholipids in porcine aorta, quantified by enzymatic analysis30. Wehmeyer et al.25 showed no positive staining of membrane lipids in amniotic membrane, but the lipid alkyl chains were still present, according to results from differential scanning calorimetry.
Regarding DNA removal, studies remain controversial. Some authors claim successful results using scCO2 with the addition of ethanol or other modifiers (arguably forming a non-supercritical pressurized fluid)21,27,30. Other publications concluded that it was not possible with scCO2, even if aqueous or ethanol modifiers were used17,29. These discrepancies could result from differences between studies with respect to the origin, type, compactness, thickness and size of the tissue, as well as from differences in tissue pretreatment, composition and physical state of the fluid (supercritical vs non-supercritical pressurized fluids).
Notably, some of the published works claiming supercritical fluid decellularization are presumably not treating the sample with supercritical but rather one-phase pressurized fluids composed of CO2 and cosolvent. These fluids have higher polarizability than supercritical mixtures and better mass transfer properties than neat cosolvents31. They occur when excess of cosolvent is added to the scCO2, and specific pressure and temperature conditions are used. The physical state of the solvent mixture is crucial for the outcome of any extraction process and may be of most importance for the decellularization process in particular. A more appropriate use of nomenclature is advisable in the quest of decreasing the controversy found in the literature.
In this study, we examine the efficiency of one-phase pressurized CO2-cosolvent fluids and neat supercritical CO2 in the decellularization of porcine pulmonary arteries, paying special attention to lipid removal. Several treatment conditions (15.0–30.0 MPa, 90–120 min, 37–40 °C) were explored using a design of experiments (DoE) approach. The amount and identity of the lipids removed from pulmonary artery by pressurized CO2-cosolvent fluids was studied by ultrahigh performance supercritical fluid chromatography (UHPSFC) coupled to quadrupole time-of-flight mass spectrometry (QTOF-MS/MS) followed by statistical analysis. Two different methods were used to evaluate the removal of DNA from the tissue, i.e. DNA quantification and staining of nuclei. Finally, the integrity of the ECM after pressurized treatment was evaluated in terms of tissue hydration, matrix morphology and mechanical properties.
Materials and Methods
Biological material
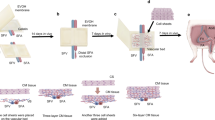
Lungs from outbred pigs were provided by the Thoracic Surgery research group (Lund University, Sweden). Pulmonary arteries (PA) were excised, rinsed in PBS and cut into pieces of 5 mm2 size. PA sections (n = 250 from two porcine lungs) were randomized and kept at −80 °C until use. Immediately before treatment, samples were de-frosted at room temperature and the tissue dried for three seconds using absorbing paper (Whatman N°4) (Fig. 1). All methods were performed in accordance with the relevant guidelines and regulations. The study was approved by the local research ethical committee at Lund University (2015–174).

Workflow for decellularization of porcine pulmonary arteries. Multiple linear regression for DoE (MLR) and principal component analysis (PCA) were used as statistical techniques for data analysis.
Reagents and standards
Ultrapure carbon dioxide (99.9993% purity) was provided by AGA GAS AB (Växjö, Sweden). Ethanol (95% v/v) and limonene (97% purity), used as cosolvents for the pressurized treatment, were purchased from Solveco (Rosersberg, Sweden) and Sigma- Aldrich (St. Louis, MO), respectively. Formaldehyde 4% aqueous solution buffer (VWR, Leuven, Belgium) was used as histological tissue fixative. Ultrapure water (18 Ω/cm) was dispensed by Milli-Q devices from Merck Millipore (Darmstadt Germany). Methanol (LC-MS grade) and dichloromethane (stabilized with 0.002% methyl-2-butene) were provided by VWR Chemicals (Fontenay-sous-bois, France). Ammonium formate (≥99% purity), and the lipid standards glyceryl tripalmitate (TG 16:0/16:0/16:0; ≥99% purity), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (PC 16:0/16:0; ≥99% purity), sphingomyelin from chicken egg yolk (SM 18:1/16:0; ≥98%) and stearic acid (≥98.5% purity) were purchased from Sigma-Aldrich. Internal standards, N-oleoyl(d9)-D-erythro-sphingosylphosphorylcholine (SM 18:1-d9; ≥99% purity), 1-pentadecanoyl-2-oleoyl(d7)-sn-glycero-3-phosphocholine (DPPC, PC 15:0–18:1-d7; ≥99% purity), 1-oleoyl(d7)-rac-glycerol (MG 18:1-d7; ≥99% purity), 5Z,8Z,11Z,14Z-eicosatetraenoic-16,16,17,17,18,18,19,19,20,20,20-d11 acid (arachidonic acid d11 ≥99% purity) and 1,3-dipentadecanoyl-2-oleyol(d7)-glycerol (TG 15:0–18:1-d7-15:0; ≥99% purity) were from Avanti Polar Lipids Inc. (Alabaster, AL).
For histological stainings, Mayer’s Hematoxylin and 0.2% Eosin Y solution were acquired from Histolab (Gothenburg, Sweden) and for staining of elastic fibers a Modified Verhoeff Van Gieson Elastic Stain Kit (HT25A) from Sigma-Aldrich was purchased. For decellularization of reference tissue sodium deoxycholate (SDC, ICN Biomedicals Inc., Aurora, OH, USA) and sodium dodecyl sulfate (SDS, >98% purity, VWR, Radnor, PA, USA) was used.
For the preparation of TEM samples, xylene was purchased from Histolab (Gothenburg, Sweden), acetone 99.8% for Analysis Emsure® ACS from MilliporeSigma (Billerica, MA), Poly/Bed® 812 Embedding Media from Polysciences, Inc. (Warrington, PA), 4% Uranyl acetate from Agar Scientific (Stansted Essex, England, UK) and lead citrate from Merck (Darmstadt, Germany).
Supercritical and pressurized carbon dioxide mediated decellularization
Tissue pieces were individually loaded in a sample holder specially designed by Prototypverkstaden (Lund, Sweden) (Fig. 2). Samples were then treated using an in-house constructed equipment, as previously reported32. Briefly, the sample holder was placed in a stainless-steel extraction vessel with a volume of 80 mL (Ångström Laboratory, Uppsala University, Uppsala, Sweden), mounted on top of a magnetic stirrer (VWR, Leuven, Belgium) inside a GC-oven (HP 5890 GC, Hewlett-Packard Co., Palo Alto, CA). A thermocouple was used to control the temperature inside the vessel and a high-pressure syringe pump (Isco 260D, Teledyne Technologies Inc., NE) delivered liquid carbon dioxide from a dip-tube CO2 cylinder to the bottom of the vessel. Depressurization of the vessel was performed using an upper valve, which exclusively released CO2, and a lower valve, which released cosolvent. For safety reasons, a burst disk (maximum pressure of 40 MPa, Zook Enterprises, Sheffield, UK) was placed between the vessel and the upper CO2 vent valve.

Details of the sample holder specially design for decellularization of pulmonary arteries. Ten pieces of tissue were loaded per decellularization condition.
The extraction vessel was preheated, followed by addition of 15 mL of ethanol:water (95:5, v/v) or limonene at the bottom of the vessel, when cosolvent was used. The water (5% in ethanol) was introduced to avoid tissue dehydration to the maximum possible extent, and it also helps to increase the polarizability of the mixture. Then, the vessel was closed, tubing connected, and oxygen purged by pumping CO2 for 1 min to avoid oxidation of the substrate. Subsequently, the system was pressurized to yield the desired pressure. During the decellularization process, the pump was set to maintain a constant pressure to compensate for undetectable leaks. After depressurization, cosolvent remaining in the vessel was removed by passing a constant flow of scCO2 rinse (8.5 MPa, 37 °C, 8 mL/min, 10 min). Samples destined for histological evaluation (n = 3) were immersed in 5 mL of 10% formalin for 12 h, rinsed with 10 mL of 70% EtOH for 30 min (x3) and then kept in 10 mL of 70% EtOH at 4 °C until examination (Fig. 1). Samples to be used for lipid analyses were freeze-dried and kept at −80 °C until extraction.
Two full-factorial design, one for each cosolvent, with three center points, were created in MODDE 10.1 (Sartorius Stedim Biotech, Malmö, Sweden) to investigate the impact of pressure (15.0–30.0 MPa), time (90–120 min) and temperature (35–40 °C) on tissue decellularization (Table 1). Higher pressures are obtained by adding more CO2 to the mixture, thus modifying the composition of the mixture. To simplify the DoE, we have investigated pressure as a variable while we have given composition a fictitious constant value, reflected as the average molar fraction.
Total amount of remaining lipids per lipid class (μg of lipid class per mg of freeze-dried tissue) and remaining DNA were used as response variables to estimate the delipidation and DNA removal efficacy, respectively. PA pieces were subjected to neat scCO2 treatment for 120 min at 30.0 MPa and 40 °C.
Enzymatic DNA removal
Tissue samples subjected to neat scCO2 and pressurized CO2-cosolvent fluids were allowed to equilibrate in enzyme buffer (20 mM tris(hydroxymethyl)aminomethane, 20 mM NaCl, 2 mM MgCl2) for 30 min, followed by treatment with 90 U/mL of benzonase endonuclease for 30 min at 37 °C, 1 mL per 5 mm2 sample. Samples were subsequently washed three times in PBS during a total of 44 h, followed by fixation in formalin or quantification of residual DNA.
Evaluation of nuclei removal
The nuclei remaining in the specimen after treatment were estimated by hematoxylin and eosin (H&E) staining and quantification of residual DNA.
Hematoxylin and eosin staining of treated pulmonary arteries from pigs
Formalin fixed samples were dehydrated, embedded in paraffin and sectioned with a thickness of 4 µm. Sections corresponding to the central portion of the sample were selected for H&E staining. Untreated tissue as well as tissue submitted to neat scCO2 were used as references.
DNA quantification
Residual double stranded DNA (dsDNA) were quantified by fluorescent nuclei acid staining using the Quant-iTTM PicoGreenTM dsDNA Assay Kit (Molecular Probes, Inc., Eugene, OR). Samples were lyophilized and homogenized using a Omni Tissue Homogenizer (Omni, Kennesaw, GA) followed by incubation with 200 U/mL of Proteinase K (Sigma-Aldrich) for 16 h at 37 °C. Samples were then centrifuged at 2000 × g for 10 min and dsDNA quantified in the supernatant (ng dsDNA/initial wet weight of the sample) according to manufacturer’s instructions.
Fresh tissue submitted to detergent based decellularization33 were used as positive controls. Briefly, pieces of PA were treated with a combination of 0.5% sodium deoxycholate (SDC) and 0.5% sodium dodecyl sulfate (SDS) for 24 h at room temperature with constant shaking. Samples were subsequently washed three times in phosphate buffered saline (PBS) for a total of 42 h, followed by fixation in formalin or quantification of residual DNA.
Extraction of lipid residues from treated tissues
Lipids were recovered by a dichloromethane/methanol/water-based extraction method adapted for porcine pulmonary arteries34. Briefly, freeze-dried tissue pieces (see Supplementary Fig. S1) (in randomly assembled batches of 20 samples) were disrupted in a Qiagen TissueLyser (Qiagen GmbH, Hilden, Germany) for 10 min (1 min per cycle) at 25 Hz, followed by extraction of lipids as previously described in detail34. Extracts were dried under a stream of nitrogen gas, weighted for gravimetric analysis and stored at −80 °C until analysis.
Lipid analysis
Lipid fractions (n = 4 per condition) were dissolved in 50 µL of CHCl3:MeOH (1:1, v/v) and analyzed on an Acquity Ultra Performance Convergence Chromatography (UPC2, Waters, MA, USA) system equipped with an Acquity UPC2 Torus DIOL column (130 Å, 1.7 µm, 3 mm ×100 mm, Waters, MA, USA) and fitted with a Van Guard Torus DIOL (130 Å, 1.7 µm, 2.1 mm ×5 mm, Waters, MA, USA) pre-column. Chromatography was performed as previously described in detail34, with a few modifications. The cosolvent (methanol containing 10 mM of ammonium formate) gradient was: 0 min, 2%; 2 min, 2%; 4 min 13%; 7 min, 27%; 7.5 min, 35%; 8.5 min, 35%; 9 min 2%; 11 min 2%. The flow rate was 1.6 mL/min.
Mass spectrometric detection was performed using a Xevo-2G quadrupole time-of-flight mass spectrometer (QTOF-MS; Waters, MA, USA). Make-up solvent (10 mM ammonium formate in methanol) was supplied at 0.25 mL/min and back-pressure regulated using two T-pieces, placed between the chromatographic system and the mass spectrometer (split ratio about 1:100)34,35. The capillary voltage was set at 3.0 kV and 2.5 kV for positive and negative electrospray ionization mode, respectively. The sampling cone voltage was set at 40 V, the cone gas flow rate at 100 L/h and the drying gas flow rate at 800 L/h, with a source and drying temperature of 120 °C and 200 °C respectively. The mass spectrometer was operated in MSE mode with a scanning range of m/z 150−1000, with a resolution of at least 20000 at m/z 500–900 using leucine-enkephalin (1500 ng/mL at 5 µL/min) for internal calibration. Data were processed in MassLynx v4.1(Waters, MA, USA) and Mzmine236.
Lipids were identified by exact mass using LipidMaps® Lipidomics gateway (San Diego, CA) and published data34,37 retention times and by fragments.
Absolute quantification was performed using an external calibration curve. Calibrant mixtures were composed by tripalmitin, DPPC, sphingomyelin and stearic acid in CHCl3:MeOH (1:1, v/v) with concentrations ranging from 10 to 50 ppm. Samples, blanks and calibration mixtures (50 µL) were spiked with 17 µL of the internal standard (IS) stock solution, which was composed of a mix of TG 15:0–18:1-d7-15:0, MG 18:1-d7, PC 15:0–18:1-d7, SM 18:1-d9 and arachidonic-d11 acid at 200 ppm in CHCl3:MeOH (1:1, v/v). The final concentration of each IS in samples and calibrant mixtures was 50.7 ppm.
Limit of detection (LOD) and limit of quantification (LOQ) were defined as three and ten times the signal-to-noise-ratio (S/N), respectively, for the analyte/internal standard-ratio and were calculated separately per lipid species (n = 3 per lipid species). Lipid species with levels below their respective LOD were considered absent and those with levels below LOQ were not quantified and hence not used for calculation of total amounts of lipid within the assessed lipid classes. A weighted calibration curve (1/Y) was used for TG and FAs, due to significant heteroscedasticity of the data38.
All analyses were conducted in a single randomized batch with alternation between positive and negative electrospray ionization. Calibration standards (n = 3) were analyzed prior to the first sample in the sequence and 8 blanks were evenly distributed in the batch.
Investigation of extracellular matrix integrity
The evaluation of the ECM integrity after treatment was approached by (1) the degree of tissue dehydration expressed as water retention (%; mg of retained water per 100 mg of water content in fresh tissue), (2) staining of elastin and collagen fibers of treated tissue, using the Modified Verhoeff Van Gieson Elastic Staining39, (3) transmission electron microscopy and (4) length-tension studies.
Transmission electron microscopy (TEM)
Biopsies (2 mm sections) were taken out from four different paraffin blocks containing samples of native, detergent treated, pressurized CO2-EtOH-H2O treated or pressurized CO2-limonene treated. The samples had previously been sectioned for H&E staining and all had been treated with benzonase nuclease, except for the native control. The biopsy was dewaxed in xylene, washed in ethanol, stained with 0.05% methylene blue in ethanol, rinsed in ethanol, acetone, followed by 1:1 mixture of Polybed-acetone and finally embedded in pure Polybed 812. The polymerised block was sectioned with a Leica UC7 ultramicrotome (Leica Microsystems GmbH, Wetzlar, Germany) and sections were mounted on a pioloform coated copper Maxtaform H5 grid. The section was contrasted with 4% Uranyl acetate followed with 1% lead citrate. Images of the samples were analysed in a Tecnai BioTWIN transmission electron microscope (FEI Company, OR, USA) at two different magnifications.
Wire myography experiments for length-tension studies
Length-tension studies were carried out using wire myography. The samples studied were 2 mm long segments of porcine pulmonary artery either non-treated (native) or previously submitted to pressurized CO2-limonene, pressurized CO2-EtOH-H2O and detergent. The pressurized treatment corresponded to the central points in the experimental design. The samples were tied to pins in three Myograph Systems (610 M and 620 M from Danish Myotechnology a/s, Aarhus, Denmark)40 using silk thread (6–0). The temperature inside the myograph chambers was maintained at 37 °C. Zero basal tension was ascertained in Hank’s Balanced Salt Solution (HBSS, Sigma-Aldrich). Following equilibration, preparations were stretched in pre-specified steps and force was measured after 3 min. This procedure was repeated 10 times to obtain length-tension curves for each individual sample. Length and dry weight of each sample was used to obtain an approximation of the cross-sectional area that was used to normalize force.
Statistical and chemometric analysis
The full factorial designs were evaluated using multi-linear regression in MODDE 10.1 (Sartorius Stedim Biotech). Principal component analysis (PCA) was performed in SIMCA-P 12.0.1 (Sartorius Stedim Biotech). Differences between groups were evaluated by analysis of variance (ANOVA), with Tukeys test post hoc, and precision was estimated using an F-test. Significance was defined as p < 0.05. In this work, the applicability of scCO2 and pressurized CO2-cosolvent fluids to decellularize porcine pulmonary arteries was studied.
Results and Discussion
Bio-scaffolds have been produced from animals and used for transplantations in humans11. A prerequisite for such an approach is that all genetic material and lipids can be removed, while maintaining the integrity of the ECM fibers17,21,30. In this work, the applicability of scCO2 and pressurized CO2-organic solvent mixtures for decellularization of porcine pulmonary arteries was studied.
Lipid species in pulmonary arteries
First, we examined which lipid classes were most abundant in untreated tissue using UHPLC/QTOF-MS. In agreement with previous studies, we found high levels of several lipid species within triacylglycerides (TGs; [M+NH4]+), phosphatidylcholines (PCs; [M+H]+), sphingomyelins (SMs; [M+H]+), and fatty acids (FAs; [M − H]−)34, and relatively low levels of cholesteryl ester (CEs; [M+Na]+), monoacylglycerides (MGs; [M+H]+), phosphatidylethanolamines (PEs; [M+H]+) (Supplementary Table S1). TG 52:1, TG 52:2, PC 34:1, PC 38:4, SM 34:1, FA 16:0, FA 18:0 and FA 18:1 (Supplementary Fig. S2) were the most abundant lipid species, which is in line with previously published results34. The total content of TG, PC, SM and FA was consequently used to quantify residual lipids in treated tissue.
Delipidation of pulmonary arteries by pressurized CO2
Next, we examined the effects of temperature, time and pressure on tissue delipidation, using a design of experiments (DoE) approach. However, we could not detect any significant effects of these parameters on any of the response variables with either of the solvent combinations. Hence, as TG, PC, SM, FA, and total extractable lipid residues did not differ between conditions, we pooled data for the different solvent combinations to gain power in our further analyses.
Subsequently, we compared total lipid residues in treated and untreated tissue, as determined by gravimetric analysis. Pressurized CO2-EtOH-H2O treatment efficiently reduced tissue lipid levels (p < 0.01) (see Supplementary Fig. S3). Unexpectedly, samples submitted to both scCO2 and pressurized CO2-limonene showed higher levels of residual lipids than untreated samples (p < 0.01) (Supplementary Fig. S3). Since no limonene was detected during mass spectrometric analysis (the intensity of limonene adducts were below respective spectrometry base lines), these results are not due to residual limonene being extracted in the dichloromethane fraction. An interpretation is that the treatment with scCO2 and pressurized CO2-limonene did not lead to a significant removal of lipids but it improved accessibility of lipids in subsequent extraction by dichloromethane.
To generate a more comprehensive depiction of the delipidation process, we studied in detail the lipid profiles generated by mass spectrometric analysis. TGs were partially reduced for all treatments and were found at detectable levels in all samples. All PCs and SMs were also found at detectable levels in all samples, except for PC 34:0 and PC 32:1 which were undetectable after pressurized CO2-EtOH-H2O treatment, and SM 41:1 which was absent in most samples treated with this solvent combination (Fig. 3a). Overall, pressurized CO2-EtOH-H2O treatment was found to be the most efficient solvent combination for delipidation, resulting in a majority of TG, PC and SM lipid species to be reduced to levels below their respective LOQs. Therefore, concentrations of no quantifiable lipids species were ranged from 0.027, 0.919 and 1.253 ppm (i.e. TG 52:0, PC(O-34:1)/PC(P-34:0), SM 40:0) to 0.101, 4.088 and 2.381 ppm (i.e. TG 54:3, PC 32:1, SM 34:1) for TGs, PCs and SMs respectively.

Maps of lipid species presence in untreated tissue (fresh) and samples subjected to neat scCO2, pressurized CO2-EtOH-H2O and pressurized CO2-limonene treatment for (a) data generated in positive electrospray ionization and (b) negative electrospray ionization modes. Columns represent samples clustered by treatment (at different temperatures, pressures and times) and lines correspond to lipid species sorted by class, from non-polar to polar lipids, as shown in Supplementary Table S1. Pink boxes indicate that the respective lipid specie was reduced to levels below its respective LOD (upper panel) and LOQ (lower panel). Boxes from lipids present in enough amount to be detected and/or quantified are not colored. *Outliers identified by PCA.
Pressurized CO2-limonene, on the other hand, mainly reduced levels of TG 48:2 (Fig. 3a).
Pressurized CO2-EtOH-H2O was also found to be the most efficient solvent combination to reduce levels of FAs (Fig. 3b). FAs 18:1, 22:3, 22:5 and 22:6 were all absent. In addition, all long- and very long-chained FAs (C > 18), regardless their degree of unsaturation, showed levels below their respective LOQs (Fig. 3b, Supplementary Table S1).
Hence, the choice of cosolvent exerted a much more dramatic influence on PA delipidation, as compared to pressure, temperature and extraction time (Fig. 3). The extensive delipidation observed for pressurized CO2-EtOH-H2O, compared to scCO2 and pressurized CO2-limonene, are in line with results obtained for porcine retina32. The relative permittivity of CO2 is low, which makes it appropriate to dissolve mostly non-polar compounds of low molecular weight41. The static relative permittivity, and therefore the polarizability, of scCO2 can be increased with the addition of cosolvents. As an example, supercritical CO2-cosolvent fluids have been effective in the extraction of edible lipids like triacylglycerols and fatty acids42. However, there is no evidence that the addition of such small amounts of cosolvent is enough to dissolve more polar lipids, like the ones known to be present in pulmonary artery34. As an alternative, higher amounts of cosolvents than what is soluble in supercritical CO2 can be used. Under controlled conditions of pressure and temperature, this leads to a one-phase pressurized CO2-cosolvent fluid (see Supplementary Fig. S4), however not in the supercritical regime. Such fluids are rarely studied31, but they offer an even higher range of polarizability than supercritical mixtures, expanding the type of extractable lipids32. Mass transfer properties of such pressurized fluids are also modified with respect to the neat cosolvent, in favor of better extraction power. Furthermore, a one-phase pressurized CO2-cosolvent fluid prevents direct contact between the tissue and the liquid organic solvent, which would otherwise cause cytotoxic effects. By selecting ethanol as cosolvent in the pressurized fluid, we are increasing the amount of hydrogen-bonding interactions that can be created between solvent and solute. This results in higher solubilization of the polar lipids present in pulmonary arteries. In contrast to ethanol, limonene is not a polar molecule, but it is generally used to dissolve lipids in industrial applications. In the case of limonene as cosolvent, dispersion forces become stronger than for neat CO2, but these intermolecular interactions are still not strong enough to achieve full lipid removal of the less polar lipids (i.e. TGs) nor to dissolve the most polar lipid classes.
A PCA was calculated to visualize the impact of treatments on lipid profiles (described variation, R2 = 0.83; predictive ability Q2 = 0.76) (Fig. 4). The score scatter plot (Fig. 4a) revealed a clear separation between the untreated and treated PA along principal component (PC) 2, and a separation of CO2-EtOH-H2O treated PA from PA treated with CO2-limonene or neat scCO2 along PC1. The loading plot (Fig. 4b), revealed that differences in TG levels did not affect clustering as much as the other species, possibly because all treatments were partially effective at removing TGs from the tissue. The biggest impact on the clustering was due to the more polar lipids, with the biggest difference most clearly observed for PCs and SMs. CO2-EtOH-H2O was more efficient than the other conditions in extracting polar lipids from the tissue, which corresponds well with its differentiated position along PC1 in Fig. 4a. These results are in agreement with the discussion above, based on the polarizability of the solvent mixtures and the type of intermolecular interactions present.

Principal component analysis calculated for lipid data from untreated, scCO2, pressurized CO2-EtOH-H2O and pressurized CO2-limonene treated porcine pulmonary arteries. (a) Cross-validated score-scatter plot colored by treatment. The number 1 next to the data point indicates samples selected for tissue staining. (b) Loading plot showing how levels of lipid species impact on the clustering observed in the score-scatter plot. Circles indicate the average ± standard deviation of the different lipid classes.
In line with the results from the PCA, univariate analysis of the lipid data revealed pressurized CO2-EtOH-H2O to be the most efficient of the tested methods for PA delipidation (Fig. 5a–c). Levels of TGs were reduced by all delipidation methods (p < 0.01), with CO2-EtOH-H2O treatment decreasing TG by 93.2% (p < 0.01) (Fig. 5a). This reduction was more significant than the reduction observed with CO2-limonene (81.0%). The correspondence between TG removal rates observed for scCO2 and pressurized CO2-limonene is likely governed by the similar low polarity of these fluids. Levels of PCs and SMs differed more dramatically between treatment conditions and were mainly reduced after pressurized CO2-EtOH-H2O treatment (97.8% and 94.6%, respectively; p < 0.01). Unexpectedly, the total FA content was found to increase after treatment with neat scCO2 (p < 0.01) and pressurized CO2-limonene (p < 0.05) (Fig. 5d), in line with the unexpected results from the gravimetric analysis. Similar trends have been observed for other tissue treated with pressurized CO2-based fluids32. The reason for this observation may be a result of the pressurized CO2 (at both supercritical or pressurized state) impacting on FA availability and facilitating subsequent extraction with dichloromethane. This effect is also observed for saturated FAs in samples treated with pressurized CO2-EtOH-H2O. The fact that we can appreciate this effect is due to that saturated FAs were not removed by the pressurized treatment to the same extent as other FAs (Fig. 6).

Representation of total (a) TG, (b) PC, (c) SM and (d) FA content (μg lipid/mg dried tissue) of untreated tissue and tissue subjected to neat scCO2, pressurized CO2-EtOH-H2O and pressurized CO2-limonene treatment. Data are presented as mean ± standard deviation for n = 5 (untreated and neat scCO2) and n = 42 (CO2 with cosolvent). Data were compared using ANOVA with Tukey’s test post hoc. *p < 0.05 and **p < 0.01.

Presence of fatty acids (%) in tissues after treatment. ARA, arachidonic acid. Untreated samples expressed as 100%. * And ** denote significant differences, p < 0.05 and p < 0.01 respectively, compared with their respective fatty acids group of the untreated samples. # And ## denote significant differences, p < 0.05 and p < 0.01 respectively, compared with their respective fatty acids group of the pressurized CO2-EtOH-H2O samples.
Pressurized CO2-EtOH-H2O was found to more efficiently remove fatty acids, as compared to neat scCO2 and pressurized CO2-limonene, presumably due to the possibility to form hydrogen bondings with the carboxylic acid moieties and OH…π−bonds with carbon-carbon double bonds43. The type of FA that remains in the tissue may affect the success of a subsequent recellularization. Notably, monounsaturated fatty acids (MUFAs) and polyunsaturated fatty acids (PUFAs), the most polar FA subclasses, were more efficiently removed by pressurized CO2-EtOH-H2O, as compared with the other treatments (Fig. 6). Levels of saturated FAs were more similar between conditions. Details on individual lipid species can be found in Supplementary Fig. S5. These are very promising results, considering that unsaturated FAs have been reported to impair proper cell adhesion, whereas the saturated FA stearic acid (FA 18:0) has been shown to induce cell adhesion14.
Notably, total levels of the pro-inflammatory fatty acid arachidonic acid (FA 20:4)15 and arachidonic acid containing lipids such as TG 54:4, TG 54:5, PC 38:4 and PC 38:544,45, were reduced by 48% after treatment of the tissue with pressurized CO2-EtOH-H2O, as compared to untreated samples. ScCO2 treatment resulted in a less effective removal of pro-inflammatory lipids reduction (13.9%) followed by pressurized CO2-limonene (30.9%). Whether the reduction in arachidonic acid (FA 20:4) observed after pressurized CO2-EtOH-H2O treatment can lower proinflammatory responses after transplantation remain to be examined.
Removal of DNA from pulmonary arteries by pressurized CO2
Following delipidation studies, we set out to examine whether any of the treatments also removed nuclei from the tissue, as has previously been suggested30, and questioned17 for porcine aorta. We did not find any evidence of DNA removal, assessed by both dsDNA quantification and H&E staining, using any of the pressurized CO2 conditions tested. In the case of scCO2, these results were expected due to the high polarity and molecular weight of DNA. In the case of CO2-cosolvent mixtures, these results are in agreement with the work by Casali et al.17 but contradict that of Sawada et al.30. The former used a mixture of CO2-cosolvents in the supercritical state, which is not comparable to one-phase pressurized CO2-cosolvent fluids. The latter is more comparable to the fluid conditions used in this work, although their mixture contains higher amounts of ethanol. It is not clear at this point if this small difference in composition explains the discrepancies in results.
Considering the unmatched Hanssen Solubility Parameter (HSP values ([δD, δP, δH]) of [19.8, 20.1, 11.2]46 and [15.4, 8.7, 18.8]47 estimated respectively for intact DNA and a CO2-EtOH mixture of XCO2 = 0.31 at 40 °C and 9.3 MPa, it seems unlikely that one-phase pressurized CO2-ethanol fluids are able to dissolve DNA. It is likewise, in the case of limonene (HSP of [17.2, 1.8, 4.3] for neat limonene).
However, after nuclease treatment, one third of the pressurized CO2-limonene treated tissues showed a notable nuclei removal in H&E staining (see example in Fig. 7g), suggesting the absence of dsDNA. A reduction up to 93.4% of the dsDNA content compared with fresh tissues (for samples submitted to 15 MPa and 40 °C for 90 min) was achieved, resulting in an improvement of a 12.2% compared to the results from detergent-enzyme treatment. The effect of treating the sample with pressurized CO2-EtOH-H2O and neat scCO2 fluids prior to nuclei removal by detergent-enzyme treatment was moderate and null, respectively. There is no theory that can currently explain the promising effect of pressurized CO2-limonene fluid as facilitator of denaturalization of DNA by enzymes.

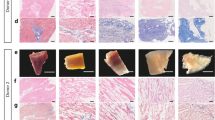
Hematoxylin and Eosin staining (a–h) and TEM images at two different magnifications (i-l and m-p, showing 2 μm and 500 nm scale bars respectively) of porcine pulmonary artery treated with pressurized CO2-EtOH-H2O and CO2-limonene. Representative images of (a,i,m) native artery, (b) artery treated with pressurized CO2-EtOH-H2O (30.0 MPa, 35 °C, 90 min), (c) artery treated with pressurized CO2-limonene (15.0 MPa, 40 °C, 120 min), (d) artery treated with a mixture of sodium deoxycholate (SDC) and Sodium dodecyl sulfate (SDS) and not subjected to pressurized CO2 mixtures, (e) artery subjected to neat scCO2 (30.0 MPa, 40 °C, 120 min) followed by endonuclease treatment, (f–h) same treatments as in b, c, and d with the addition of subsequent endonuclease treatment. (j–l) and (n–p) correspond to TEM images at low and high magnification, respectively, of the respective samples above, i.e. f, g and h. Collagen fibers in cross section (black arrowheads), longitudinally sectioned collagen fibers (white arrowheads) and cell nucleus (asterisk).
Assessment of extracellular matrix integrity
Tissue dehydration is an important parameter determining the suitability of the tissue as a bio-scaffold, and a low water content has been suggested to impair the mechanical properties of the tissue48. However, the minimum hydration grade to preserve the functionality of a certain tissue remains unknown.
All explored fluid combinations exerted a dehydration effect. Higher water retention was observed for pressurized CO2-limonene and pressurized CO2-EtOH-H2O treatments, i.e. 16.8% and 15.0% respectively, compared to neat scCO2 treatment (11.4%). These results suggest that the presence of limonene and EtOH-H2O prevent the tissue from suffering extreme dehydration, which is in line with a previous study17.
We also conducted a histological examination of overall tissue structure and the integrity of elastic fibers, using a limited set of samples as indicated in Fig. 4a. This examination did reveal an overall conserved morphology with parallel aligned continuous elastic fibers without any differences between treated and untreated tissue. The spacing between elastic fibers tended to be decreased after treatment, consistent with extraction of lipids and other cellular material. The change in spacing between fibers was more pronounced for tissue samples treated with detergents (Supplementary Fig. S6).
Tissue ultrastructure was evaluated in the same samples used in the histological evaluation by reprocessing paraffin embedded samples for TEM. Tissues were stained and imaged at up to 60000 times magnification. None of the pressurized treatments exerted a distinct effect on the ECM (Fig. 7i-p). Fibrils exhibited thickness and organization comparable to the native tissue regardless of treatment with pressurized CO2-EtOH-H2O, CO2-limonene or detergents. Collagen fiber organization appeared intact as far as can be judged by the density and parallel appearance of the collagen fibrils. These observations were supported by length-tension studies. Our results did not show any clear differences in length-tension relationships, and thus presumably stiffness, due to sample treatments or compared to the native samples (Supplementary Fig. S7).
Decellularization and tissue integrity remains key for the production of bio-scaffolds. However, detergent and ezyme-based decellularization does not completely remove all immunogenic reactions. Previous studies have shown that decellularized xeno-transplanted tissue may still provoke immunogenic responses such as antibody formation in patients that have received decellularized porcine valves49. The immunogenic response appears to be induced by the ECM protein collagen VI, albumin and αGal epitopes49,50. These findings indicate that the ECM itself possess immunogenicity after decellularization by detergents and enzymes. Overall, the results in this work revealed that pressurized CO2 fluids make an impact in the removal of lipids from pulmonary artery, and in the removal of DNA when added to the battery of decellularization strategies available. Further studies are needed to evaluate if treatment with pressurized CO2 fluids has also a positive effect on reducing the immunogenic properties of ECM.
Conclusion
Pressurized CO2-EtOH-H2O fluid was found to be the most efficient pressurized solvent combination to achieve lipid removal from pulmonary artery tissue. It led to the efficient removal of TGs, PCs, SMs and most FAs including pro-inflammatory lipids, and a less efficient removal of saturated FAs. Pressurized CO2-limonene showed a low delipidation efficiency, similar to neat scCO2. Further studies are needed to evaluate whether the close to complete delipidation achieved in this work results in low immune responses, cellular invasion and functional recellularization. DNA removal was more efficient with pressurized CO2-limonene after endonuclease treatment. Importantly, this, as well as the other tested treatments, preserved ECM integrity.
References
Benjamin, E. J. et al. Heart disease and stroke statistics-2017 update: a report from the american heart association. Circulation 135, e146–e603 (2017).
Williams, B. et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 39, 3021–3104 (2018).
Thenappan, T., Chan, S. Y. & Weir, E. K. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am. J. Physiol. Circ. Physiol. 315, H1322–H1331 (2018).
Sandoval Zarate, J. Hipertensión arterial pulmonar. Archivos de cardiología de México 76, 69–75 (2006).
Michelakis, E. Spatio-temporal diversity of apoptosis within the vascular wall in pulmonary arterial hypertension. Circ. Res. 98, 172–175 (2006).
World Health Organization & Spanish Transplant Organization. Summary of 2016 activity data. ONT-WHO Global observatory on donation and transplantation (2016).
Comission, E. Journalist Workshop on organ donation and transplantation. Recent facts and figures (2014).
El-Sherbiny, I. M. & Yacoub, M. H. Hydrogel scaffolds for tissue engineering: Progress and challenges. Glob. Cardiol. Sci. Pract. 2013, 316–342 (2013).
Pashneh-Tala, S., MacNeil, S. & Claeyssens, F. The tissue-engineered vascular graft-past, present, and future. Tissue Eng. Part B. Rev. 22, 68–100 (2016).
Frantz, C., Stewart, K. M. & Weaver, V. M. The extracellular matrix at a glance. J. Cell Sci. 123, 4195–4200 (2010).
Gilbert, T. W., Sellaro, T. L. & Badylak, S. F. Decellularization of tissues and organs. Biomaterials 27, 3675–3683 (2006).
Yu, S., Guo, Z., Johnson, C., Gu, G. & Wu, Q. Recent progress in synthetic and biological studies of GPI anchors and GPI-anchored proteins. Curr. Opin. Chem. Biol. 17, 1006–1013 (2013).
Seong, J., Wang, Y., Kinoshita, T. & Maeda, Y. Implications of lipid moiety in oligomerization and immunoreactivities of GPI-anchored proteins. J. Lipid Res. 54, 1077–1091 (2013).
Curtis, A. S., Chandler, C. & Picton, N. Cell surface lipids and adhesion. III. The effects on cell adhesion of changes in plasmalemmal lipids. J. Cell Sci. 18, 375 LP–384 (1975).
Mori, D. N., Kreisel, D., Fullerton, J. N., Gilroy, D. W. & Goldstein, D. R. Inflammatory triggers of acute rejection of organ allografts. Immunol. Rev. 258, 132–144 (2014).
Cissell, D. D., Hu, J. C., Griffiths, L. G. & Athanasiou, K. A. Antigen removal for the production of biomechanically functional, xenogeneic tissue grafts. J. Biomech. 47, 1987–1996 (2014).
Casali, D. M., Handleton, R. M., Shazly, T. & Matthews, M. A. A novel supercritical CO2-based decellularization method for maintaining scaffold hydration and mechanical properties. J. Supercrit. Fluids 131, 72–81 (2018).
Choi, J. S. et al. Decellularized extracellular matrix derived from human adipose tissue as a potential scaffold for allograft tissue engineering. J. Biomed. Mater. Res. Part A 97A, 292–299 (2011).
Liao, J., Joyce, E. M. & Sacks, M. S. Effects of decellularization on the mechanical and structural properties of the porcine aortic valve leaflet. Biomaterials 29, 1065–1074 (2008).
Petersen, T. H. et al. Tissue-engineered lungs for in vivo implantation. Science 329, 538–541 (2010).
Seo, Y., Jung, Y. & Kim, S. H. Decellularized heart ECM hydrogel using supercritical carbon dioxide for improved angiogenesis. Acta Biomater. 67, 2708–281 (2017).
Hwang, J. et al. Molecular assessment of collagen denaturation in decellularized tissues using a collagen hybridizing peptide. Acta Biomater. 53, 268–278 (2017).
Reing, J. E. et al. The effects of processing methods upon mechanical and biologic properties of porcine dermal extracellular matrix scaffolds. Biomaterials 31, 8626–8633 (2010).
Zvarova, B. et al. Residual detergent detection method for nondestructive cytocompatibility evaluation of decellularized whole lung scaffolds. Tissue Eng. Part C. Methods 22, 418–428 (2016).
Wehmeyer, J. L., Natesan, S. & Christy, R. J. Development of a sterile amniotic membrane tissue graft using supercritical carbon dioxide. Tissue Eng. Part C. Methods 21, 649–659 (2015).
Balestrini, J. L. et al. Sterilization of lung matrices by supercritical carbon dioxide. Tissue Eng. Part C. Methods 22, 260–269 (2016).
Hennessy, R. S. et al. Supercritical carbon dioxide–based sterilization of decellularized heart valves. JACC Basic to Transl. Sci. 2, 71–84 (2017).
Zambon, A. et al. Dry acellular oesophageal matrix prepared by supercritical carbon dioxide. J. Supercrit. Fluids 115, 33–41 (2016).
Antons, J. et al. Decellularised tissues obtained by a CO2-philic detergent and supercritical CO2. Eur. cells Mater. 36, 81–95 (2018).
Sawada, K., Terada, D., Yamaoka, T., Kitamura, S. & Fujisato, T. Cell removal with supercritical carbon dioxide for acellular artificial tissue. J. Chem. Technol. Biotechnol. 83, 943–949 (2008).
Cunico, L. P. & Turner, C. Chapter 7 supercritical fluids and gas-expanded liquids in The application of green solvents in separation processes (eds. Pena-Pereira, F. & Tobiszewski, M.) 155–214 (Elsevier, 2019).
Gil-Ramirez, A., Spangenberg, A., Spégel, P. & Rodriguez-Meizoso, I. Pressurized carbon dioxide combined with aqueous ethanol as cosolvent induces efficient delipidation of porcine retina for their use as bioscaffolds. J. CO2 Util. 34, 700–708 (2019).
Cebotari, S. et al. Detergent decellularization of heart valves for tissue engineering: toxicological effects of residual detergents on human endothelial cells. Artif. Organs 34, 206–210 (2010).
Gil-Ramirez, A. et al. Efficient methodology for the extraction and analysis of lipids from porcine pulmonary artery by supercritical fluid chromatography coupled to mass spectrometry. J. Chromatogr. A 1592 (2019).
Jumaah, F. et al. A rapid method for the separation of vitamin D and its metabolites by ultra-high performance supercritical fluid chromatography–mass spectrometry. J. Chromatogr. A 1440, 191–200 (2016).
Pluskal, T., Castillo, S., Villar-Briones, A. & Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395 (2010).
Lísa, M. & Holčapek, M. High-throughput and comprehensive lipidomic analysis using ultrahigh-performance supercritical fluid chromatography–mass spectrometry. Anal. Chem. 87, 7187–7195 (2015).
da Silva, C. P., Emídio, E. S. & de Marchi, M. R. R. Method validation using weighted linear regression models for quantification of UV filters in water samples. Talanta 131, 221–227 (2015).
Kazlouskaya, V. et al. The utility of elastic Verhoeff-Van Gieson staining in dermatopathology. J. Cutan. Pathol. 40, 211–225 (2013).
Rippe, C. et al. Hypertension reduces soluble guanylyl cyclase expressionin the mouse airte via the Notch signaling pathway. Sci. Rep. 7, 1334, https://doi.org/10.1038/s41598-017-01392-1 (2017).
Leeke, G. et al. Solubilities of 4-phenyltoluene, phenylboric acid, biphenyl, and iodobenzene in carbon dioxide from measurements of the relative permittivity. J. Chem. Eng. Data 50, 1370–1374 (2005).
Martínez, J. & de Aguiar, A. Cde Extraction of triacylglycerols and fatty acids using supercritical fluids - Review. Current Analytical Chemistry 10, 67–77 (2014).
Heger, M., Mata, R. A. & Suhm, M. A. Soft hydrogen bonds to alkenes: the methanol–ethene prototype under experimental and theoretical scrutiny. Chem. Sci. 6, 3738–3745 (2015).
Denning, G. M., Figard, P. H., Kaduce, T. L. & Spector, A. A. Role of triglycerides in endothelial cell arachidonic acid metabolism. J. Lipid Res. 24, 993–1001 (1983).
Nelson, J. R. & Raskin, S. The eicosapentaenoic acid:arachidonic acid ratio and its clinical utility in cardiovascular disease. Postgrad. Med. 131, 268–277 (2019).
Hansen, C. Hansen Solubility Parameters- A user´s handbook. (CRC Press, Inc, 2007).
Al-Hamimi, S., Abellan Mayoral, A., Cunico, L. P. & Turner, C. Carbon dioxide expanded ethanol extraction: solubility and extraction kinetics of α-pinene and cis-verbenol. Anal. Chem. 88, 4336–4345 (2016).
Nicolle, S. & Palierne, J.-F. Dehydration effect on the mechanical behaviour of biological soft tissues: Observations on kidney tissues. J. Mech. Behav. Biomed. Mater. 3, 630–635 (2010).
Böer, U. et al. Antibody formation towards porcine tissue in patients implanted with crosslinked heart valves is directed to antigenic tissue proteins and alpha Gal epitopes and is reduced in healthy vegetarian subjects. Xenotransplantation. 24, 12288, https://doi.org/10.1111/xen.12288 (2017).
Boeer, U. et al. Immunogenicity of intensively decellularized equine carotid arteries is conferred by the extracellular matrix protein collagen type VI. Plos One. 9, 105964, https://doi.org/10.1371/journal.pone.0105964 (2014).
Acknowledgements
All members of the research team would like to thank Igelösa Life Science AB (Igelösa, Lund, Sweden) for supplying the porcine lungs. We would like to thank Lina Gefors at Lund University Bioimaging Centre (LBIC) for technical help with the TEM analysis. This work has received funding from European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Fellowship Grant Agreement No. H2020-MSCA-IF-2016-746137. Open access funding provided by Lund University.
Author information
Authors and Affiliations
Contributions
I.R.M. conceived the project. The text was written by A.G.R., P.S. and I.R.M., with feedback from all authors. I.R.M. and A.G.R. designed the work, with contributions from all authors. A.G.R. conducted the lipid analysis with input from P.S. and I.R.M. O.R. conducted the DNA removal, histology and matrix integrity analysis with input from A.K.L.C. and G.W.T. K.S., K.S, A.K.L.C and O.R. conducted and analyzed the length-tension studies.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gil-Ramírez, A., Rosmark, O., Spégel, P. et al. Pressurized carbon dioxide as a potential tool for decellularization of pulmonary arteries for transplant purposes. Sci Rep 10, 4031 (2020). https://doi.org/10.1038/s41598-020-60827-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-60827-4
This article is cited by
-
An overview of the production of tissue extracellular matrix and decellularization process
Cell and Tissue Banking (2024)
-
Biofabrication, biochemical profiling, and in vitro applications of salivary gland decellularized matrices via magnetic bioassembly platforms
Cell and Tissue Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
