
Abstract
In Tanzania, chloroquine was replaced by sulphadoxine- pyrimethamine (SP) as a first-line for treatment of uncomplicated malaria. Due to high resistance in malaria parasites, SP lasted for only 5 years and by the end of 2006 it was replaced with the current artemisinin combination therapy. We therefore, set a study to determine the current genotypic mutations associated with Plasmodium falciparum resistance to artemisinin, partner drugs and chloroquine. Parasites DNA were extracted from dried blood spots collected by finger-prick from Tanzanian malaria infected patients. DNA were sequenced using MiSeq then genotypes were translated into drug resistance haplotypes at Wellcome Sanger Institute, UK. About 422 samples were successful sequenced for K13 gene (marker for artemisinin resistance), the wild type (WT) was found in 391 samples (92.7%) whereby 31 samples (7.3%) had mutations in K13 gene. Of 31 samples with mutations, one sample had R561H, a mutation that has been associated with delayed parasite clearance in Southeast Asia, another sample had A578S, a mutation not associated with artemisinin whilst 29 samples had K13 novel mutations. There were no mutations in PGB, EXO, P23_BP and PfMDR1 at position 86 and 1246 (markers for resistance in artemisinin partner drugs) but 270 samples (60.4%) had mutations at PfMDR1 Y184F. Additionally, genotyped PfCRT at positions 72–76 (major predictors for chroquine resistance), found WT gene in 443 out of 444 samples (99.8%). In conclusion, this study found mutations in K13-propeller gene and high prevalence of chloroquine susceptible P. falciparum in Southeast of Tanzania.
Similar content being viewed by others


Introduction
Artemisinin-based combination therapy (ACT) is recommended by World Health Organization (WHO) to its partner states1,2 as the first and second-line treatment for uncomplicated Plasmodium falciparum malaria as well as chloroquine-resistant Plasmodium vivax3. In Tanzania chloroquine (CQ) was replaced by sulphadoxine- pyrimethamine (SP) as first-line treatment and amodiaquine as second-line for uncomplicated malaria, due to high resistance SP lasted for only five years and by the end of 2006 it was replaced with the current ACT4. Reversibly, an extended use of artemisinin (ART)-based combination therapy in malaria control and elimination programs has resulted to an emergence of P. falciparum resistant to ART derivatives in Southeast Asia5. The risk of ART-resistant parasites reported to spread from western Cambodia to the Greater Mekong Subregion and to Africa6. This is an urgent concern for global health7. The spread of resistant P. falciparum to previous first-line treatment of malaria (chloroquine and sulfadoxine-pyrimethamine) in nearly all endemic countries originated in almost similar fashion8.
The discovery of mutations in the propeller domain of the kelch (K13) gene were marked as candidate molecular markers and has remained to be the key predictor of ART resistance9. Several mutations at K13 are therefore associated with ART resistance10, i.e. K13 M476I mutation was first investigated in Tanzanian F32 parasites that were exposed in vitro to escalating concentrations of ART for more than 5 years11,12. In addition to that genomic analysis of Cambodian isolates identified four prevalent K13 mutations (Y493H, R539T, I543T and C580Y) that were associated with ART resistance12,13. In this regard, the list of K13 has kept on increasing and regularly updated by World Health Organization (WHO)3. Additionally, evidence from research reported parasite genetic background (PGB), is the mutations that allowed the emergence of K13 mutations, these mutations include; V127M and D128Y/H in the PfARPS10 (PF3D7_1460900) protein, D193Y in ferredoxin (PfFD, PF3D7_1318100), N326S and I356T in PfCRT (PF3D7_0709000), and T484I in PfMDR2 (PF3D7_1447900) and they are expressed as concatenated haplotype form (VDDNIT) as a reference allele (wild type)14.
Moreover, P. falciparum multi-drug resistance gene 1 (PfMDR1, PF3D7_0523000) and particularly, single nucleotide polymorphisms (SNPs) resulting in an amino acid change in codons 86 (N86Y), 184 (Y184F), and 1246 (D1246Y) have been associated with changes in parasite susceptibility to various drugs, including ACT. Mutations at position 86 and 1246 have been associated with parasite resistance to CQ and amodiaquine15 while mutations at positions 86, 184, and 1246 increase susceptibility of mefloquine and lumefantrine16. Genome‐wide association study (GWAS), a single nucleotide polymorphism (SNP) in a putative exonuclease gene (PfEXO, Pf3D7_1362500) was associated with an increased tolerance of piperaquine.
On the other hand, gene amplification of a section of chromosome 14 involving the genes plasmepsin 2 and plasmepsin 3, P2/3 breakpoint (P23_BP) has been associated with an increased resistance to piperaquine17. Additionally, CQ resistance transporter (PfCRT) and PfMDR1 both located on the food vacuole of the parasite involved in CQ resistance18. However, the CQ transporter PfCRT is a stronger predictor of CQ resistance than PfMDR119. Nevertheless, in areas where usage has been strictly regulated, withdrawal of CQ have resulted in dramatic decreases in the prevalence of CQ-resistant parasites4,20,21.
Therefore, it was necessary to conduct a molecular surveillance of gene mutations associated with P. falciparum resistance to artemisinin, partner drugs and chloroquine, one decade since ART-based combination therapy was introduced as a first line treatment and 18 years after CQ withdrawal for treatment of uncomplicated malaria in Tanzania4.
Results
kelch13 (K13) propeller polymorphisms
A total of 489 samples were genotyped but 67 (13.7%) could not be detected/missing genotypes. Of 422 successful sequenced samples, wild type (WT) K13 gene was found in 391 samples (92.7%) whereby 31 samples (7.3%) had mutations in K13 gene. Of those mutations, one sample had R561H, a mutation that has been associated with delayed parasite clearance, another sample had A578S, a mutation not associated with clinical or in vitro resistance to artemisinin whilst 24 nonsynonymous mutations are not yet listed (uncharacterized) in WHO artemisinin resistance report of 2018 (Table 1).
Prevalence PGB, EXO/P23_BP and PfMDR1 mutations
Generally, 489 samples were genotyped but the total number per every marker differed based on the number genotypes could be detected. All genotypes for PGB and EXO/P23_BP, markers for ART and piperaquine resistance respectively were WT. There were 270 samples (60%) with mutations at PfMDR1 Y184F; a marker for lumefantrine, amodiaquine and mefloquine drug (Table 2).
Prevalence of PfCRT polymorphisms
Of 443 genotyped samples and analyzed for the PfCRT 76, 442 samples (99.8%) contained the wild type (WT)/susceptible (K76) while only one (0.2%) threonine (76T) was detected. The PfCRT haplotypes at positions 72–76, CMNVK were detected in 442 samples (99.8%). while the resistant haplotype CVIET was detected in only one (0.2%) samples (Table 3). The PfMDR1 mutation at position N86Y is the first in the 3 amino‐acid haplotype (NYD) which enhances resistance to CQ was not detected in the all samples (100%) (Table 2).
Discussion
To the best of authors’ knowledge this is the first study to report mutations at K13-propeller, associated with ART resistance from the southeast of Tanzania. We report resistance patterns, one decade since ART-based combination therapy was introduced as the first and second line treatment of malaria whilst 18 years after CQ withdrawal for treatment of uncomplicated malaria in Tanzania. Generally, this study found a prevalence of 7.3% for K13 mutations, these mutations contained those which are found in WHO list3, and those reported elsewhere10 and undocumented non-synonymous K13 mutations. Prevalence obtained in this study was higher than the one reported the polymorphisms of P. falciparum K13-propeller gene among migrant workers returning to Henan Province, China from Africa, the study found the frequency of the K13-propeller 6.50% in Central Africa, followed by East Africa (5.26%), West Africa (4.55%) and South Africa (4.55%)6 and study by Kamau et al., 201422 reported that, allele frequencies of K13-propeller polymorphisms in P. falciparum parasites from sub-Saharan Africa ranged between 1% and 3%. In contrary to that, the recent findings from Uganda23, Kenya24 as well as Tanzania25 reported no evidence of K13 mutations. The differences in study periods between the studies could be the cause of the observed discrepancies in prevalence.
K13 mutations at position R561H and A578S, are two mutations previously described by WHO as validated3 and not associated26 with ART resistance, respectively. Surprisingly, both two mutations were detected as recombinant which contained WT gene, i.e. WT R561H and WT A578S WT W565C. These mutations especially, the validated mutation R561H cause a delayed parasite clearance10,27. Mutation in K13 gene has also been reported from the study conducted in southern Rwanda28. More importantly, this study documented 24 K13 mutations which currently don’t form part of WHO ART resistance markers list of 20183. On the other hand, all genotypes for PGB and EXO/P23_BP, markers for ART and piperaquine resistance respectively found no mutation.
High prevalence (60.4%) PfMDR1Y184F mutations was detected from Tanzanian samples. These finding are similar from the study conducted in Equatorial Guinea which found high prevalence of PfMDR1 Y184F mutations in P. falciparum isolates. Nevertheless, there are limited evidences which associated the PfMDR1Y184F mutation with lumefantrine and mefloquine16 susceptibility. Furthermore, the study reported that PfMDR1 at amino acids 86 and 184, demonstrate resistance to the ACT partner drug amodiaquine and the former first-line agent CQ. In contrast, N86Y increases parasite susceptibility to the partner drugs lumefantrine and mefloquine, and the active artemisinin metabolite dihydroartemisinin. The PfMDR1 N86Y plus Y184F isoform moderately reduces piperaquine potency in strains expressing an Asian/African variant of the CQ resistance transporter PfCRT16. On the other hand, the current findings suggest that CQ-susceptible P. falciparum parasites have reemerged and are now predominant in Tanzania (sub-Saharan Africa) where CQ was withdrawn in 2001. These findings were similar from the recent study conducted in Zambia29, the neighboring country to southwest. In both two countries CQ were withdrawn as a first line treatment in 20014 and 200321 in Tanzania and Zambia, respectively. These findings were contrary to the country where high levels of CQ resistance have persisted due to incomplete withdrawal of CQ30. The study of 2019, conducted in Nigeria revealed a high prevalence of PfCRT mutant genotypes and haplotypes and low frequency of PfMDR1 mutant genotypes, 11 years after the switch in malaria treatment policy from CQ to artemisinin combination therapy (ACT) in Nnewi, Nigeria. The study suggested that continual circulation and spread of CQ-resistant P. falciparum parasites in the study area due to the continued use of unrecommended CQ30.
Moreover, this study found a complete deletion of PfMDR1 mutation in all samples at position N86Y, a mutation which is associated with an enhanced resistance to chloroquine. These findings are in line with those reported in a study conducted in Zambian29. Another study conducted in Malawi reported a slower decline in prevalence of mutations in PfMDR1 than PfCRT suggesting that PfMDR1 mutations may be less deleterious to parasite fitness than are PfCRT mutations31. However, mutations in PfMDR1 by themselves are insufficient to confer CQ-resistance32 also the combination of PfMDR1 mutations and PfCRT mutations, provided no added advantage to CQ treatment failure than PfCRT mutations alone33. Furthermore, PfMDR1 mutations do not add to the predictive value of PfCRT mutations for CQ treatment failure34.
Since the design of the current study was cross sectional, there was a limitation in establishing the association between the molecular resistance markers and clinical/treatment outcomes of patients. However, the relationship between malaria treatment outcomes and resistance markers have been well described elsewhere3,10.
In conclusion, K13-propeller mutations associated with artemisinin resistance were found in Tanzanian samples. Mutations included K13 R561H and K13 A578S haplotypes validated to cause artemisinin resistance and not associated with artemisinin resistance, respectively. Twenty four K13 non-synonymous mutations not yet listed by WHO (unclassified) are reported. Further phenotypic studies are warranted to investigate the unclassified K13 mutations. Additionally, the return of chloroquine-susceptible P. falciparum malaria, 18 years after the removal of chloroquine drug pressure in Tanzania is documented. In this regard, chloroquine may be considered for malaria prevention, i.e. sickle cell disease children or the reintroduction in future, ideally in combination with other antimalarial drugs, especially in areas where disappearance of chloroquine resistance is evident while safe and affordable alternatives antimalarials are limited.
Materials and Methods
Study design, area, period and population
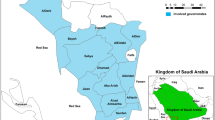
Surveillance of molecular markers for ART and partner drugs (piperaquine, lumefantrine, amodiaquine, mefloquine and chloroquine) resistance was conducted between April and August 2019 at Kibiti Health Center (KHC), Kibiti District, Tanzania (Fig. 1). Kibiti District is found along the Indian ocean35 and has malaria prevalence of 10.2%36 where P. falciparum is responsible for more than 95% of all malaria cases37. Patients attending clinic at Kibiti Health Center (KHC) who presented with symptoms suggestive of malaria infection were recruited in the study. The symptoms such as fever, general body weakness and headache were confirmed by the attending physician38. Patients screened for malaria and those who tested positive using CareStart Malaria HRP2/pLDH test (Access Bio, Ethiopia) were requested to participate. Then positive samples by rapid tests were subjected to blood smear (BS) microscopy for confirmation. A total of 489 dried blood samples (DBS) from patients tested positive with BS microscopy were subjected to DNA extraction and genotyping.

A map of the study site. Left is the map of Kibiti District while on the right side is the map of Tanzania, located in Eastern Africa. Tanzania is bordered by the Indian Ocean, Kenya and Uganda to the north, Rwanda, Democratic Republic of the Congo, and Burundi to the east, and Zambia, Malawi, and Mozambique to the south (not shown on the map). The study site map was originally generated using ArcGIS software version 10.7.1 (https://www.esri.uconn.edu/software/arcgis-student/).
DBS preparation
DBS were prepared according to MalariaGEN SpotMalaria, DBS collection protocol39. A sterilized patient’s finger was pricked to allow blood drops where four blood spots from each patient were prepared, two on each paper card. The blood spots were allowed to air dry and placed in the desiccant sachet for storage.
DNA extraction
DNA from the DBS was extracted following QIAamp DNA Investigator Kit for isolation of total DNA from filter papers (Qiagen, Valencia, CA, USA) and as previously described by Oyola et al.40.
Genotyping of antimalarial resistance markers
Molecular genotyping of ART, partner drugs and chloroquine (K13, PGB, EXO, P23_BP, MDR1 and CRT) were performed by Wellcome Sanger Institute, UK.
Briefly, targets for genotyping were identified and multiplex PCR primers were designed using a modified version of the mPrimer software41 and the exact design of the primer sequences will be described elsewhere (Goncalves, manuscript in preparation). Primers were designed to amplify products between 190–250 bp and were combined into 3 pools. A two-step protocol was used to first amplify the target regions of the parasite genome, followed by a second PCR to incorporate sequencing and multiplexing adapters. Batched samples (384) were sequenced in a single MiSeq lane combining all PCR products. Samples were de-plexed using the multiplexing adapters and individual CRAM files were aligned to a modified amplicon reference genome. Genotyping was done using bcftools as well as custom scripts to filter and translate genotypes into drug resistance haplotypes.
Statistical analysis
Laboratory information on Microsoft Excel Sheet (Redmond, WA) were exported directly to Statistical Package for Social Sciences version 25 (SPSS Software, Chicago Inc., USA) for data analysis. Genotypes were expressed as frequencies and percentages.
Ethics approval and consent to participate
Ethical approval to conduct this study was sought from Muhimbili University of Health and Allied Sciences Ethical Review Board (Ref. DA.282/298/01A.C/) and National Institute for Medical Research (Ref. NIMR/HQ/R.8A/Vol.IX/3107). Permission to conduct the study at KHC was obtained from both Kibiti District Medical Officer and KHC Medical Officer In-charge. Written informed consent after explaining the purpose of the study was requested before enrollment of participants. Additionally, all methods were carried out in accordance with relevant guidelines, regulations and good laboratory practice.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
References
World Health Organization (WHO). World Malaria Report.; https://www.who.int/publications-detail/world-malaria-report-2019 (2019).
Hussein Mwinyi, H. the United Republic of Tanzania Standard Treatment Guidelines and Essential Medicines List Ministry of Health and Social Welfare Fourth Edition.; (December):42–43., http://www.who.int/selection_medicines/country_lists/Tanzania_STG_052013.pdf (2013).
Subregion, G. M. Artemisinin resistance and artemisinin-based combination therapy efficacy;(August). (2018).
Mohammed, A. et al. Trends in chloroquine resistance marker, Pfcrt-K76T mutation ten years after chloroquine withdrawal in Tanzania. Malar J. 12(1), 1, https://doi.org/10.1186/1475-2875-12-415 (2013).
Ariey, F. et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 505(7481), 50–55, https://doi.org/10.1038/nature12876 (2014).
Yang, C. et al. Polymorphisms of Plasmodium falciparum k13-propeller gene among migrant workers returning to Henan Province, China from Africa. BMC Infect Dis. 17(1), 1–8, https://doi.org/10.1186/s12879-017-2634-z (2017).
Newman, R. D. & Plowe, C. V. The Threat of Artemisinin-Resistant Malaria. 2011–2013 (2011).
Mita, T., Tanabe, K. & Kita, K. Spread and evolution of Plasmodium falciparum drug resistance. Parasitol Int. 58(3), 201–209, https://doi.org/10.1016/j.parint.2009.04.004 (2009).
Ariey, F., Witkowski, B., Amaratunga, C. & Beghain, J. Anne- Claire Langlois, Nimol Khim, Saorin Kim, Valentine Duru, Christiane Bouchier, Laurence Ma, Pharath Lim, Rithea Leang, Socheat Duong, Sokunthea Sren and DM. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. 1(1), 23–30, https://doi.org/10.1007/s40778-014-0003-z.Genome (2016).
Wwarn, K & Group, G. S. Association of mutations in the Plasmodium with parasite clearance rates after artemisinin-based treatments — a WWARN individual patient data meta-analysis 3, 1–20 (2019).
Noya, O, Katz, N & Pointier, J. P. Neglected Tropical Diseases - Latin America and the Caribbean, 11–44, https://doi.org/10.1007/978-3-7091-1422-3 (2015).
Witkowski, B. et al. Increased tolerance to artemisinin in plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob Agents Chemother. 54(5), 1872–1877, https://doi.org/10.1128/AAC.01636-09 (2010).
Amaratunga, C., Witkowski, B., Khim, N., Menard, D. & Fairhurst, R. M. Artemisinin resistance in Plasmodium falciparum. Lancet Infect Dis. 14(6), 449–450, https://doi.org/10.1016/S1473-3099(14)70777-7 (2014).
Miotto, O. et al. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet. 47(3), 226–234, https://doi.org/10.1038/ng.3189 (2015).
Venkatesan, M. et al. Polymorphisms in Plasmodium falciparum chloroquine resistance transporter and multidrug resistance 1 genes: Parasite risk factors that affect treatment outcomes for P. falciparum malaria after artemether-lumefantrine and artesunate-amodiaquine. Am. J. Trop. Med. Hyg. 91(4), 833–843, https://doi.org/10.4269/ajtmh.14-0031 (2014).
Veiga, M. I. et al. Globally prevalent PfMDR1 mutations modulate Plasmodium falciparum susceptibility to artemisinin-based combination therapies. Nat Commun., 7(May), https://doi.org/10.1038/ncomms11553 (2016).
Amato, R. et al. Genetic markers associated with dihydroartemisinin–piperaquine failure in Plasmodium falciparum malaria in Cambodia: a genotype–phenotype association study. Lancet Infect Dis. 17(2), 164–173, https://doi.org/10.1016/S1473-3099(16)30409-1 (2017).
Zhang, H. & Roepe, P. M. The antimalarial drug resistance protein Plasmodium falciparum chloroquine resistance transporter binds chloroquine. Biochemistry. 43(26), 8290–8296, https://doi.org/10.1021/bi049137i (2004).
Pulcini, S. et al. Mutations in the Plasmodium falciparum chloroquine resistance transporter, PfCRT, enlarge the parasite’s food vacuole and alter drug sensitivities. Sci Rep. 5(August), 1–16, https://doi.org/10.1038/srep14552 (2015).
Laufer, M. K. et al. Return of Chloroquine‐Susceptible Falciparum Malaria in Malawi Was a Reexpansion of Diverse Susceptible Parasites. J. Infect Dis. 202(5), 801–808, https://doi.org/10.1086/655659 (2010).
Mwanza, S. et al. The return of chloroquine-susceptible Plasmodium falciparum malaria in Zambia. Malar J. 15(1), 1–6, https://doi.org/10.1186/s12936-016-1637-3 (2016).
Kamau, E. et al. K13-propeller polymorphisms in plasmodium falciparum parasites from sub-saharan Africa. J. Infect Dis. 211(8), 1352–1355, https://doi.org/10.1093/infdis/jiu608 (2015).
Conrad, M. D. et al. Polymorphisms in K13 and falcipain-2 associated with artemisinin resistance are not prevalent in Plasmodium falciparum isolated from Ugandan children. Plos One. 9(8), https://doi.org/10.1371/journal.pone.0105690 (2014).
Muwanguzi, J. et al. Lack of K13 mutations in Plasmodium falciparum persisting after artemisinin combination therapy treatment of Kenyan children. Malar J. 15(1), 1–6, https://doi.org/10.1186/s12936-016-1095-y (2016).
Kakolwa, M. A. et al. Efficacy and safety of artemisinin-based combination therapy, and molecular markers for artemisinin and piperaquine resistance in Mainland Tanzania ACTRN12615000159550 ACTRN. Malar J. 17(1), 1–10, https://doi.org/10.1186/s12936-018-2524-x (2018).
Collet, L. et al. A Worldwide Map of Plasmodium falciparum K13-Propeller Polymorphisms. N. Engl. J. Med. 374, 2453–2464, https://doi.org/10.1056/NEJMoa1513137 (2016).
Putaporntip, C. et al. Natural selection of K13 mutants of Plasmodium falciparum in response to artemisinin combination therapies in Thailand. Clin Microbiol Infect. 22(3), 285.e1–285.e8, https://doi.org/10.1016/j.cmi.2015.10.027 (2016).
Tacoli, C. et al. Artemisinin resistance-associated K13 polymorphisms of plasmodium falciparum in Southern Rwanda, 2010–2015. Am. J. Trop. Med. Hyg. 95(5), 1090–1093, https://doi.org/10.4269/ajtmh.16-0483 (2016).
Sitali, L. et al. En - route to the ‘ elimination’ of genotypic chloroquine resistance in Western and Southern Zambia, 14 years after chloroquine withdrawal., 1–8 (2019).
Ikegbunam, M. N. et al. Analysis of Plasmodium falciparum Pfcrt and Pfmdr1 genes in parasite isolates from asymptomatic individuals in Southeast Nigeria 11 years after withdrawal of chloroquine. Malar J. 18(1), 1–7, https://doi.org/10.1186/s12936-019-2977-6 (2019).
Kublin, J. G. et al. Reemergence of Chloroquine‐Sensitive Plasmodium falciparum Malaria after Cessation of Chloroquine Use in Malawi. J Infect Dis. 187(12), 1870–1875, https://doi.org/10.1086/375419 (2003).
Michael, B. et al. Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature. 403(12), pages 906–909, https://doi.org/10.1002/j.1551-8833.1954.tb20256.x (2000).
Plasmodium, B. C. a Mol Ecul Ar Marke R F or Ch Loroquine-R Es Ista Nt Fa Lcipa Rum Ma L a Ria a Molecular Marker for Chloroquine-Resistant. English J. 344(4), 257–263, http://www.ncbi.nlm.nih.gov/pubmed/11172152 (2001).
Jelinek, T. et al. Diagnostic value of molecular markers in chloroquine-resistant falciparum malaria in southern Mauritania. Am. J. Trop. Med. Hyg. 67(5), 449–453, https://doi.org/10.4269/ajtmh.2002.67.449 (2002).
TMIS. Tanzania Malaria Indicator Survey (TMIS) 2017., https://dhsprogram.com/pubs/pdf/MIS31/MIS31.pdf (2017).
Kilonzi, M. et al. Comparison of malaria treatment outcome of generic and innovator’s anti-malarial drugs containing artemether–lumefantrine combination in the management of uncomplicated malaria amongst Tanzanian children. Malar J. 18(1), 133, https://doi.org/10.1186/s12936-019-2769-z (2019).
Bwire, G. M. et al. Diagnostic performance of CareStartTM malaria HRP2/pLDH test in comparison with standard microscopy for detection of uncomplicated malaria infection among symptomatic patients, Eastern Coast of Tanzania. Malar J. 18(1), 354, https://doi.org/10.1186/s12936-019-2990-9 (2019).
The united republic of Tanzania standard treatment guidelines and essential medicines list (2013).
MalariaGEN Resource Centre members based at the Wellcome Trust Sanger Institute U. Protocol for collecting dried blood spots from malaria patients, for the purposes of sequencing Plasmodium parasites, https://www.malariagen.net/network/capacity-building/methods#DBS .
Oyola, S. O. et al. Whole genome sequencing of Plasmodium falciparum from dried blood spots using selective whole genome amplification. Malar J. 15(1), 1–12, https://doi.org/10.1186/s12936-016-1641-7 (2016).
Shen, Z. et al. MPprimer: A program for reliable multiplex PCR primer design. BMC Bioinformatics. 11, https://doi.org/10.1186/1471-2105-11-143 (2010).
Acknowledgements
This publication uses data generated using Tanzanian samples in collaboration with MalariaGEN SpotMalaria Project (https://www.malariagen.net/projects/spotmalaria); the project is coordinated by the MalariaGEN Resource Centre with funding from Wellcome (206194, 090770). The authors would like to thank the staff of Wellcome Sanger Institute Sample Management, Genotyping, Sequencing and Informatics teams for their contribution. Furthermore, we thank Sonia Goncalves for providing the statement on the genotyping of antimalarial resistance markers. The author (GMB) acknowledge the training on antimalarial resistance markers when he attended Global Health Fellowship 2018, Novartis Institutes for Biomedical Research, Emeryville, CA, USA. Special thanks to Judith Straimer and the whole Malaria team at Novartis Institute for Tropical Diseases. This study was funded by Swedish International Development Cooperation Agency (Sida), Sweden through Muhimbili University of Health and Allied Sciences. The funder did not participate in the design of the study, data collection, analysis, interpretation, and manuscript preparation.
Author information
Authors and Affiliations
Contributions
G.M.B. participated in conception, study design, data collection, analysis and manuscript writing. M.K. and W.P.M. participated in data analysis and manuscript writing. B.N. and A.A.R.K. participated in research design and revising the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons License, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons License, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons License and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this License, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bwire, G.M., Ngasala, B., Mikomangwa, W.P. et al. Detection of mutations associated with artemisinin resistance at k13-propeller gene and a near complete return of chloroquine susceptible falciparum malaria in Southeast of Tanzania. Sci Rep 10, 3500 (2020). https://doi.org/10.1038/s41598-020-60549-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-60549-7
This article is cited by
-
In vitro delayed response to dihydroartemisinin of malaria parasites infecting sickle cell erythocytes
Malaria Journal (2024)
-
Efficacy of artemether-lumefantrine and dihydroartemisinin-piperaquine and prevalence of molecular markers of anti-malarial drug resistance in children in Togo in 2021
Malaria Journal (2024)
-
Efficacy of artesunate-amodiaquine for treatment of uncomplicated Plasmodium falciparum malaria in mainland Tanzania
Malaria Journal (2024)
-
Molecular surveillance of Kelch 13 polymorphisms in Plasmodium falciparum isolates from Kenya and Ethiopia
Malaria Journal (2024)
-
Emergence, transmission dynamics and mechanisms of artemisinin partial resistance in malaria parasites in Africa
Nature Reviews Microbiology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
