
Abstract
The presence of advanced fibrosis is an important measure of the severity of chronic liver disease. Prior works that have examined the gut microbiome as a novel biomarker for advanced fibrosis have only examined patients with nonalcoholic fatty liver disease. Therefore, our goal was to examine the gut microbiome across varying etiologies of liver disease to create a predictive model for liver fibrosis based upon a microbial signature. Stool samples were obtained from patients with chronic liver disease (n = 50) undergoing FibroScan (ultrasound elastography) at the VA Greater Los Angeles Healthcare System. Healthy control patients (n = 25) were also recruited as a reference population. Fecal samples underwent 16S ribosomal RNA sequencing. Using differentially abundant microbes, a random forest classifier model was created to distinguish advanced fibrosis from mild/moderate fibrosis. The findings were then validated in a separate cohort of chronic liver disease patients (n = 37). Etiologies for liver disease included non-alcoholic liver disease (58.0%), hepatitis C (26.0%), hepatitis B (10.0%), and alcohol (6.0%). Microbiome composition was distinct in liver patients with advanced fibrosis compared to those with minimal fibrosis and healthy controls (p = 0.003). In multivariate negative binomial modeling, 26 bacterial taxa were differentially abundant in patients with advanced fibrosis as compared to those with minimal/moderate fibrosis (q-value < 0.05). A random forests classifier based on these taxa had an AUROC of 0.90 to predict advanced fibrosis. Prevotella copri, which was enriched in patients with advanced fibrosis, was the most strongly predictive microbe in the classifier. The classifier had an AUROC of 0.82 for advanced fibrosis in the validation cohort and Prevotella copri remained the strongest predictive microbe for advanced fibrosis. There is a distinct microbial signature for patients with advanced fibrosis independent of liver disease etiology and other comorbidities. These results suggest that microbial profiles can be used as a non-invasive marker for advanced fibrosis and support the hypothesis that microbes and their metabolites contribute to hepatic fibrosis.
Similar content being viewed by others

Introduction
Chronic liver disease is one of the most common medical conditions worldwide that affects as many as 840 million people with an estimated rate of mortality of 2 million deaths per year1. From 1999 to 2016, deaths from chronic liver disease in the US increased by 65 percent and deaths from liver cancer doubled2. This escalation has been attributed to factors such as increased alcohol use in younger Americans, increased intravenous drug use and rapidly rising rates of obesity in our society2.
Over the several years, there is a growing body of research investigating the role of the gut microbiome in disease pathology, and in particular, chronic liver disease3. The human microbiome has nearly 37 trillion cells and contains more than 100 times the number of genes than the human body4. While one of the principal function of the human microbiome is to ferment and create short-chain fatty acids from indigestible fiber, recent research studies have shown that the microbiome is also a key regulator of such liver diseases as NAFLD and primary sclerosing cholangitis5,6,7,8,9,10.
One of the most challenging aspects of chronic liver disease is the identification of patients with liver fibrosis. The development of advanced fibrosis is a major predictor of liver-related morbidity and mortality11,12,13. Early identification of advanced fibrosis using non-invasive testing is a growing area of research in the field of hepatology12,14,15. The characterization of gut microbial biomarkers for advanced fibrosis has been a novel area of ongoing research. For example, Qin and colleagues in 2014 reported that an intestinal microbial signature was present in individuals with cirrhosis in a Chinese cohort as compared to healthy controls16. This study included different causes of cirrhosis including hepatitis C, hepatitis B, NAFLD, and alcoholic liver disease. Loomba et al. in two separate studies was able to identify and validate a distinct microbial signature that was related to advanced fibrosis in patients with NAFLD17,18. However they did not explore other etiologies of chronic liver disease, so it is unclear at this time if this signature holds true for other causes of liver disease in western society. Given the association of the microbiome with chronic liver disease and cirrhosis, the aim of this study was to determine if specific fecal microbial profiles can be used as non-invasive biomarkers for advanced fibrosis in patients with varying etiologies of chronic liver disease.
Methods
Patient recruitment and stool collection
Patients with a diagnosis of chronic liver disease and undergoing ultrasound elastography were recruited prospectively from the VA Greater Los Angeles Healthcare System (VA) from 6/2017 to 6/2018. Chronic liver disease included patients with chronic hepatitis C virus (HCV) infection, chronic hepatitis B virus (HBV) infection, liver disease due to chronic alcohol use, primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), Wilson’s disease, autoimmune hepatitis, hemochromatosis, and NAFLD. Patients were excluded if they were treated with antibiotics or probiotics within 3 months of enrollment, had only acute liver injury without any underlying chronic liver disease, treated HCV infection with sustained virologic response without any other forms of chronic liver disease, were on a specialized diet (e.g. gluten free, vegan, vegetarian, high protein), had a personal history of GI surgeries, irritable bowel syndrome or inflammatory bowel disease. Stool was collected within 7 days of their ultrasound elastography and placed into 95% ethanol and stored at −80°C until processing. Patient information including age, gender, race/ethnicity, and comorbidities were also collected. For race and ethnicity, there were 5 categories with Hispanic as a separate category (i.e. non-Hispanic white, non-Hispanic black, Hispanic, Asian, and other). Co-morbidities were collected in order to calculate the Charlson comorbidity index, a validated score that assesses overall health and risk of 1-year all-cause mortality19. Stool samples from heathy control patients without any evidence of chronic liver disease were also collected. The study was approved by the Veteran’s Affair Greater Los Angeles Healthcare System Institutional Review Board. All methods herein were performed in accordance with relevant guidelines and regulations. Verbal and written informed consent for study participation was obtained from all patients.
Liver ultrasound elastography
All patients with chronic liver disease underwent an ultrasound elastography using the FibroScan touch 502 machine (Echosens, MA, USA). All ultrasound elastographies were performed by trained technicians with over 100 scans of experiences each. Medium (M) and extra-large (XL) probes were utilized depending on the patient’s body habitus according to manufacturer’s protocol. Controlled attenuation parameter (CAP) score and liver stiffness were collected as non-invasive measurements of hepatic steatosis and fibrosis, respectively. All measurements were done at least 10 times at the same spot with interquartile range/median value less than 30% as per manufacturers guidelines. A CAP score of between 238 and 260 was given a steatosis grade of S1 representing 11–33% of fatty change in the liver, a score between 260 and 290 was given a grade of S2 representing 34–66% of fatty change, and a score higher than 290 was given a grade of S3 representing 67% or more of fatty change as per manufacturer’s guideline. Standard cutoffs of liver stiffness as measured in kilopascals based on etiology of liver disease was used to determine extent of liver fibrosis (F0/F1 to F4)20. Minimal fibrosis was defined as a score consistent with F0-F2 and advanced fibrosis was defined as a score consistent with F3-F4, similar to prior published studies17.
16S rRNA sequencing
DNA was extracted from ethanol preserved stool using the Powersoil kit as per the manufacturer’s instructions (MO BIO, Carlsbad, CA, USA). The V4 region of 16S ribosomal RNA was amplified and underwent paired end sequencing on an Illumina HiSeq 2500 (San Diego, CA, USA) as previously described21. The 253 base-pair reads were processed using QIIME 1.9.1 (San Diego, CA, USA) with default parameters22. The average sequence depth per sample was 45,560. Operational taxonomic units (OTUs) were picked against the May 2013 version of the Greengenes database, prefiltered at 97% identity. After removing OTUs that were present in fewer than 10% of all samples, 1479 OTUs remained for analysis. Raw 16S rRNA sequence data were deposited under National Center for Biotechnology Information BioProject PRJNA542724.
Statistical analysis
For demographic data, means are expressed along with their standard deviations and comparisons between means were performed using the Student’s t-test. Categorical data were compared using the Pearson’s chi-squared test.
For 16S rRNA sequencing data, alpha diversity metrics that included Chao1 (a metric for species richness), Faith’s phylogenetic diversity, and Shannon Index (a metric that incorporates both species richness and species evenness) were computed using QIIME. The statistical significance of differences in alpha diversity metrics was calculated using a two-tailed t-test. Beta diversity, a metric of differences between samples, was calculated using the square root of the Jensen-Shannon divergence and visualized by principal coordinates analysis in R23. Univariate Adonis, a permutational analysis of variance, was performed using 10,000 permutations to test for differences in the square root of the Jensens-Shannon divergence across the following variables: age, gender, race/ethnicity, BMI, control/patient cohort, fibrosis as a binary categorical variable, steatosis grade, etiology of liver disease, and Charlson’s comorbidity index. Only variables with a p-value < 0.1 were used for the final multivariate analysis. This included steatosis grade, Charlson’s comorbidity index, and fibrosis. Differential abundance testing was evaluated using DESeq2 in R, which employs an empirical Bayesian approach to shrink dispersion and fit non-rarified count data to a negative binomial model24. Variables listed in the multivariate analysis of DESeq2 were the same variables listed above for the multivariate Adonis analysis. P-values for differential abundance were converted to q-values to correct for multiple hypothesis testing (<0.05 for significance). All authors had access to the study data and had reviewed and approved the final manuscript.
Random forests classifier
A random forests classifier to predict advanced fibrosis was created in R using the randomForest package (https://cran.r-project.org/web/packages/randomForest) with 1001 trees and mtry = 225. Features inputted into the random forest classifier were those associated significantly with advanced fibrosis as determined by multivariate DESeq2 models. The accuracy of the random forest classifier was estimated using a 10-fold cross-validation.
Predicted metagenomics
Metagenomic data of each sample was inferred from 16S rRNA sequencing data by using PICRUSt 1.1.3 (http://picrust.github.io/picrust), a well validated tool designed to impute metagenomic data from 16S rRNA compositional data26. 16S rRNA sequencing data was inputted into PICRUSt and normalized by copy number using default parameters. The subsequent metagenes were then categorized by function using the KEGG database. Differences in predicted metagenes by advanced fibrosis were identified using DESeq2 with p-values adjusted for multiple hypothesis testing.
Validation cohort
The findings of the random forest classifier were validated in a separate cohort of NAFLD patients recruited at the VA from January 1st, 2019 to October 1st, 2019. Inclusion and exclusion criteria were the same as above. All patients underwent stool collection and liver ultrasound elastography as described above. Demographic data, race, ethnicity, and comorbidities were collected. In addition, all patients within this cohort filled out a validated diet questionnaire, the NIH Diet History Questionnaire III (DHQIII), at the time of their stool collection27.
Synopsis
This is one of the few studies that have examined the microbiome as a novel biomarker for advanced fibrosis. Unlike prior works that have only examined patients with nonalcoholic fatty liver disease, this study included patients from various races and etiologies of liver disease. The study highlights how the gut microbiome may play a role in fibrosis progression.
Results
Patient and healthy control characteristics
Fifty patients with chronic liver disease and 25 healthy controls were recruited. Etiologies for liver disease included non-alcoholic liver disease (58.0%), hepatitis C (26.0%), hepatitis B (10.0%), and alcohol (6.0%) (Table 1). Nineteen patients had advanced fibrosis and 7/19 (36.8%) had F4 fibrosis. The healthy control cohort were younger on average than the patients with chronic liver disease and comprised of more females (p-value < 0.001). The average Charlson’s Comorbidity Index for the liver disease cohort was 4.33 ± 2.31. There was no difference in Charlson’s Comorbidity Index between patients with advanced fibrosis as compared to those without advanced fibrosis. There was no difference in race/ethnicity between any groups and there was no statistical difference in etiologies of chronic liver disease by fibrosis stage.
Microbial profiles differs by fibrosis stage and etiology of liver disease
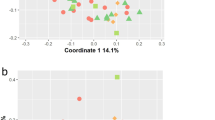
In univariate analysis of beta diversity, only 3 variables had a p-value < 0.1: steatosis grade, Charlson’s comorbidity index, and the presence of advanced fibrosis. Therefore, these variables were used for the multivariate analysis. As demonstrated in the principal coordinates analysis plot (Fig. 1A), the microbial profile of patients with advanced fibrosis differed significantly as compared to those with minimal or no fibrosis or healthy controls (p = 0.003), while adjusting for the other covariates. In regards to alpha diversity metrics, patients with NAFLD and minimal or no fibrosis had a lower Chao1 index (species richness) and a lower Faith’s Phylogenetic Diversity as compared to healthy controls and NAFLD patients with advanced fibrosis (Fig. 1B). There was no statistically significant difference in the Shannon Index (species richness/evenness) in any of the group comparisons.

Patients with Advanced Fibrosis have Distinct Microbial Composition and Diversity Compared to Other Liver Disease Patients or Healthy Controls. (A) Beta diversity visualized by principal coordinates analysis plot of all patients colored by fibrosis stage or control group. (B) Alpha diversity metrics by etiology of chronic liver disease and fibrosis stage. Chao1 is a metric of species richness, Faith’s Index is a metric of phylogenetic diversity, and Shannon index is a metric of species richness/evenness. *Represents comparison with p-value < 0.05.
The average taxonomic composition of chronic liver disease patients divided by etiology is summarized in Fig. 2A on a phylum and genus level. The composite taxonomic summary of all patients with advanced fibrosis, minimal or no fibrosis, or healthy controls is shown in Fig. 2B. Patients with alcoholic liver disease with F0-F2 fibrosis had a higher relative abundance of Bacteroidetes than any other group. Examining all patients with advanced fibrosis, there was a statistically higher abundance of Prevotella as compared to either healthy control or patients with F0-F2 disease as determined by differential abundance analysis adjusting for covariates.

Taxonomic Profiles Categorized by Etiology of Chronic Liver Disease and Fibrosis Stage. (A) Taxonomic profiles at the phylum and genus levels, divided by etiology of chronic liver disease and fibrosis stage. (B) Taxonomic profiles by phylum and genus of patients with advanced fibrosis (F3/F4), liver patients with minimal/no fibrosis (F0-F2), and healthy controls. EtOH: Alcohol, HBV: Hepatitis B virus, HCV: Hepatitis C virus, NAFLD: nonalcoholic fatty liver disease.
Differential abundance analysis adjusting for covariates was also performed to compare patients with different etiology of liver disease to healthy controls (Fig. 3). Because there were only 8 patients with alcoholic liver disease or HVC infection, the analysis only focused on patients with chronic HCV infection and NAFLD adjusting for fibrosis and the other covariates listed above. Patients with HCV disease as compared to controls differed significantly across 25 different OTUs (a taxonomic unit roughly corresponding to species). An undefined species belonging to the family Rikenellaceae, two undefined species in the genus Bacteroides, and an undefined species in the genus Dialister made up the OTUs with the largest relative abundance (Fig. 3A). NAFLD patients had 34 separate OTUs that were differentially abundant from healthy controls (Fig. 3B). The species with the highest relative abundance included Prevotella copri, an undefined species in the family Ruminococcaceae and an undefined species in the family Rikenellaceae. All 3 of these species were underrepresented in patients with NAFLD. Comparing NAFLD to HCV patients, there were 10 OTUs that were differentially abundant between the two groups. Prevotella copri, an undefined species belonging to the genus Bacteroides, and an undefined species of the order Clostridiales made three most abundant OTUs. Prevotella copri was higher in patients with NAFLD adjusting for fibrosis stage, while the other two OTUs were higher in patients with HCV (Fig. 3C).

Microbial Communities Differ by Etiology of Chronic Liver Disease. DESeq2 differential abundance analysis comparing (A) HCV patients to control, (B) NAFLD patients to control, and (C) NALFD patients to HCV patients controlling for fibrosis. HCV: Hepatitis C virus, NAFLD: nonalcoholic fatty liver disease.
Between patients with advanced fibrosis vs. minimal or no fibrosis, 26 OTUs were differentially abundant. The two most highly abundant differential OTUs were Prevotella copri and an undefined species belonging to the genus Bacteroides, both of which were elevated in patients with advanced fibrosis (Fig. 4A). Examining differences between fibrosis stage within patients with HCV and with NAFLD, there were 12 OTUs and 23 OTUs that were differentially abundant, respectively. While Prevotella copri did have a higher relative abundance in HCV patients with advanced fibrosis, it did not reach statistical significance. Instead, two undefined species in the family Ruminococcaceae and Akkermansia muciniphila were the three differential OTUs with the highest abundance; all three were elevated in patients with HCV with advanced fibrosis (Fig. 4B). In NAFLD patients, Prevotella copri was the predominant species and it was elevated in patients with advanced fibrosis (Fig. 4C).

Microbial Communities Differ by Fibrosis Stage. DESeq2 differential abundance analysis comparing (A) advanced fibrosis patients to minimal/no fibrosis patients, (B) HCV patients with advanced fibrosis (F3/F4) to HCV patients without advanced fibrosis (F0-F2), and (C) NAFLD patients with advanced fibrosis to NAFLD patients without advanced fibrosis. HCV: Hepatitis C virus, NAFLD: nonalcoholic fatty liver disease.
Predicted metagenomic profile differs by fibrosis
Metagenomic profiles were predicted for each sample from 16S rRNA compositional data using PICRUSt. The predicted metagenomic profile that differed between patients with advanced fibrosis as compared to those patients with minimal or no fibrosis is summarized in Fig. 5. The average weighted Nearest Sequenced Taxon Index (NTSI) per sample was 0.08. Low scores indicate availability of closely related reference genomes and thus a higher quality of predictions26. While there was no overall large difference of the predicted metagenome between samples by fibrosis stage as represented by the principal coordinates analysis in Fig. 5A (p = 0.34), patients with advanced fibrosis did have a trend to have more bacterial genes present per sample (Fig. 5B, p = 0.09). From 16S rRNA compositional data, DESeq2 analysis of PICRUSt predicted metagenes showed 168 metagenes that were statistically differentially expressed in patients with advanced fibrosis as compared to those with minimal or no fibrosis. Categorizing these metagenes into functional categories showed 9 pathways that are different between the two groups. The pathways that were most different between the two groups were those involved in mineral absorption, arachidonic acid metabolism, carbohydrate digestion and absorption, and linoleic acid metabolism (Fig. 5C).

Predicted Metagenomic Differences by Fibrosis Stage. (A) Principal coordinates analysis plot of predicted metagenomic profiles between samples by fibrosis stage. (B) Number of predicted genes present per sample by fibrosis stage. Solid bar represents the mean and the box represents 1 standard deviation. *p = 0.09. (C) Differential abundance analysis (q < 0.05) of predicted metagenes for advanced fibrosis categorized by KEGG pathways predicted by PICRUSt from 16S rRNA data.
A microbial signature predicts advanced fibrosis
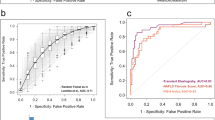
Using the 26 OTUs that were differentially abundant between patients with advanced fibrosis and patients with minimal or no fibrosis, a random forest classifier was created with high accuracy for predicting advanced fibrosis. The area under the receiver operating characteristic curve (AUROC) was 0.90 in 10-fold cross-validation (Fig. 6A). The contribution of each OTU to the classifier was expressed as variable importance score, which measures the decreased accuracy of the classifier if that feature was removed (Fig. 6B). The species with the greatest variable importance score was Prevotella copri followed by two undefined OTUs belonging to the genus Lachnobacterium and family Ruminococccaceae.

A Distinct Microbial Signature Can Accurately Identify Patients with Advanced Fibrosis. (A) Receiver operating characteristic curve of the random forests classifier for identifying patients with advanced fibrosis. (B) Importance scores for features included in the random forests classifier for predicting advanced fibrosis.
A separate cohort validates the finding that a distinct microbial signature predicts advanced fibrosis
In the validation cohort, there was no statistical difference between patients with advanced fibrosis as compared to minimal or no fibrosis in regards to age, gender, comorbidities, race, or dietary patterns (Table 2). Similar to the original cohort, a distinct microbial profile exists for patients with advanced fibrosis as compared to those with minimal or no fibrosis (Fig. 7). In univariate analysis of beta diversity, only age and advanced fibrosis had a p-value < 0.1. Therefore, these two variables were used for multivariate analysis. Adjusting for age, the microbial profile of patients with advanced fibrosis differed significantly as compared to those with minimal or no fibrosis as demonstrated in the principal coordinate analysis plot (p = 0.002). There was no statistical difference in Shannon index between patients with advanced fibrosis or those with minimal to no fibrosis in the validation cohort.

Validation of an advanced fibrosis microbial signature in a prospective study of NAFLD patients. (A) Principal coordinate analysis plot of beta diversity between patients with minimal to no fibrosis (F0-F2) versus patients with advanced fibrosis (F3/F4) within the validation cohort. (B) Shannon index of the validation cohort between patients with F0-F2 fibrosis and F3/F4 fibrosis. (C) Validation of the random forests classifier as depicted by a receiver operating characteristic curve. (D,E) Taxonomic profiles by phylum and genus of patients with minimal/no fibrosis (F0-F2) and patients with minimal/no fibrosis (F0-F2) within the validation cohort. (F) DESeq2 differential abundance analysis comparing advanced fibrosis patients to minimal/no fibrosis patients in the validation cohort.
The average taxonomic composition by fibrosis category is summarized in Fig. 7D,E, highlighting increased Prevotella in the advanced fibrosis group. Differential abundance testing demonstrated that 7 OTUs differed between patients with advanced fibrosis vs. minimal or no fibrosis. Of these, Prevotella copri was the most abundant and it was the only one that was enriched in those with advanced fibrosis. Applying the same random forest classifier trained on the initial cohort, microbiome composition had an AUROC of 0.82 for differentiating advanced vs. minimal or no fibrosis based on 10-fold cross-validation (Fig. 7C).
Discussion
This study yielded several important findings. In patients with chronic liver disease, we show that those with advanced stages of fibrosis have a distinct microbiome signature compared to those with lesser stages of fibrosis. This held true regardless of etiology of the liver disease and after adjusting for other covariates. These differences are characterized by an increase in the genus Prevotella and a decrease in Bacteroides. Furthermore, by using these microbial differences, a highly accurate model based on stool analysis can be created to identify those with advanced fibrosis.
We also show that microbial signatures differ across different etiologies of chronic liver disease. Similar to prior published works28,29, chronic HCV infection is associated with a decrease in the order Clostridiales and family Ruminococcaceae in patients with advanced fibrosis. This study also builds on prior data from NAFLD patients. Within our cohort the most abundant species that were significantly different between healthy controls and NAFLD patients while adjusting for the level of fibrosis were Prevotella copri, an undefined species in the family Ruminococcaceae, and an undefined species in the family Rikenellaceae. This is similar to other prior works showing a reduction of Ruminococcus and Prevotella in non-cirrhotic NAFLD patients18,30. Prevotella’s reduction in non-cirrhotic NAFLD patients as compared to healthy controls is likely related to diet. Diets that are high in fats and animal protein as compared to diets that are rich in fiber have been shown to increase Bacteroides and decrease Prevotella31,32. This finding is therefore in line with previous works that have linked the gut microbiome to diet and non-cirrhotic fatty liver disease.
Though the idea of using stool as a novel biomarker for advanced fibrosis was recently explored and validated, it was only done in patients with NAFLD and did not include other etiologies18. In our cohort of racially diverse patients with varying etiologies of chronic liver disease, we show that the idea of using stool analysis to identify patients with advanced fibrosis is not only feasible but potentially highly accurate. While several other non-invasive methods are currently available for the diagnosis of advanced fibrosis including magnetic resonance elastography (MRE), transient elastography, and lab-based models, these modalities can have reduced accuracy in patients with diabetes or severe obesity33. Therefore, we propose that stool analysis can be a potentially accurate method when other modalities are limited. Combination of stool testing with other non-invasive tests including Fib-4 and NAFLD fibrosis scoring may also prove to be an important clinical tool to identify those patients who are more likely to progress to advanced fibrosis or cirrhosis.
In our model, we show that Prevotella copri was the predominant species predictive of advanced fibrosis. This was also true in our validation cohort as well. While Prevotella copri is still present in normal healthy controls, it is significantly higher in patients with advanced fibrosis, a trend that is consistent across all etiologies of chronic liver disease. This is similar to Qin et al. who showed that Prevotella was enriched in patients with cirrhosis as compared to healthy controls16. Prevotella copri is of great interest as it has been extensively studied in other inflammatory diseases34. It encodes a unique superoxide reductase which may provide resistance to or even the use of host-derived reactive oxygen species produced during inflammation35. Mice colonized with P. copri have increased inflammation in a colitis model induced by dextran sulfate sodium36. In vitro models have shown that P. copri can stimulate IL-6, IL-23, and IL-17, all cytokines associated with pro-inflammatory Th17 responses37. This has led many to believe that P. copri is a potential driver of inflammation and can even induce such inflammatory diseases as rheumatoid arthritis34. In a recent publication, Prevotella copri was also seen as the main bacteria associated with advanced fibrosis in NAFLD pediatric patients38. Our analysis also shows a distinct bacterial metagenomic profile for patients with advanced fibrosis. In our analysis, we show that the pathways that were most different between patients with advanced fibrosis compared to those without were related to mineral absorption, arachidonic acid metabolism, carbohydrate digestion and absorption, and linoleic acid metabolism. In mouse models of liver steatosis, linoleic acid was shown to be protective against inflammation by affecting PPAR-α and NF-κβ signaling39. The observed associations of P. copri and these functional pathways with advanced fibrosis provide preliminary evidence that the gut microbiome may contribute to the progression of liver fibrosis. Therefore, it can be both a useful non-invasive biomarker as well as a potential target for future interventions.
We acknowledge that there were several limitations to this study. For example, we relied on FibroScan rather than liver histology to make the diagnosis of hepatic fibrosis. With the wide adoption of non-invasive testing for fibrosis, the use of liver biopsy is becoming less frequent. However, FibroScan is becoming a more widely accepted and accurate method for detecting the presence of hepatic fibrosis40. While other papers have mentioned that obesity might be a limitation of FibroScan, our facility and technicians had access to and were familiar with the XL probe, which has been proven to have improved diagnostics in obese patients40. Another limitation is that this is a single center VA study and so the generalizability of this study to other settings is still uncertain. While the multivariate analyses did not control for all factors that could affect the microbiome, including diet and medications, the corroboration of our findings in a separate validation cohort that accounted for diet strengthens the findings of our study. Furthermore, while we did attempt to represent a wide array of chronic liver disease, the majority of our patients had chronic HCV or NAFLD. A complete representation of all etiologies of chronic liver disease was unable to be accomplished due to the rarity of less common etiologies including autoimmune disease, Wilson’s disease, hemochromatosis, PSC, PBC, and alpha-1 antitrypsin deficiency. Therefore, future studies will be needed in order to confirm that these findings apply to other chronic liver disease etiologies. Because this study is cross-sectional, it is unable to establish causality between the gut microbiome and hepatic fibrosis. Planned future studies will include the use of fecal metabolomics to examine the differential pattern of microbial derived metabolites in patients with advanced fibrosis and the use of animal models with microbial transplant or single bacteria gavage to understand the causal relationship between the gut microbiome and hepatic fibrosis.
In conclusion, there is a distinct microbial signature for patients with advanced fibrosis independent of liver disease etiology and other comorbidities. These results suggest that microbial profiles can be used as a non-invasive marker for advanced fibrosis and support the hypothesis that microbes and their metabolites contribute to hepatic fibrosis. Future studies should focus on the mechanism by which these microbial differences may contribute to the progression of fibrosis and if the models presented here are valid in other clinical subgroups.
References
Byass, P. The global burden of liver disease: a challenge for methods and for public health. BMC Med. 12, 159 (2014).
Tapper, E. B. & Parikh, N. D. Mortality due to cirrhosis and liver cancer in the United States, 1999-2016: observational study. BMJ 362, k2817 (2018).
Lynch, S. V. & Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 375, 2369–2379 (2016).
Sender, R., Fuchs, S. & Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 14, e1002533 (2016).
Woodhouse, C. A., Patel, V. C., Singanayagam, A. & Shawcross, D. L. Review article: the gut microbiome as a therapeutic target in the pathogenesis and treatment of chronic liver disease. Aliment. Pharmacol. Ther. 47, 192–202 (2018).
Chen, J., Thomsen, M. & Vitetta, L. Interaction of gut microbiota with dysregulation of bile acids in the pathogenesis of nonalcoholic fatty liver disease and potential therapeutic implications of probiotics. J. Cell. Biochem. 120, 2713–2720 (2019).
Chu, H., Williams, B. & Schnabl, B. Gut microbiota, fatty liver disease, and hepatocellular carcinoma. Liver Res. 2, 43–51 (2018).
Baffy, G. Potential mechanisms linking gut microbiota and portal hypertension. Liver Int. 39, 598–609 (2019).
Teschke, R. Alcoholic Liver Disease: Alcohol Metabolism, Cascade of Molecular Mechanisms, Cellular Targets, and Clinical Aspects. Biomedicines 6 (2018).
Hov, J. R. & Karlsen, T. H. The Microbiome in Primary Sclerosing Cholangitis: Current Evidence and Potential Concepts. Semin. Liver Dis. 37, 314–331 (2017).
Angulo, P., Machado, M. V. & Diehl, A. M. Fibrosis in nonalcoholic Fatty liver disease: mechanisms and clinical implications. Semin. Liver Dis. 35, 132–45 (2015).
Dulai, P. S. et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 65, 1557–1565 (2017).
Ekstedt, M. et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 61, 1547–54 (2015).
Decaris, M. L. et al. Identifying nonalcoholic fatty liver disease patients with active fibrosis by measuring extracellular matrix remodeling rates in tissue and blood. Hepatology 65, 78–88 (2017).
Kim, R. G. et al. Magnetic resonance elastography identifies fibrosis in adults with alpha-1 antitrypsin deficiency liver disease: a prospective study. Aliment. Pharmacol. Ther. 44, 287–99 (2016).
Qin, N. et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 513, 59–64 (2014).
Loomba, R. et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 25, 1054–1062.e5 (2017).
Caussy, C. et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 10, 1406 (2019).
Quan, H. et al. Updating and validating the Charlson comorbidity index and score for risk adjustment in hospital discharge abstracts using data from 6 countries. Am. J. Epidemiol. 173, 676–82 (2011).
Bonder, A. & Afdhal, N. Utilization of FibroScan in clinical practice. Curr. Gastroenterol. Rep. 16, 372 (2014).
Tong, M., Jacobs, J. P., McHardy, I. H. & Braun, J. Sampling of intestinal microbiota and targeted amplification of bacterial 16S rRNA genes for microbial ecologic analysis. Curr. Protoc. Immunol. 107, 7.41.1–11 (2014).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–6 (2010).
Preheim, S. P. et al. Computational methods for high-throughput comparative analyses of natural microbial communities. Methods Enzymol. 531, 353–70 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Breiman, L. Random Forests. Mach. Learn. 45, 5–32 (2001).
Langille, M. G. I. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–21 (2013).
Millen, A. E., Midthune, D., Thompson, F. E., Kipnis, V. & Subar, A. F. The National Cancer Institute diet history questionnaire: validation of pyramid food servings. Am. J. Epidemiol. 163, 279–88 (2006).
Zaneveld, J. R., McMinds, R. & Vega Thurber, R. Stress and stability: applying the Anna Karenina principle to animal microbiomes. Nat. Microbiol. 2, 17121 (2017).
Aly, A. M., Adel, A., El-Gendy, A. O., Essam, T. M. & Aziz, R. K. Gut microbiome alterations in patients with stage 4 hepatitis C. Gut Pathog. 8, 42 (2016).
Boursier, J. et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63, 764–75 (2016).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–7 (2012).
Wu, G. D. et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–8 (2011).
Blond, E. et al. EASL-EASD-EASO clinical practice guidelines for the management of non-alcoholic fatty liver disease in severely obese people: do they lead to over-referral? Diabetologia 60, 1218–1222 (2017).
Larsen, J. M. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology 151, 363–374 (2017).
Scher, J. U. et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2, e01202 (2013).
Elinav, E. et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–57 (2011).
Maeda, Y. et al. Dysbiosis Contributes to Arthritis Development via Activation of Autoreactive T Cells in the Intestine. Arthritis Rheumatol. (Hoboken, N.J.) 68, 2646–2661 (2016).
Schwimmer, J. B. et al. Microbiome Signatures Associated With Steatohepatitis and Moderate to Severe Fibrosis in Children With Nonalcoholic Fatty Liver Disease. Gastroenterology 157, 1109–1122 (2019).
Tapia, G. et al. N-3 long-chain PUFA supplementation prevents high fat diet induced mouse liver steatosis and inflammation in relation to PPAR-α upregulation and NF-κB DNA binding abrogation. Mol. Nutr. Food Res. 58, 1333–41 (2014).
Eddowes, P. J. et al. Accuracy of FibroScan Controlled Attenuation Parameter and Liver Stiffness Measurement in Assessing Steatosis and Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology. https://doi.org/10.1053/j.gastro.2019.01.042 (2019).
Acknowledgements
Grant support from the Barbara and Joel Marcus Fellowship Seed grant (T.D.), NIH T32 DK 07180 (T.D.), and VA Career Development Award IK2CX001717 (J.P.J.).
Author information
Authors and Affiliations
Contributions
T.S.D., J.P., J.J. contributed equally to the design of the study. T.S.D., W.K., V.L., K.L., M.H. were responsible for sample processing. T.S.D., W.K., J.P. and J.J. contributed to the writing and editing of the manuscript. T.S.D. and J.J. were responsible for data analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dong, T.S., Katzka, W., Lagishetty, V. et al. A Microbial Signature Identifies Advanced Fibrosis in Patients with Chronic Liver Disease Mainly Due to NAFLD. Sci Rep 10, 2771 (2020). https://doi.org/10.1038/s41598-020-59535-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-59535-w
This article is cited by
-
Gut microbiome-based machine learning for diagnostic prediction of liver fibrosis and cirrhosis: a systematic review and meta-analysis
BMC Medical Informatics and Decision Making (2023)
-
Frequentmers - a novel way to look at metagenomic next generation sequencing data and an application in detecting liver cirrhosis
BMC Genomics (2023)
-
Characterization of gut microbiome composition in Iranian patients with nonalcoholic fatty liver disease and nonalcoholic steatohepatitis
Scientific Reports (2023)
-
Characteristics of gut microbiota in captive Asian elephants (Elephas maximus) from infant to elderly
Scientific Reports (2023)
-
Efficacy of Biejiajian Pill on Intestinal Microbiota in Patients with Hepatitis B Cirrhosis/Liver Fibrosis: A Randomized Double-Blind Controlled Trial
Chinese Journal of Integrative Medicine (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
