
Abstract
Since the emergence of deadly pathogens and multidrug-resistant bacteria at an alarmingly increased rate, bacteriophages have been developed as a controlling bioagent to prevent the spread of pathogenic bacteria. One of these pathogens, disease-causing Vibrio parahaemolyticus (VPAHPND) which induces acute hepatopancreatic necrosis, is considered one of the deadliest shrimp pathogens, and has recently become resistant to various classes of antibiotics. Here, we discovered a novel vibriophage that specifically targets the vibrio host, VPAHPND. The vibriophage, designated Seahorse, was classified in the family Siphoviridae because of its icosahedral capsid surrounded by head fibers and a non-contractile long tail. Phage Seahorse was able to infect the host in a broad range of pH and temperatures, and it had a relatively short latent period (nearly 30 minutes) in which it produced progeny at 72 particles per cell at the end of its lytic cycle. Upon phage infection, the host nucleoid condensed and became toroidal, similar to the bacterial DNA morphology seen during tetracycline treatment, suggesting that phage Seahorse hijacked host biosynthesis pathways through protein translation. As phage Seahorse genome encodes 48 open reading frames with many hypothetical proteins, this genome could be a potential untapped resource for the discovery of phage-derived therapeutic proteins.
Similar content being viewed by others


Introduction
Vibrio is a genus of motile Gram-negative bacteria that possesses a curved-rod cell shape with a single flagellum. Vibrios are abundant and diverse bacteria that are typically found in marine habitats. The genus Vibrio consists of 14 recognized clades and at least 86 different species1. While some of them are not pathogenic, many can cause serious health effects in both human and aquatic life. Due to the continuously rising ocean temperature, the composition of vibrio in the ocean microbiome has been reported to be higher than usual2,3,4. This vibrio-rich environment might increase the incident of a vibrio outbreak in the near future posing risks to global health2.
Vibrio parahaemolyticus, which is one of the disease-causing Vibrio species, is pathogenic to both humans and marine animals5. Consumption of raw seafoods contaminated with the bacteria can cause acute gastroenteritis5,6. This opportunistic bacterium is also able to infect through an open wound which can lead to sepsis and, in rare cases, subsequent death in immunocompromised patients7,8. Moreover, V. parahaemolyticus that has acquired a plasmid encoding the deadly binary toxins PirAvp/PirBvp is even more virulent9. The V. parahaemolyticus strain harboring the plasmid has been found to cause a newly emerging disease in shrimp, known as acute hepatopancreatic necrosis disease (AHPND)9. Moreover, the AHPND-causing plasmid is also found to be transferable among other vibrios, increasing the chance of the disease spreading regionally and globally10. Unsurprisingly, the spread of AHPND has been reported in many countries, including China, Vietnam, Malaysia, Thailand, Mexico, the Philippines, and South America11,12,13. Because of its efficient transferability, the gross impact of the infection is also a concern. The infection from AHPND-causing V. parahaemolyticus (VPAHPND) in cultured shrimp results in a near 100% mortality rate within a week after the first symptoms appear14. Altogether, VPAHPND has easily become a leading cause in tremendous reduction of shrimp farming yield, which could lend itself to global financial detriments in key shrimp aquaculture industries.
To prevent these detriments, antibiotics are top candidate for control agents because of their ease of use and high accessibility in many countries. However, the heavy misuse of antibiotics undoubtedly contributes to the emergence of multidrug-resistant (MDR) bacteria. This also accelerates the spread of multidrug resistant genes to other bacteria via well documented genetic element transfers within the microbial community15. As a result, over the last decade, MDR-vibrios (MDR-V) have been rapidly emerging worldwide and have been found in the United States, China, India and South East Asian countries including Thailand16,17, with the most recent emergence in Nigeria and Malaysia this past year18,19,20. In particular, MDR-V isolated from cultured animals are now strongly resistant to ampicillin and tetracycline, and moderately resistant to nalidixic acid16,17. Specifically, VPAHPND strains have been reported to be resistant to ampicillin, tetracycline, and erythromycin21,22. Thus, alternative measures are desperately needed in order to control vibrio outbreaks and prevent further antibiotic resistance development.
Bacteriophage therapy has been under a recent spotlight as an alternative therapeutic method that helps minimize the extensive use of antibiotics, delay the emergence of antibiotic resistance, and combat the existing MDR bacteria23. In aquaculture, phage application using various potent lytic phages has been proven successful in preventing vibriosis by V. harveyi, V. alginolyticus, V. coralliilyticus, V. anguillarum, V. cyclitrophicus, V. splendidus as well as V. parahaemolyticus24,25. During the past few years, many phages, such as VP882, Vp1, Vpms1, A3S, VpKK5, and VVP1, have been isolated from natural resources exhibiting potent antibacterial activity against V. parahaemolyticus26. However, due to the high specificity of phage, this renders their use too narrow and thus impractical. There have been recent studies to address this, one of which utilized a phage cocktail comprising three lytic phages simultaneously (VP-1, VP-2, and VP-3) that inactivated V. parahaemolyticus more efficiently than the individual phages alone27. Additionally, a series of reports from Jun JW et al. revealed that pVp-1 showed a bactericidal activity toward MDR-V. parahaemolyticus and a broad-host range against VPAHPND strains obtained from diverse regions. Moreover, its application on VPAHPND-infected penaeid shrimp in a lab-scale tank revealed high effectiveness in both prophylactic and therapeutic aspects indicating the potential method of phage therapy, so there is evidence that phage therapy can overcome the specificity concern28,29,30.
With the ultimate goal of extending the variety of phages targeting VPAHPND and to provide more untapped resources for antimicrobial discovery from phage-encoded products, we report here a novel vibriophage isolated from seawater that was able to kill VPAHPND strain TM. Exploiting fluorescence microscopy techniques based on bacterial cytological profiling (BCP) principle31, we observed a mechanism of pre-killing (MOK) of this vibriophage in which the phage intercepts host protein translation machinery during the period of infection before host cell lysis. Our study suggests the discovery of a possible therapeutic agent derived from the phage that inhibits protein synthesis of this pathogenic bacterium.
Results
Morphological and biological properties of phage Seahorse
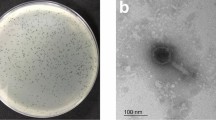
Bacteriophages that target VPAHPND were enriched and isolated from seawater collected from a local shrimp farming area in Thailand. A phage selected for this study actively lysed VPAHPND and produced a 2–3 mm plaque with a 0.5 mm-clear spot at the center surrounding by the halo-turbid area (Fig. 1a). As observed in negative staining by transmission electron microscopy (TEM), the phage belongs to the order Caudovirales and the family Siphoviridae as it has an icosahedral capsid with a long non-contractile tail with short tail fibers (Fig. 1b). In order to visualize the phage at a higher resolution and at a near-native state, we imaged the phage using cryo-electron tomography (cryo-ET). Our cryo-ET images indicated that the phage had a capsid of ~65 nm in diameter and a tail of ~125 nm in length (Fig. 1c–e; n = 3). The phage capsid seemed to be decorated with other proteins which may likely constitute the head fiber similar to those found in Bacillus subtilis phage Phi29 and adenoviruses32,33,34. Our cryo-ET images of the phage also revealed the presence of capsid fibers (the shaft with a knob) of ~10 nm in length at the vertices of the capsid (Purple arrows; Fig. 1c). We also see densities on the capsid facets (~3 nm) which may correspond to minor capsid proteins similar to those found in adenoviruses (Orange arrows; Fig. 1c)34,35. These are believed to enhance capsid stability by forming strong protein-protein interactions. Further biochemical and structural studies are needed to characterize and resolve these protein densities unambiguously. Based on the 3-dimensional phage structure under Cryo-ET (Movie S1), we designated this vibriophage as “Seahorse”.

Morphological and biological properties of phage Seahorse. (a) Plaque morphology of phage Seahorse. An individual plaque is shown in the right panels. Scale bar equals to 2 mm. Morphology of phage Seahorse as determined by Negative staining and TEM (b) and Cryo-ET (c,d). Different slices (c,d) through the same Cryo-ET showing the structure of phage Seahorse. Arrows indicate capsid, capsid fiber, minor capsid protein, tail and tail fibers. Scale bar equals to 50 nm. (e) Schematic of phage Seahorse as visualized by Cryo-ET with the capsid size and the tail length indicated. (f–i) Biological studies of phage Seahorse; Adsorption assay (f), One-step growth curve (g), phage viability in different pH (h) and different temperature (i). The experiments (f–i) were conducted in at least 3 independent biological replicates and the data are represented as the mean ± standard deviation.
To gain more information on the biological properties of phage Seahorse, we tested its host range and conducted a one-step growth curve, a phage tolerance test and measured phage adsorption rate. Out of 26 different bacterial strains tested, phage Seahorse exhibited a narrow host spectrum and specifically infected V. parahaemolyticus strain TM that causes AHPND or VPAHPND (Table 1). A phage adsorption assay revealed that more than 95% of the phage were rapidly adsorbed onto the host cell within 15 minutes (Fig. 1f). The one-step growth curve showed that the phage propagated in the cell during the latent period for at least 30 minutes and resulted in a burst size of 72 virions per cell (Figs. 1g, S1, and Table S2). Additionally, the phage was highly tolerant to a wide range of pH and temperatures (Fig. 1h,i). Figure 1h revealed that the phage was able to infect the host with the highest infectivity at pH 6–7 and the infectivity was found to be completely lost at pH 1–3 (Fig. 1h). A thermal stability study showed that the phage was still active at temperatures between 4 °C–50 °C while the phage pre-treated with high temperatures above 60 °C significantly lost their infectivity (Fig. 1i).
Genome features and annotation of phage Seahorse
The complete genome of Seahorse was 45,171 bp long with a GC content of 42.59% encoding 48 putative open reading frames (ORFs) and 3 tRNA genes (Fig. 2 and Table 2). These predicted ORFs were scattered throughout the phage Seahorse genome with different gene arrangements; 35 ORFs in the forward direction and 13 ORFs in the reverse direction (Fig. 2). Out of the total predicted ORFs, 22 ORFs were assigned a putative function according to the significant hits in the indicated databases with E-values less than 10−4 while the rest were identified as hypothetical proteins (Table 2). Among them, more than 80% of start codons in the ORFs were ATG followed by TTG (14.58%) and CTG (2.08%). We classified the 22 ORFs into 7 main groups according to function; (1) replication, transcription and translation, (2) DNA metabolism and modification, (3) virion structure and assembly, (4) phage regulation, (5) Nin regions, (6) lysis protein, and (7) other phage-related proteins (Fig. 2). In addition, we did not observe any antimicrobial resistance-coding genes or putative toxins from the phage genome.

Genome map of phage Seahorse. The genome size is 45,171 base pairs long and the positions in term of base pairs are indicated by the number on the outermost circle. The grey scale on the innermost circle indicates GC content. The open reading frames (ORFs) are annotated and color-coded by their putative function; blue: DNA replication and transcription, and translation, pink: DNA metabolism and modification, purple: phage regulation, yellow: virion structural and assembly, green: Nin region, red: lysis protein, black: others, and grey: hypothetical proteins. The direction of arrows indicates gene arrangement in the genome.
As shown in Table 2, we found that the majority of predicted ORFs were virion structural proteins including head morphogenesis protein, tail tape measurement protein, and tail tubular protein as well as ATPase and both small and large subunits of the terminase enzyme, which is involved in DNA encapsidation. A phylogenetic tree of large subunits of terminase of phage Seahorse revealed the close relationship to the temperate vibriophage MAR10 (Fig. S2). Phage Seahorse encoded a set of crucial enzymes that are involved in DNA replication and transcription (e.g. DNA helicase, ribonuclease, and transcriptional regulator), and DNA metabolism and modification (e.g. nucleoside triphosphate pyrophosphohydrolase and adenine methylase). Some ORFs were predicted as a transposase which is involved in phage DNA integration into the host genome while others were categorized as High frequency lysogenization C and Rha family proteins, which also serve a role in the regulation of lysogenic life cycle of phages, all of which suggest that phage Seahorse is indeed a temperate phage36,37,38. This annotation was further validated by a lysogeny experiment and a host cell lysis profile, both confirming that phage Seahorse has an ability to lysogenize the host (Fig. S3). Moreover, we also identify N-acetylmuramoyl-L-alanine amidase, an enzyme that degrades the peptidoglycan layer in bacterial cell walls39.
Phage Seahorse infection triggers the condensation of host nucleoid
To investigate how phage Seahorse hijacks and kills the host VPAPHND, a single cell-leveled assay was used to visualize the bacterial cells upon the phage infection. We first focused on a 30-minute post infection (mpi) window because the one-step growth curve suggests that the phage replicates inside the host for only approximately 30 minutes before cell lysis (Fig. 1g). Fluorescence microscopy of Seahorse infected VPAPHND revealed a nonuniformly distributed nucleoid at time zero (0 mpi, lower panel; Fig. 3a), identical to the uninfected VPAPHND control (0 mpi, upper panel; Fig. 3a). Over intervals of 10 mpi, this distributed nucleoid became more condensed as infection progressed until 30 mpi in which it appeared as a single sphere (30 mpi, lower panel; Fig. 3a). During late infection (after 30 mpi), some unlysed infected cells contained multiple nucleoids exhibiting an archetypal toroid shape (upper panel; Fig. 3b). Similar to a previous study in Pseudomonas chlororaphis phage 201Phi2-1, the bacterial host cells lysed at a late time point resulting in the release of phage particles appearing as puncta (blue) surrounding cell debris (red) suggesting the complete lytic cycle of the phage (lower panel; Fig. 3b)40. We observed no evidence of condensed DNA (“blob”) or toroid formation in the control cells (Figs. 3a and S4; n = 1,782).

Single cell-leveled assay revealing the formation of blob and toroid of host DNA inside the phage-infected cells. Bacterial cells were grown in liquid culture to log phase and infected by phage Seahorse at MOI 5. At desired time points, the bacterial cells were harvested and fixed. For live cell imaging, the cells were inoculated on an agar pad after the phage infection. Prior to fluorescence microscopy, cell membrane (red) and nucleoid (blue/grey) were stained by FM4–64 and DAPI, respectively. (a) Fluorescence images of fixed bacterial cells in the presence of mock lysate (upper panel) and phage lysate (lower panel) at various time points. (b) Still images of phage-infected cells during late infection; 45 mpi (upper panel) and 60 mpi (lower panel). (c) Live cell images of phage-infected cells. SYTOX-green as impermeable DNA staining dye was used as an indicator of live cells. (d) Time-lapse imaging of phage-infected cells over the course of 30 minutes. Nucleoid (green) as stained by SYTO 16 condensed and became blob shaped as early as 25 minutes. Dashed lines indicate cell borders. Scale bars equal to 1 micron.
To rule out the possibility that the observed nucleoid condensation is caused by host cell membrane leakage, previously seen in pore-forming molecules such as nisin and calcimycin31, we investigated the membrane integrity of infected cells by testing cell permeability to the DNA staining dye, SYTOX-green. Our result showed that the DNA blob in the infected cells was not stained by SYTOX-green, indicating that the cell membrane remained intact throughout infection, further supporting that phage Seahorse is the cause of the blob DNA formation, not the cell membrane leakage (Fig. 3c).
To confirm that the condensed nucleoid seen in the phage-infected cell is host bacterial DNA, we performed time-lapse fluorescence microscopy over a 30-minute infection period using live cell permeant SYTO 16 DNA dye. At the beginning of infection, the host nucleoid (green) appeared diffuse, similar to the uninfected cell control (Fig. 3d, Movie S2). Over the first 10 minutes, the nucleoid decondensed, seen by the diffusion and reduction of signal within the cell. Beginning at 15 mpi, the DNA nucleoid condensed and eventually appeared in a sphere at the midcell by 30 mpi, identical to the structure seen the infected cell shown in Fig. 3a. This time-lapse observation mirrors the morphological change in the DNA of fixed infected cells conducted over the same time course as shown in Fig. 3a. Altogether, these results suggest that phage Seahorse possibly interferes with the host cellular machineries in which it triggers the morphology change of host DNA.
Inhibition of protein translation resulted in blob and toroid structure formation of the bacterial chromosome
As previously reported by Nonejuie et al.31, fluorescence microscopy-based method, bacterial cytological profiling (BCP), can be used to identify biosynthetic pathways of bacteria that are inhibited based on characteristic cell morphological changes. We therefore applied the principle of BCP to investigate which metabolic pathway of VPAHPND is targeted during the phage Seahorse infection by fluorescence microscopy. Since phages are believed to hijack host DNA replication, RNA transcription, and protein translation pathways during the lytic cycle41, we focused on antibiotics that also inhibit these major pathways. Ciprofloxacin, rifampicin, and tetracycline were used as inhibitors to replication, transcription, and translation respectively. Fluorescence microscopy results of VPAHPND treated with antibiotics showed that each antibiotic treatment led to a unique morphological change in VPAHPND, similar to what previously observed in gram-negative Escherichia coli and Acinetobactor baumannii treatments31,42. Upon ciprofloxacin treatment, inhibition of DNA replication resulted in cell elongation and DNA pooling at the midcell while treatment with rifampicin resulted in DNA decondensation (Fig. 4a). Tetracycline-treated cells exhibited the signature condensed and toroidal-shaped DNA at 30 minutes but more prominently at 60 minutes post treatment with an intact cell membrane (Figs. 4a and S5). This morphology upon tetracycline treatment is notably similar to the condensed DNA morphology of the bacterial cells infected by phage Seahorse (lower panel; Fig. 3a).

Fluorescence microscopy showed unique morphology of bacterial cells during the antibiotic treatment targeting different cellular pathways. Bacterial cells during the log phase were treated with antibiotics (ciprofloxacin, rifampicin, and tetracycline) at 5X MIC for indicated period of time. At desired time points, the cells were fixed and stained with FM4–64 (red) and DAPI (blue) prior to fluorescence microscopy. (a) Fluorescence images of fixed bacterial cells after the treatment with different antibiotics at 30 minutes (upper panel) and 60 minutes (lower panel). (b) Time-course still images of tetracycline-treated bacterial cells. Scale bars equal to 1 micron.
The production of “condensed” DNA upon infection instead of perfect “toroidal” shaped DNA urged us to ask if the condensed DNA morphology is truly the result of protein translation inhibition (lower panel, 10–30 mpi; Fig. 3a). The archetypal DNA shape of protein translation inhibition in many studies is a “toroid”31,42,43, but DNA shape alteration during very early translation inhibition by an antibiotic (less than one hour) has never been reported. Thus, time-course analysis of toroidal DNA formation in VPAHPND during tetracycline treatment was performed. This resulted in a more “condensed” DNA morphology that changed over time with the eventual appearance of the signature “toroid” DNA at 30 minutes post treatment and it became increasingly prominent at 60 minutes (Fig. 4b). Due to the fact that the phage Seahorse had a short latent period and completed its lytic cycle by 30 mpi, it is then reasonable to assume that the toroid DNA morphology was merely undetectable in infected cells because the host cell lysed before toroids were formed. Simply put, if it was able to progress beyond 30 mpi, the nucleoid would have resembled a toroid shape seen in Fig. 4b (45 mpi; Fig. 3b). Altogether, these results suggest that DNA condensation morphology observed in phage Seahorse-infected VPAHPND is likely due to the protein translation inhibition caused by the phage hijacking mechanism.
Discussion
A new strain of V. parahaemolyticus, called VPAPHND, emerged in 2009 as a devastating pathogen to shrimp, causing acute hepatopancreatic necrosis disease (AHPND) or early mortality syndrome (EMS). The infection resulted in mass production losses in southern China and within several years, the incidence of VPAPHND infection had expanded to other neighboring countries such as Vietnam, Malaysia and Thailand11. The disease contributes to 100% mortality of shrimp within one week and the infected animals present an atrophied and discolored hepatopancreas due to the Photorhabdus insect-related (Pir) binary toxins (PirAvp/PirBvp), which are encoded on the virulent plasmid of the pathogen. The toxins degenerate the tubule epithelial cells of the hepatopancreas leaving the diseased animals susceptible to additional bacterial infections44,45.
Due to the emergence of VPAHPND and its multidrug resistant isolates, effective tools to control and combat these pathogens are urgently needed. Most recently, Angulo et al. revealed a number of studies and research reports on using phages as a biocontrol for the wide spread of AHPND26. Until now, even though many phages have been reported to target V. parahaemolyticus, only phage pVp-1 has been found to effectively kill VPAHPND. Phage pVp-1 is highly potent and lyses more than 90% out of the VPAHPND strains that were isolated from Vietnam, Mexico, Costa Rica, Honduras, and Central America Countries29. However, the bacterial strain isolated from Thailand is not targeted by the pVp-1 phage. In this study, we successfully isolated a novel phage (named “Seahorse”) that specifically infects the VPAHPND strain isolated in Thailand. This study also elucidates that the phage is able to hijack protein translation machinery of the host bacteria. In comparison to other phages that infect vibrios (Table S2)46,47,48,49,50,51,52, phage Seahorse has relatively big burst size with a shorter latency period than phages VP-1, VP-2, VP-3, VpKK5, and VhKM4 that replicate in V. parahaemolyticus27,29,53,54,55,56. It is also tolerant to the wide range of pHs and temperatures compared to other vibriophages suggesting unique survivability in harsh natural environments. These infectious characteristics not only add phage Seahorse to the library of phage targeting VPAHPND but also make it a potential candidate for a biocontrol agent.
Bacteriophage requires a bacterial host cell to replicate, manipulating the native biosynthesis machinery of the host to assemble its own progeny. During its reproduction process, various phage-encoding proteins are produced to redirect or inhibit the host metabolism at the molecular level to benefit its fitness. These phage-derived proteins not only target the major metabolic pathways but are also capable of lysing the host cell membrane. Due to nature of these effects, these proteins can be considered antimicrobial agents similar to that of antibiotics with possible therapeutic applications39. Phage Seahorse was classified in the family Siphoviridae due to characteristics of the flexible, noncontractile tail with a non-enveloped icosahedral capsid. In addition, at high resolution near a native-state by Cryo-ET, unique spikes and fibers (a shaft with a sphere at the tip) on the phage capsid were observed. This is not the first time that this structure has been reported32,33,34,35. These are most likely structurally diverse glycoproteins encoded by the phage and thought to promote tethering of the viral capsid to receptors present on the host cell surface. For instance, in the case of human adenoviruses, the fibers have a long shaft with a knob at the distal end of the capsid whereas, in phage Phi29 and some bovine adenoviruses, they consist of only a protruding stem without a terminal sphere32,34,35,57,58. Moreover, phage Sf6 that infects Shigella flexneri has an identical shaft-knob structure at the tail terminus called the “tail needle knob”59. Due to the narrow host spectrum of phage Seahorse, the capsid spikes and fibers located on the surface of the phage capsid serve a potential role in host specificity and recognition.
Unfortunately, in a therapeutic context, phage Seahorse is not appropriate for application due to the ability to enter lysogenic cycle which renders the phage unsafe for treatment. Phage Seahorse appeared phylogenetically related to the temperate phage MAR10 that belongs to the genus Ssp2virus and targets V. parahaemolyticus (Fig. S2)60,61. This reassures our conclusion that phage Seahorse is temperate and also suggests the possible viral family it belongs to. Moreover, due to the lack of bioinformatics in phage related databases, the majority of the ORFs annotated in the phage Seahorse genome were predicted as unknown. As they might produce unwanted products or other unknown virulence factors, the therapeutic application is not warranted unless the fundamental knowledge in phage biology is better established or the phage is engineered to strictly enter only the lytic cycle62.
However, despite the therapeutic shortcomings of phage Seahorse itself, phage genomes generally are considered an untapped resource for antimicrobials due to the metabolic hijacking ability and lytic capacity of the proteins they encode63,64. For example, from our genome analysis, albeit its relatively small genome, we were able to identify a lysis-related enzyme, N-acetylmuramoyl-L-alanine amidase, from ORF45. This lysis enzyme is involved in the cleavage of a very common bond present in most bacterial cell membranes, thus serving a crucial role in cell lysis39. In fact, it has been reported that the application of recombinant phage lysis enzymes from vibriophages can target a wider spectrum of bacterial hosts as compared to the parental phages65,66. Therefore, there is a strong possibility of finding other antibacterial protein candidate from this small phage in the future. Determining whether or not other hypothetical proteins found in the phage Seahorse genome and other newly discovered phage exhibit antibacterial activity needs further investigation.
With the lysis-related enzyme in mind, we set out to investigate whether the phage Seahorse genome encoded other proteins that targeted major host metabolic pathways during its lytic cycle. Bacterial morphological changes have been used as indicators for various physiological states of bacteria elicited by genetic alteration or stress response31,67,68. BCP exploits these cell morphological change patterns under different stresses to identify the specific mechanism being targeted by an antibiotic that causes bacterial growth inhibition. This study, for the first time, applied the principle of BCP technique to identify the underlying effected pathway and found that phage Seahorse likely inhibits protein translation of VPAHPND at the early stage of infection. This finding is concomitant with other reported hijacking mechanisms that use phage-host protein-protein interactions to inhibit host machineries (i.e. replication, transcription and translation) with the effect of disarming host defenses and producing its own proteins for reproduction69. Thus, the hijacking model of host protein machinery in favor of phage protein production right after infection is plausible. For example, Pseudomonas aeruginosa phage PaP3 strongly suppresses host protein synthesis through the reduction of ribosome to preserve energy70. Our finding does not directly indicate that native host replication and transcription are not also inhibited during the infection. To date, BCP has never been applied to study replication, transcription and translation inhibition simultaneously or chronologically. Thus, it is possible that host replication and transcription machineries were inhibited but only the profound protein translation inhibition phenotype was detected. Whether or not a temporal hijacking mechanism is presented during different stages of infection requires further investigation.
Our study revealed the formation of host toroidal nucleoids that are likely the result of encoded phage proteins that hijack host translation machinery before cell lysis, as we called it: “Mechanism of pre-killing (MOK)”. This study suggests that the phage Seahorse genome contained at least one product that was involved in the inhibition towards a protein biosynthesis pathway. Further investigation into which of the phage-derived proteins target important pathways of the bacterial host will be needed in order to identify these antimicrobial proteins for development as therapeutics against pathogens. Localization profiling of phage proteins inside the host, as we previously reported71,72, could also be utilized to study Seahorse-infected cells to gain a better understanding of how individual phage-encoded proteins temporally and spatially function within the host. These investigations could help overcome Seahorse’s therapeutic shortcomings while identifying novel antimicrobial agents at the molecular level.
Material and Methods
Phage isolation, purification, and preparation
Overnight cultures of VPAHPND were prepared by inoculating the bacteria, previously grown on Tryptic Soy Agar supplemented with 1.5% Sodium chloride (TSA-1.5%NaCl), into 5 ml of Tryptic Soy Broth supplemented with 1.5% Sodium chloride (TSB-1.5%NaCl) and allowed to incubate at 30 °C, 200 rpm for at least 16 hours. One milliliter of seawater was added to 25 ml of TSB-1.5%NaCl, 250 μl of 100 mM CaCl2, and 2.5 ml of VPAHPND overnight culture. Phage was enriched by growing at 30 °C, 200 rpm for 48 hours. Phage was next harvested by centrifugation at 9,000 rpm for 10 minutes followed by collecting and filtering the supernatant using a 0.45 μm filter. Next, phage purification was performed using double-layer agar method. Briefly, 10-fold serial dilutions of phage were prepared using SM buffer. Ten microliters of each diluent were added to 100μl of overnight culture of VPAHPND, mixed and allowed to stand for 10 minutes. To this, 5 ml of melted 0.35% top agar of TSA-1.5%NaCl was added, mixed and poured onto a TSA-1.5%NaCl plate. The plates were incubated overnight at 30 °C. Putative translucent plaques were identified and picked and resuspended in 100 μl of SM buffer. This purification step was repeated 3 times. A high-titer phage lysate was prepared described by Chaikeeratisak et al.72. Briefly, 5 ml SM buffer was added to a near-confluent plate and incubated at 30 °C for at least 5 hours. The solution was aspirated into a tube and centrifuge at 9,000 rpm for 5 minutes. Finally, the supernatant was filtered using a 0.45 μm filter and stored at 4 °C.
This work has been reviewed and approved by Chulalongkorn University-Institutional Biosafety Committee (CU-IBC) in accordance with the levels of risk in pathogens and animal toxins listed in the Risk Group of Pathogen and Animal Toxin (2017) published by Department of Medical Sciences (Ministry of Public Health), the Pathogen and Animal Toxin Act (2015) and Biosafety Guidelines for Modern Biotechnology BIOTEC (2016) with approval number: SC CU-IBC-006/2018.
Transmission electron microscopy and cryo-electron tomography
3 μl of phage titer was deposited on QUANTIFOIL 200 mesh holey carbon R 2/1 gold grids, glow-discharged using PELCO easiGlow (Ted Pella). The grids were blotted using Whatman No. one filter paper and plunge-frozen into a liquid ethane/propane mixture cooled by liquid nitrogen using a custom-built device (Max Planck Institute for Biochemistry, Munich). Tilt series were collected on grids clipped onto autogrids (Thermo Scientific) in a 300 keV Titan Krios (Thermo Scientific) fitted with a K2 Summit 4k x 4k pixel direct electron detector and a GIF Quantum post-column energy filter (Gatan) using a nominal magnification of 42 kx or a pixel size of 3.4 Å and −5 μm defocus. Tilt series were acquired using SerialEM in low dose mode, typically from −40o to +40o every 2–3 degrees with a total dose of 50–70 e/Å2. The tilt series were aligned and dose-weighted according to the cumulative dose using MotionCor2 and reconstructed in IMOD software using weighted back-projection.
Conventional phage study
To evaluate phage adsorption, VPAHPND culture (OD600 ~ 0.4) was infected with phage particles at MOI 0.01 (The OD600 of 1 = 1 × 109 CFU/ml73) and incubated at 30 °C. At each time point of 0, 1, 2, 5, 7.5, 10, 15, 20, 25, and 30 minutes, 100 μl of the samples were collected and diluted 10-fold in SM buffer. After centrifugation at 15,000 × g for 2 minutes at 4 °C, the supernatant was harvested and the number of free phages was determined by double-layer agar method. For the one-step growth curve analysis, VPAHPND was infected at MOI 0.01 at 30 °C for 15 minutes, then the cell suspension was centrifuged at 12,000 × g for 5 minutes. The pellet was resuspended in 10 ml of TSB-1.5% NaCl. The mixture was then incubated with vigorous shaking at 200 rpm, 30 °C for 2 hours. Throughout the period of shaking, the samples of the untreated group and the chloroform-treated group were taken every 10 minutes to evaluate total virions by double-layer agar method.
For pH stability, 100 μl of phage lysate was mixed with 900 μl of SM buffer in a pH range 2 to 10 and incubated at 30 °C for 1 hour. For temperature stability, 50 μl of phage lysate was incubated for 1 hour at different temperatures; 4, 20, 25, 30, 37, 40, 50, 60, and 70 °C. For both tests, phage infectivity was determined by performing a spot test. These experiments were performed in triplicate.
To determine host spectrum of the phage, a spot test was performed to test the infectivity of the isolated phage against different 26 bacterial strains that we obtained and were kindly offered from different sources as indicated in Table 1. Briefly, overnight cultures were prepared as described above. 500 μl of each culture was mixed with 5 ml of 0.35% molten top agar (TSA-1.5% NaCl) and immediately poured on an agar plate (TSA-1.5% NaCl). After the cell lawn was solidified, 5 μl of each diluent of 10-fold serially diluted phage was spotted on the surface of the top agar. The plates were allowed to dry and next incubated overnight at 30 °C. The clearing zones were then evaluated for the infection ability of the phage toward the bacterial host.
Phylogenetic tree construction
DNA sequences of the terminase large subunit of various phages were obtained through GenBank. These sequences included accession number and phage names as follows: Vibriophage KVP40: NC_005083, Vibriophage CP-T1: NC_019457.1, Vibriophage pVp-1: NC_019529.1, Vibriophage vB_VpaS_MAR10: NC_019713.1, Vibriophage VH7D: NC_023568.1, Enterobacteria phage 9 g: NC_024146.1, Salmonella phage Stitch: NC_027297.1, Vibriophage phi 3: NC_028895.1, Enterobacteria phage JenK1: NC_029021.1, Vibrio vulnificus phage SSP002: NC_041910, Enterobacteria phage EPS7: NC_010583.1 and phage Seahorse. The sequences were aligned using ClustalW and the phylogenetic tree was constructed using Molecular Evolutionary Genetics Analysis (MEGA) version 10.0 as described by Kumar et al.74. Using the Maximum Likelihood method, a bootstrap consensus phylogenetic tree from 100 bootstrap replications for tree construction was selected. The selected numbers of bootstrap were shown on the selected branches.
Lysogeny experiment
To isolate phage-resistant strains, bacterial colonies that appeared in a double layer-agar plate at high titer of phage were picked and further isolated. The isolated strains and original VPAHPND were tested for phage resistance by cross streaking each isolate with a drop of high titer phage lysate atop the bacterial stripes. The plate was incubated overnight at 30 °C and the result was recorded. To test whether the phage-resistant isolates were lysogen, a bacterial cell lawn of VPAHPND strain was prepared as described above. Single colony of each phage-resistant isolate was picked by a sterile toothpick and stabbed into the top agar. The plate was then incubated at 30 °C overnight and the presence of a clear zone surrounding the stab isolate was recorded the day after.
Bacterial cell lysis profile assay
VPAHPND culture at mid-log phase (OD600 ~ 0.4) was inoculated with phage lysate at MOI 0 as a control and MOI 0.01 and MOI 5 as experimental groups. The cultures were then incubated shaking at 200 RPM at 30 °C. OD600 of all cultures were monitored every 30 minutes until 10 hours of incubation. The experiment was carried out in triplicate.
Phage genome DNA extraction
The phage lysate was first dialyzed in sterile distilled water. Next, phage was precipitated by adding 2.5 ml of phage precipitant solution (30% w/v PEG-8,000, 3.3 M NaCl and sterile distilled water) to 10 ml of phage lysate (~109 pfu/ml) and stored overnight at 4 °C. The solution was then centrifuged at 10,000 rpm for 30 minutes followed by resuspending the pellet in 500 μl of 1xDNase I buffer. To degrade bacterial genomic DNA and RNA, 5U DNaseI and 25 μg RNaseA were added and incubated at 37 °C for 2 hours. Next, 25 mM EDTA was added to inhibit nuclease activity, followed by 0.5% SDS and 25 μg proteinase K, and incubated at 60 °C for 2 hours. Phenol-chloroform extraction was then performed to extract phage genomic DNA.
Phage genome sequencing and analysis
Phage genomic DNA was sequenced by Illumina MiSeq platform. All raw reads were qualified and the low qualities were eliminated. The adaptors in the filtered reads were then trimmed and assembled into contigs. To remove potential host DNA contamination, reads were mapped to the host strain sequence of V. parahaemolyticus strain ATCC17802 (GenBank accessions CP014046, CP014047) using the Geneious mapper in Geneious Prime 2019 (https://www.geneious.com). The unmapped reads were used for assembly in Geneious Prime 2019 with the Geneious assembler using high sensitivity and default parameters. A list of ORFs was generated from this contig using the ORF finder in Geneious; filtering out any ORFs less than 200 base pairs. The protein sequences of each ORFs were predicted by EMBOSS Transeq and they were annotated manually by BLASTp and PSI-BLAST (cut-off e-value < 10−4) against various databases: NCBI’s non-redundant (nr) protein sequences, InterPro 75.0, NCBI conserved domain and ACLAME. In addition, to confirm the predicted function, RAST sever, Prodigal and PHASTER were used as well. To determined antimicrobial resistance coding genes and putative toxins, RESFINDER and VirulenceFinder were used, respectively. Aragon and tRNAScanSE were used to identify tRNAs. The map of genome was drawn by Artemis and DNA plotter.
Single cell-infection assay
VPAHPND culture (OD600 ~ 0.4) was infected with phage at MOI 5 and the infected cells were incubated at 30 °C. At each time point; 0, 10, 20 and 30 minutes, the samples were harvested by centrifugation at 9,000 rpm for 2 minutes and the supernatant discarded. As described by Chaikeeratisak et al.72, phage-infected cells were fixed at a final concentration of 4% paraformaldehyde and incubated at room temperature for 15 minutes. The fixed cells were centrifuged and the pellets were washed with 500 μl of 1x PBS three times. The cells were resuspended in 1x PBS before loading 3 μl onto an agarose pad (1.2% agarose in 20% TSB-1.5% NaCl) that contained fluorescent dyes (2 μg/ml FM 4–64 and 2 μg/ml DAPI). The samples were visualized under DeltaVision Ultra High-Resolution Microscope. For live cells, the cells were harvested at desired time points, and inoculated on an agarose pad as described above. The nucleoid was stained with either 0.5 μM SYTOX-green or 0.5 μM SYTO 16, prior to fluorescence microscopy.
Minimal inhibitory concentration
Minimal inhibitory concentrations (MIC) were determined for the following antibiotics: Ciprofloxacin, Rifampicin, and Tetracycline, which were all used in the fluorescence microscopy experiment shown in Table S1. The antibiotics were respectively serially diluted in a 96 well plate using a microdilution method42. Overnight cultures of VPAHPND were diluted 100-fold in TSB-1.5% NaCl and allowed to grow on a roller at 30 °C until exponential growth (OD600 of 0.2) was observed. The culture was further diluted 100-fold in TSB-1.5% NaCl into wells of the same 96-well plate that contained different concentrations of the respective antibiotic. The culture was further incubated at 30 °C for 24 hours. MICs for each antibiotic were determined as the lowest concentration dilution of that antibiotic capable of inhibiting growth of the bacteria.
Fluorescence microscopy
Overnight cultures of VPAHPND were diluted 100-fold in TSB-1.5% NaCl and incubated at 30 °C on a roller until exponential phase of growth was obtained. Antibiotics were added to the culture at concentrations of 5 times the MIC. For live cell imaging, cultures were incubated at 30 °C on a roller for 60 minutes followed by staining with fluorescent dyes; FM 4–64 (2 μg/ml), DAPI (2 μg/ml) and SYTOX-green (0.5 μM). Cultures were then harvested by centrifugation at 6,000 g for 30 seconds and resuspended in 30 μl of supernatant. 3 μl of sample was loaded onto agarose pad (1.2% agarose containing 20% TSB-1.5% NaCl) on concave glass slides and fluorescence microscopy was performed, following consistent imaging parameters throughout all of the experiments. For fixed cell imaging, cultures were incubated on a roller at 30 °C for 30 and 60 minutes for treatment with ciprofloxacin and rifampicin, while 10, 20, 30 and 60 minutes for treatment with tetracycline. After the completion of each treatment, cultures were fixed as described above. Cultures were then harvested by centrifugation at 9,000 rpm for 2 minutes followed by washing the pellet with 1x PBS for 3 times. After centrifugation, the pellet was resuspended in 30 μl of 1x PBS and added to an agarose pad as described above. Fluorescence microscopy was performed using consistent imaging parameters for all experiments.
Data availability
All data generated or analyzed in this study are included in this article and its supplementary information files. Nucleotide sequence of the phage Seahorse genome was deposited in GenBank database with the accession number MN512538.
References
Sawabe, T. et al. Updating the Vibrio clades defined by multilocus sequence phylogeny: proposal of eight new clades, and the description of Vibrio tritonius sp. nov. Front. Microbiol. 4, 414 (2013).
Vezzulli, L. et al. Climate influence on Vibrio and associated human diseases during the past half-century in the coastal North Atlantic. Proc. Natl. Acad. Sci. 113, E5062 (2016).
Froelich, B., Gonzalez, R., Blackwood, D., Lauer, K. & Noble, R. Decadal monitoring reveals an increase in Vibrio spp. concentrations in the Neuse River Estuary, North Carolina, USA. Plos One 14, e0215254 (2019).
Siboni, N., Balaraju, V., Carney, R., Labbate, M. & Seymour, J. R. Spatiotemporal Dynamics of Vibrio spp. within the Sydney Harbour Estuary. Front. Microbiol. 7, 460 (2016).
Su, Y. C. & Liu, C. Vibrio parahaemolyticus: A concern of seafood safety. Food Microbiol. 24, 549–558 (2007).
Iwamoto, M., Ayers, T., Mahon, B. E. & Swerdlow, D. L. Epidemiology of seafood-associated infections in the United States. Clin. Microbiol. Rev. 23, 399–411 (2010).
Velazquez-Roman, J., León-Sicairos, N., Hernandez-Diaz, L. & Canizalez-Roman, A. Pandemic Vibrio parahaemolyticus O3:K6 on the American continent. Front. Cell. Infect. Microbiol. 3, 110 (2014).
Ghenem, L., Elhadi, N., Alzahrani, F. & Nishibuchi, M. Vibrio Parahaemolyticus: A Review on Distribution, Pathogenesis, Virulence Determinants and Epidemiology. Saudi J. Med. Med. Sci. 5, 93–103 (2017).
Lee, C. T. et al. The opportunistic marine pathogen Vibrio parahaemolyticus becomes virulent by acquiring a plasmid that expresses a deadly toxin. Proc. Natl. Acad. Sci. 112, 10798 (2015).
Restrepo, L. et al. PirVP genes causing AHPND identified in a new Vibrio species (Vibrio punensis) within the commensal Orientalis clade. Sci. Rep. 8, 13080 (2018).
Thitamadee, S. et al. Review of current disease threats for cultivated penaeid shrimp in Asia. Aquaculture 452, 69–87 (2016).
Soto-Rodriguez, S. A., Gomez-Gil, B., Lozano-Olvera, R., Betancourt-Lozano, M. & Morales-Covarrubias, M. S. Field and Experimental Evidence of Vibrio parahaemolyticus as the Causative Agent of Acute Hepatopancreatic Necrosis Disease of Cultured Shrimp Litopenaeus vannamei in Northwestern Mexico. Appl. Environ. Microbiol. 81, 1689 (2015).
Restrepo, L., Bayot, B., Betancourt, I. & Pinzón, A. Draft genome sequence of pathogenic bacteria Vibrio parahaemolyticus strain Ba94C2, associated with acute hepatopancreatic necrosis disease isolate from South America. Genomics. Data 9, 143–144 (2016).
Lai, H. C. et al. Pathogenesis of acute hepatopancreatic necrosis disease (AHPND) in shrimp. Fish Shellfish Immunol. 47, 1006–1014 (2015).
Davies, J. & Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 74, 417–433 (2010).
Elmahdi, S., DaSilva, L. V. & Parveen, S. Antibiotic resistance of Vibrio parahaemolyticus and Vibrio vulnificus in various countries: A review. Food Microbiol. 57, 128–134 (2016).
Yano, Y. et al. Prevalence and antimicrobial susceptibility of Vibrio species related to food safety isolated from shrimp cultured at inland ponds in Thailand. Food Control 38, 30–36 (2014).
Emmanuel, E. O. & Etinosa, O. I. Multi-drug resistant Vibrio species isolated from abattoir effluents in Nigeria. J. Infect. Dev. Ctries. 11, (2017).
Tan, C. W. et al. Prevalence and Antimicrobial Susceptibility of Vibrio parahaemolyticus Isolated from Short Mackerels (Rastrelliger brachysoma) in Malaysia. Front. Microbiol. 8, 1087–1087 (2017).
Odeyemi, O. A. & Ahmad, A. Population dynamics, antibiotics resistance and biofilm formation of Aeromonas and Vibrio species isolated from aquatic sources in Northern Malaysia. Microb. Pathog. 103, 178–185 (2017).
Kongrueng, J. et al. Characterization of Vibrio parahaemolyticus causing acute hepatopancreatic necrosis disease in southern Thailand. J. Fish Dis. 38, 957–966 (2015).
Han, J. E., Mohney, L. L., Tang, K. F. J., Pantoja, C. R. & Lightner, D. V. Plasmid mediated tetracycline resistance of Vibrio parahaemolyticus associated with acute hepatopancreatic necrosis disease (AHPND) in shrimps. Aquac. Rep. 2, 17–21 (2015).
Sulakvelidze, A., Alavidze, Z. & Morris, J. G. Bacteriophage Therapy. Antimicrob. Agents Chemother. 45, 649 (2001).
Doss, J., Culbertson, K., Hahn, D., Camacho, J. & Barekzi, N. A Review of Phage Therapy against Bacterial Pathogens of Aquatic and Terrestrial Organisms. Viruses 9, 50 (2017).
Kalatzis, G. P., Castillo, D., Katharios, P. & Middelboe, M. Bacteriophage Interactions with Marine Pathogenic Vibrios: Implications for Phage Therapy. Antibiotics 7, (2018).
Angulo, C., Loera-Muro, A., Trujillo, E. & Luna-González, A. Control of AHPND by phages: a promising biotechnological approach. Rev. Aquac. 1–16 (2018).
Mateus, L. et al. Efficiency of phage cocktails in the inactivation of Vibrio in aquaculture. Aquaculture 424–425, 167–173 (2014).
Jun, J. W. et al. Bacteriophage Therapy of a Vibrio parahaemolyticus Infection Caused by a Multiple-Antibiotic–Resistant O3:K6 Pandemic Clinical Strain. J. Infect. Dis. 210, 72–78 (2014).
Jun, J. W. et al. Potential application of bacteriophage pVp-1: Agent combating Vibrio parahaemolyticus strains associated with acute hepatopancreatic necrosis disease (AHPND) in shrimp. Aquaculture 457, 100–103 (2016).
Jun, J. W. et al. Phage Application for the Protection from Acute Hepatopancreatic Necrosis Disease (AHPND) in Penaeus vannamei. Indian J. Microbiol. 58, 114–117 (2018).
Nonejuie, P., Burkart, M., Pogliano, K. & Pogliano, J. Bacterial cytological profiling rapidly identifies the cellular pathways targeted by antibacterial molecules. Proc. Natl. Acad. Sci. 110, 16169 (2013).
Xiang, Y. & Rossmann, M. G. Structure of bacteriophage ϕ29 head fibers has a supercoiled triple repeating helix-turn-helix motif. Proc. Natl. Acad. Sci. 108, 4806 (2011).
van Raaij, M. J., Mitraki, A., Lavigne, G. & Cusack, S. A triple β-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature 401, 935–938 (1999).
Cheng, L. et al. Cryo-EM structures of two bovine adenovirus type 3 intermediates. Virology 450–451, 174–181 (2014).
Liu, H. et al. Atomic Structure of Human Adenovirus by Cryo-EM Reveals Interactions Among Protein Networks. Science 329, 1038 (2010).
Kihara, A., Akiyama, Y. & Ito, K. Host regulation of lysogenic decision in bacteriophage lambda: transmembrane modulation of FtsH (HflB), the cII degrading protease, by HflKC (HflA). Proc. Natl. Acad. Sci. USA 94, 5544–5549 (1997).
Noble, J. A. et al. The Escherichia coli hflA locus encodes a putative GTP-binding protein and two membrane proteins, one of which contains a protease-like domain. Proc. Natl. Acad. Sci. USA 90, 10866–10870 (1993).
Dyson, Z. A. et al. Locating and Activating Molecular ‘Time Bombs’: Induction of Mycolata Prophages. Plos One 11, e0159957 (2016).
Borysowski, J., Weber-Dąbrowska, B. & Górski, A. Bacteriophage Endolysins as a Novel Class of Antibacterial Agents. Exp. Biol. Med. 231, 366–377 (2006).
Kraemer, J. A. et al. A Phage Tubulin Assembles Dynamic Filaments by an Atypical Mechanism to Center Viral DNA within the Host Cell. Cell 149, 1488–1499 (2012).
Parmar, K. M., Gaikwad, S. L., Dhakephalkar, P. K., Kothari, R. & Singh, R. P. Intriguing Interaction of Bacteriophage-Host Association: An Understanding in the Era of Omics. Front. Microbiol. 8, 559–559 (2017).
Htoo, H. H. et al. Bacterial Cytological Profiling as a Tool To Study Mechanisms of Action of Antibiotics That Are Active against Acinetobacter baumannii. Antimicrob. Agents Chemother. 63, e02310–18 (2019).
Nonejuie, P. et al. Application of bacterial cytological profiling to crude natural product extracts reveals the antibacterial arsenal of Bacillus subtilis. J. Antibiot. 69, 353–361 (2016).
Boonyawiwat, V. et al. Impact of farm management on expression of early mortality syndrome/acute hepatopancreatic necrosis disease (EMS/AHPND) on penaeid shrimp farms in Thailand. J. Fish Dis. 40, 649–659 (2017).
De Schryver, P., Defoirdt, T. & Sorgeloos, P. Early mortality syndrome outbreaks: a microbial management issue in shrimp farming? Plos Pathog. 10, e1003919–e1003919 (2014).
Li, F. et al. Genomic and biological characterization of the Vibrio alginolyticus-infecting “Podoviridae” bacteriophage, vB_ValP_IME271. Virus Genes 55, 218–226 (2019).
Luo, P. et al. Complete genomic sequence of the Vibrio alginolyticus bacteriophage Vp670 and characterization of the lysis-related genes, cwlQ and holA. BMC Genomics 19, 741 (2018).
Lin, Y. R., Chiu, C. W., Chang, F. Y. & Lin, C. S. Characterization of a new phage, termed ϕA318, which is specific for Vibrio alginolyticus. Arch. Virol. 157, 917–926 (2012).
Kim, S. G. et al. Isolation and characterisation of pVa-21, a giant bacteriophage with anti-biofilm potential against Vibrio alginolyticus. Sci. Rep. 9, 6284 (2019).
Katharios, P., Kalatzis, P. G., Kokkari, C., Sarropoulou, E. & Middelboe, M. Isolation and characterization of a N4-like lytic bacteriophage infecting Vibrio splendidus, a pathogen of fish and bivalves. Plos One 12, e0190083 (2017).
Li, Y. et al. Complete Genomic Sequence of Bacteriophage H188: A Novel Vibrio kanaloae Phage Isolated from Yellow Sea. Curr. Microbiol. 72, 628–633 (2016).
Jacquemot, L. et al. Therapeutic Potential of a New Jumbo Phage That Infects Vibrio coralliilyticus, a Widespread Coral Pathogen. Front. Microbiol. 9, 2501 (2018).
Lal, T. M., Sano, M. & Ransangan, J. Genome characterization of a novel vibriophage VpKK5 (Siphoviridae) specific to fish pathogenic strain of Vibrio parahaemolyticus. J. Basic Microbiol. 56, 872–888 (2016).
Lal, T. M., Sano, M. & Ransangan, J. Isolation and Characterization of Large Marine Bacteriophage (Myoviridae), VhKM4 Infecting Vibrio harveyi. J. Aquat. Anim. Health 29, 26–30 (2017).
Stalin, N. & Srinivasan, P. Characterization of Vibrio parahaemolyticus and its specific phage from shrimp pond in Palk Strait, South East coast of India. Biologicals 44, 526–533 (2016).
Matamp, N. & Bhat, S. G. Genome characterization of novel lytic Myoviridae bacteriophage ϕVP-1 enhances its applicability against MDR-biofilm-forming Vibrio parahaemolyticus. Arch. Virol. https://doi.org/10.1007/s00705-019-04493-6 (2019).
Xu, J., Wang, D., Gui, M. & Xiang, Y. Structural assembly of the tailed bacteriophage ϕ29. Nat. Commun. 10, 2366 (2019).
Yu, X. et al. Cryo-EM structure of human adenovirus D26 reveals the conservation of structural organization among human adenoviruses. Sci. Adv. 3, e1602670 (2017).
Bhardwaj, A., Molineux, I. J., Casjens, S. R. & Cingolani, G. Atomic structure of bacteriophage Sf6 tail needle knob. J. Biol. Chem. 286, 30867–30877 (2011).
Lee, H. S., Choi, S., Shin, H., Lee, J. H. & Choi, S. H. Vibrio vulnificus Bacteriophage SSP002 as a Possible Biocontrol Agent. Appl. Environ. Microbiol. 80, 515 (2014).
Alanis Villa, A., Kropinski, A. M., Abbasifar, R., Abbasifar, A. & Griffiths, M. W. Genome Sequence of Temperate Vibrio parahaemolyticus Bacteriophage vB_VpaS_MAR10. J. Virol. 86, 13851 (2012).
Dedrick, R. M. et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 25, 730–733 (2019).
Liu, J. et al. Antimicrobial drug discovery through bacteriophage genomics. Nat. Biotechnol. 22, 185–191 (2004).
Drulis-Kawa, Z., Majkowska-Skrobek, G. & Maciejewska, B. Bacteriophages and phage-derived proteins - application approaches. Curr. Med. Chem. 22, 1757–1773 (2015).
Wang, W., Li, M., Lin, H., Wang, J. & Mao, X. The Vibrio parahaemolyticus-infecting bacteriophage qdvp001: genome sequence and endolysin with a modular structure. Arch. Virol. 161, 2645–2652 (2016).
Zermeño-Cervantes, L. A., Makarov, R., Lomelí-Ortega, C. O., Martínez-Díaz, S. F. & Cardona-Félix, C. S. Recombinant LysVPMS1 as an endolysin with broad lytic activity against Vibrio parahaemolyticus strains associated to acute hepatopancreatic necrosis disease. Aquac. Res. 49, 1723–1726 (2018).
Ultee, E., Ramijan, K., Dame, R. T., Briegel, A. & Claessen, D. Chapter Two - Stress-induced adaptive morphogenesis in bacteria. In Advances in Microbial Physiology (ed. Poole, R. K.) 74, 97–141 (Academic Press, 2019).
Peters, J. M. et al. A Comprehensive, CRISPR-based Functional Analysis of Essential Genes in Bacteria. Cell 165, 1493–1506 (2016).
Roucourt, B. & Lavigne, R. The role of interactions between phage and bacterial proteins within the infected cell: a diverse and puzzling interactome. Environ. Microbiol. 11, 2789–2805 (2009).
Zhao, X. et al. Global Transcriptomic Analysis of Interactions between Pseudomonas aeruginosa and Bacteriophage PaP3. Sci. Rep. 6, 19237–19237 (2016).
Chaikeeratisak, V. et al. Assembly of a nucleus-like structure during viral replication in bacteria. Science 355, 194 (2017).
Chaikeeratisak, V. et al. The Phage Nucleus and Tubulin Spindle Are Conserved among Large Pseudomonas Phages. Cell Rep. 20, 1563–1571 (2017).
Junprung, W., Supungul, P. & Tassanakajon, A. HSP70 and HSP90 are involved in shrimp Penaeus vannamei tolerance to AHPND-causing strain of Vibrio parahaemolyticus after non-lethal heat shock. Fish Shellfish Immunol. 60, 237–246 (2017).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Klockgether, J. et al. Genome Diversity of Pseudomonas aeruginosa PAO1 Laboratory Strains. J. Bacteriol. 192, 1113 (2010).
Serwer, P. et al. Improved isolation of undersampled bacteriophages: finding of distant terminase genes. Virology 329, 412–424 (2004).
Acknowledgements
This research was supported by the Thailand Research Fund and the Office of the Higher Education Commission MRG6180027 (V.C.), the Grant for Development of New Faculty Staff, Ratchadaphiseksomphot Endowment Fund (V.C.), and National Institutes of Health grant GM129245 (J.P.). This study was supported in part by the Japan Science and Technology Agency (JST)/Japan International Cooperation Agency (JICA), Science and Technology Research Partnership for Sustainable Development, SATREPS JPMJSA1806 (V.C.). P.N. was supported by the Thailand research fund and the office of the higher education commission (MRG6080081). We acknowledge Chulalongkorn University for its support of the Center of Excellence for Molecular Biology and Genomics of Shrimp. We used the UCSD Cryo-Electron Microscopy Facility (supported by NIH grant R01-GM33050 to Dr. Timothy S. Baker and the Agouron Institute) and the fluorescence microscopy facility at the Advanced Cell Imaging Center, Institute of Molecular Biosciences, Mahidol University. We would like to thank John P. Davis and Savannah Senecal for bench-work assistance and helping with intellectual discussion, and Prof. Dr. Sirirat Rengpipat at Department of Microbiology, and Assoc. Prof. Dr. Kunlaya Somboonwiwat at Department of Biochemistry, Faculty of Science, Chulalongkorn University for offering Vibrio strains for host-range determination.
Author information
Authors and Affiliations
Contributions
K.T., M.E.E., H.H.H., K.K., and V.C. conducted experiments, analyzed and interpreted data, and prepared the figures. J.S. and J.F.N. conducted experiments and collected data. K.T., M.E.E., K.K., J.P., P.N., and V.C. contributed to writing the manuscript. V.C. conceived the project and supervised all experiments. K.T., M.E.E., H.H.H., E.V., A.T., J.P., P.N., V.C. edited and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thammatinna, K., Egan, M.E., Htoo, H.H. et al. A novel vibriophage exhibits inhibitory activity against host protein synthesis machinery. Sci Rep 10, 2347 (2020). https://doi.org/10.1038/s41598-020-59396-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-59396-3
This article is cited by
-
Isolation and characterization of a novel phage belonging to a new genus against Vibrio parahaemolyticus
Virology Journal (2023)
-
Combination of genetically diverse Pseudomonas phages enhances the cocktail efficiency against bacteria
Scientific Reports (2023)
-
Nucleus-forming vibriophage cocktail reduces shrimp mortality in the presence of pathogenic bacteria
Scientific Reports (2023)
-
Shrimp disease management for sustainable aquaculture: innovations from nanotechnology and biotechnology
Aquaculture International (2021)
-
Metaviromics coupled with phage-host identification to open the viral ‘black box’
Journal of Microbiology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
