
Abstract
Here we studied HLA blocks and haplotypes in a group of 218 Lacandon Maya Native American using a high-resolution next generation sequencing (NGS) method. We assessed the genetic diversity of HLA class I and class II in this population, and determined the most probable ancestry of Lacandon Maya HLA class I and class II haplotypes. Importantly, this Native American group showed a high degree of both HLA homozygosity and linkage disequilibrium across the HLA region and also lower class II HLA allelic diversity than most previously reported populations (including other Native American groups). Distinctive alleles present in the Lacandon population include HLA-A*24:14 and HLA-B*40:08. Furthermore, in Lacandons we observed a high frequency of haplotypes containing the allele HLA-DRB1*04:11, a relatively frequent allele in comparison with other neighboring indigenous groups. The specific demographic history of the Lacandon population including inbreeding, as well as pathogen selection, may have elevated the frequencies of a small number of HLA class II alleles and DNA blocks. To assess the possible role of different selective pressures in determining Native American HLA diversity, we evaluated the relationship between genetic diversity at HLA-A, HLA-B and HLA-DRB1 and pathogen richness for a global dataset and for Native American populations alone. In keeping with previous studies of such relationships we included distance from Africa as a covariate. After correction for multiple comparisons we did not find any significant relationship between pathogen diversity and HLA genetic diversity (as measured by polymorphism information content) in either our global dataset or the Native American subset of the dataset. We found the expected negative relationship between genetic diversity and distance from Africa in the global dataset, but no relationship between HLA genetic diversity and distance from Africa when Native American populations were considered alone.
Similar content being viewed by others

Introduction
The human major histocompatibility complex (MHC) is located within chromosomal region 6p21.3 and spans at least 3.4 Mb of DNA containing more than 400 genes. Human leukocyte antigen (HLA) loci are mapped within the MHC region as well as other immune related genes and pseudogenes1. The genetic diversity of MHC genes results from selective pressures including functional adaptation to pathogens2,3,4,5,6,7,8 with some peptide-HLA complexes being more effective in eliciting an immune response than others4. It seems likely that individuals with greater diversity at their HLA loci would have a greater chance of survival in pathogen-enriched environments5, but there is debate as to whether simple heterozygote advantage or more complex host-pathogen co-evolutionary processes are responsible for the diversity of human HLA alleles generally9,10,11,12,13,14,15. In addition to their direct role in immune responses against pathogens, HLA molecules are crucial genetic markers to study the genetic diversity of populations in the context of disease susceptibility and allotransplantation3,4,5,16,17,18,19,20.
One of the most important characteristics of the MHC region is a high degree of non-random associations between inherited alleles, known as linkage disequilibrium (LD)21,22. Extensive studies in different populations have described the existence of extended haplotypes or blocks and other relatively fixed genetic fragments within the human MHC17,23. Specific DNA blocks containing two or more MHC loci are often haplospecific for particular conserved extended haplotypes (CEHs)17. The frequency of CEHs and specific block combinations varies between major ancestral groups and/or in different continental populations, and these variations in the frequency of CEHs and blocks can be used as measurements of genetic diversity of the MHC17,18,23. It has further been suggested that a specific form of linkage disequilibrium - non-overlapping combinations between physically separate HLA loci - could be a signal of pathogen selection within HLA system24. Assessing the presence of such non-overlapping associations thus arises as a possibility to assess pathogen driven selection imprinted in the genetic structure of HLA system within populations.
Several studies have described HLA class I and class II alleles and haplotypes in different Native American groups from Mexico25,26,27,28,29,30,31,32 and Latin America33,34,35,36, including Lacandons37. The study of HLA diversity in Native Americans is relevant because evidence suggests that balancing selection at different HLA loci may be involved in the prevalence of inflammatory or infectious diseases in these populations38,39,40,41,42,43. Also, numerous examples of novel alleles have been reported in Native Americans44,45,46, suggesting that pathogen-driven selection of new mutations could be critical in the adaptation to endemic pathogens, particularly after migration4,5.
Previous studies of the Lacandon population of Chiapas State have described the genetic variability of low resolution HLA class II37, blood groups47 and single nucleotide polymorphisms (SNPs) of Cytochrome P450 2D648. Individuals from the Lacandon population were also included in a study of genomic variation across Mexico49. However, no studies have been published about the immunogenetic diversity and the possible ancestral origin of HLA conserved extended haplotypes (CEH) in a representative group of Lacandon Maya.
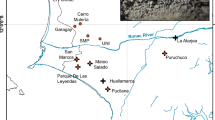
The Lacandon or Hach Winik (“the real people”) are descendants of ancient Mayan civilizations, which have been considered to have reached their cultural apex in Mesoamerica c. AD 80050,51. Their settlements are mainly distributed in the Lacandon Jungle (Fig. 1), which stretches from the State of Chiapas, Mexico, into Guatemala and into the southern part of the Yucatán Peninsula52,53. The historical landscape of Lacandon Maya includes multiple genocides related with Spaniard conquest, land disputes as well as novel infections such as smallpox, yellow fever and influenza, and a high degree of inbreeding that has resulted in high prevalence of certain genetic conditions, including albinism51,54,55.

Locations where the samples were obtained in Chiapas, Mexico. The communities where the samples were taken are represented by a star.
The aim of this study is to analyze the immunogenetic diversity and the ancestral origin of HLA CEH in Lacandon Maya using next generation high-resolution HLA typing methods, and to use such information to assess if there are signatures of evolutionary forces modifying the expected diversity within the HLA system. We also assess whether a correlation exists between the genetic diversity (as measured by the polymorphism informative content, PIC) at class I (HLA-A, -B) and class II (-DRB1) loci in Native Americans (including Lacandon Mayans) and a) pathogen and viral richness, and b) the geographic distance from Africa.
Results
HLA allele and haplotype frequencies within the Lacandon population
HLA allele frequencies were obtained by analyzing 458 haplotypes from 229 non-first degree related Lacandon Maya individuals genotyped by next generation sequencing (NGS) technologies. Complete haplotypes could be obtained for 218 individuals for a total of 436 haplotypes. The most frequent class I haplotypes, all with haplotypic frequencies (H.F.) higher than 5% were HLA-A*31:01~B*40:02~C*03:04 [Native American Most probable ancestry (MPA)], A*02:06~B*35:01~C*07:02 (Native American MPA), A*68:03~B*35:01~C*07:02 (Not previously reported), A*24:02~B*35:12~C*04:01 (Native American MPA), A*68:01~B*40:08~C*03:04 (previous reports only include mixed-ancestry populations), A*68:03~B*39:05~C*07:02 (previous reports only include mixed-ancestry populations), and A*68:03~B*39:05~C*07:02 (previous reports only include mixed-ancestry populations). Only two HLA-A~B~C~DRB1~DRB3/4/5~DQB1~DQA1~DPA1~DPB1 haplotypes exhibited frequencies greater that 5%: A*31:01~B*40:02~C*03:04-DRB1*04:11~DRB4*01:03~DQB1*03:02~DQA1*03:01~DPA1*01:03~DPB1*04:02 (H.F. = 0.0633) and A*31:01~B*40:02~C*03:04~DRB1*04:11~DRB4*01:01~DQB1*03:02~DQA1*03:01~DPA1*01:03~DPB1*04:02 (H.F. = 0.0502). These two high frequency haplotypes differ only in their HLA-DRB4 allele. The complete frequencies of HLA-A, -B, -C, -DRB1, -DRB3/4/5, -DQA1, -DQB1, -DPA1, and -DPB1 alleles in Lacandon Maya individuals are detailed in Table 1 (class I) and Table 2 (class II). HLA-B~C blocks present in Lacandon Maya are listed in Table 3, while the extension of the HLA-B~C block to the HLA-A gene is provided in Supplementary Table 2. HLA class II haplotypic diversity (HLA-DRB1~DRB3/4/5~DQA1~DQB1~DPA1~DPB1) is listed in Table 4. The CEHs in Lacandons are listed in Table 5 and their extension to HLA-A is shown in Table 6. Table 7 shows the complete class I/class II haplotypes for our Lacandon Mayans sample. Aggregate block frequency (ABF) for each ancestry can be found in Tables 3 to 6. The calculation of ABF is a previously reported and validated approach to estimate the diversity and contribution of precisely described HLA blocks of specific ancestries17,56.
Diversity of Lacandon HLA alleles
Forensic parameters of genetic diversity were calculated to assess HLA diversity in Lacandon using polymorphism information content (PIC), power of discrimination (PD), and Hardy-Weinberg equilibrium (HWE) (Table 8). In this regard, HLA-A, HLA-B and HLA-DRB1 were the most polymorphic loci with PIC values of 0.8444, 0.8227 and 0.7555 respectively; whereas HLA-DPB1 and HLA-DPA1 were the less diverse HLA loci with PIC values of 0.3547 and 0.1581, respectively. A significantly (p < 0.05) lower observed heterozygosity (OH) than expected heterozygosity (EH) was observed for HLA-B, HLA-C, HLA-DRB1, HLA-DRB3/4/5, HLA-DQA1, and HLA-DQB1 loci. In contrast, HLA-A locus exhibited a higher OH than EH value (p < 0.0001).
Genetic similarities with other populations
A PCA plot and a population phylogenetic tree were constructed using 180 populations (including the Lacandon group studied in this work) with HLA-A, HLA-B and HLA-DRB1 data from a worldwide population dataset. Figure 2 and Supplementary Fig. 1 illustrate the results of the Principal Components Analysis (PCA). The Lacandon Mayans (purple star) cluster together with other Mexican Native American and Mexican Admixed populations, including Mayans from Guatemala. In addition, a Neighbor-Joining (NJ) analysis (Fig. 3) revealed that Lacandon Mayans are more closely related to Mixe, Mixtec and Zapotec Mexican Native American populations, which are geographically speaking the closest ones to the Lacandon Mayans. It is interesting to note that using HLA genes as genetic estimators, it is possible to mimic the results (although not to the same resolution) obtained with genome-wide data49.

Principal Component Analysis for 163 populations (including the Lacandon group studied in this work). European populations are represented by green dots; African human groups correspond to yellow dots; red dots were assigned to Asian populations; Native American populations are represented by purple dots; Populations from Oceania are indicated with pink dots. Our Lacandon sample is represented by a purple star. For a view of the complete PCA, please refer to Supplementary Fig. 1. Argentina Gran Chaco Eastern Toba (n = 135) ArE; Argentina Gran Chaco Mataco Wichi (n = 49) ArM; Argentina Gran Chaco Western Toba Pilaga (n = 19) ArP; Argentina Rosario Toba (n = 86) ArT; Bolivia Aymara (n = 102) BoA; Bolivia Quechua (n = 80) BoQ; Brazil Ivaí Kaingang (n = 127) BrI; Brazil Río das cobras Guaraní (n = 98) BrG; Brazil Río das cobras Kaingang (n = 113) BrR; Brazil Terena (n = 60) BrT; Guatemala Maya (n = 132) GtM; Mexico Tarahumara (n = 44) MTa; Mexico Teenek (n = 55) MTe; Mexico Mayos (n = 60) MMa; Mexico Mazatecan (n = 89) MMz; Mexico Mixe (n = 55) MMx; Mexico Mixtec (n = 103) MMi; Mexico Zapotec (n = 90) MZa; Mexico Nahuas (n = 85) MNa; Mexico Seri (n = 34) MSe; Paraguay Guarani (n = 40) Py; Peru Lama (n = 83) PeL; Peru Titikaka Lake Uro (n = 105) PeU; USA Gila River (n = 492) USG; USA Sioux (n = 302) USS; USA Yupik (n = 252) USY; Venezuela Yucpa (n = 73) VeY. The complete list of abbreviations is included in Supplementary Table 1. Component 1 bears 35.23% of the total variance, while component 2 is explained by 16.62% of the variance. For the zoom-in of this PCA, please see Supplementary Fig. 1.

Zoom-in to the Native American cluster of the population phylogenetic tree built with 114 human groups (including Lacandon Mayan from this work). European populations are represented by green branches; African human groups correspond to yellow branches; red branches were assigned to Asian populations; Native American populations are represented by purple branches; populations from Oceania are indicated with pink branches. Our Lacandon sample is highlighted with a purple star. For a view of the complete tree, please refer to Supplementary Fig. 2. Argentina Gran Chaco Eastern Toba (n = 135) ArE; Argentina Gran Chaco Mataco Wichi (n = 49) ArM; Argentina Gran Chaco Western Toba Pilaga (n = 19) ArP; Argentina Rosario Toba (n = 86) ArT; Brazil Terena (n = 60) BrT; Mexico Lacandon (n = 228) Lac; Mexico Mixe (n = 55) MMx; Mexico Mixtec (n = 103) MMi; Mexico Seri (n = 34) MSe; Mexico Tarahumara (n = 44) MTa; Mexico Zapotec (n = 90) MZa; Paraguay Guarani (n = 40) Py; Peru Titikaka Lake Uro (n = 105) PeU; USA Gila River (n = 492) USG; USA Sioux (n = 302) USS; USA Yupik (n = 252) USY; Venezuela Yucpa (n = 73) VeY. References for all populations of this dataset are listed in Supplementary Table 1. For the zoom-out of this phylogenetic tree, please see Supplementary Fig. 2.
Non-overlapping associations between HLA alleles
Plots of the frequencies of all possible HLA-A~B, HLA-B~C and HLA-B~DRB1 allele combinations are shown in Fig. 4, and plots for all HLA class I and class II associations can be found in Supplementary Fig. 3. Visual inspection suggests a high degree of non-overlap between HLA-B and -C in particular. Figure 5 displays the \({f}_{adj}^{\ast }\) metric (a parameter used to rank the strength of non-overlapping associations between different pairs of HLA loci) for all possible pairwise combinations of HLA loci with HLA-A (4a), with HLA-B (4b) and with HLA-DRB1 (4c), showing also the distribution of \({f}_{adj}^{\ast }\) values obtained when the alleles at the relevant loci are randomized (retaining their population frequencies within the dataset). The heatmap in Fig. 4 (4d) illustrates how many standard deviations above the mean randomized \({f}_{adj}^{\ast }\) value the actual Lacandon \({f}_{adj}^{\ast }\) value is for each indicated pair of loci. HLA-B and -C exhibit the highest degree of non-overlap by this measure, followed by HLA-A and -B, HLA-DRB1 and HLA-DRB3/4/5, HLA-A and -C, and HLA-DRB3/4/5 and HLA-DQA1. All \({f}_{adj}^{\ast }\) values for each pair of HLA loci in the dataset are displayed in Supplementary Table 4.

HLA associations in the Lacandon Maya sample from SE Mexico. (A) HLA-A~B associations, (B) HLA-B~C associations and (C) HLA-B~DRB1 associations.

HLA \({f}_{adj}^{\ast }\) scores. The red crosses in panels (A–C) illustrate non-overlap measured using \({f}_{adj}^{\ast }\) for (A) HLA-A and all other HLA loci in the Lacandon dataset; (B) HLA-B and all other HLA loci in the dataset and (C) HLA-DQB1 and all other HLA loci in the dataset. For each pair of loci we also generated 5000 different random combinations of the alleles found at each locus, and calculated \({f}_{adj}^{\ast }\) scores obtained from those randomized combinations. The \({f}_{adj}^{\ast }\) scores from the random combinations are shown as blue markers. These individual markers overlay one another at the scale used here, but illustrate the distribution of randomized scores. The heatmap in panel (D) indicates by how many standard deviations of the randomized \({f}_{adj}^{\ast }\) score distribution the actual \({f}_{adj}^{\ast }\) for a pair of loci in the Lacandon dataset exceeds the mean of the randomized \({f}_{adj}^{\ast }\) distribution for that pair.
Assessment of the correlation between pathogen richness and HLA diversity
We calculated PIC values for 122 populations as an estimator of genetic diversity for HLA-A, HLA-B and HLA- DRB1 high resolution data (see Supplementary Table 1). We extracted pathogen and viral richness data from the GIDEON database57. We first tested the correlation between genetic diversity for each gene and geographic distance from Africa. Distance was calculated from East Africa to the location of each sample set analyzed. The general outlooks for (a) pathogen richness; (b) viral richness; (c) HLA class I (represented by its most polymorphic gene, HLA-B) genetic diversity; and (d) HLA class II (represented by its most polymorphic gene, HLA-DRB1) genetic diversity are shown in Fig. 6. We ran linear regressions for the PIC values for all three HLA loci vs. the geographic distance from Africa (Supplementary Fig. 4). We found a similar tendency for the three genes analyzed: a general decrease in diversity when distance from Africa increases (HLA-A: r2 = 0.5444; HLA-B: r2 = 0.2787; HLA-DRB1: r2 = 0.3352; all these r2 values correspond to regressions ran after removing outliers). We then ran general linear models including both distance from Africa and either pathogen richness (Supplementary Table 3) or viral richness (Supplementary Table 4) as predictor variables. No relationships were apparent between HLA-A or HLA-B diversity and either pathogen or viral richness. The 95% confidence interval for the gradient of the relationship between HLA-DRB1 diversity and both pathogen and viral richness suggests a negative relationship (Supplementary Tables 3 and 4), but these relationships do not retain significance following a Bonferroni correction. Furthermore, only the negative relationship between viral richness and HLA-DRB1 diversity in the non-Native American populations retains a 95% confidence interval < 0 in the analysis excluding outliers.

Worldwide distribution of pathogen and viral richness and genetic diversity for HLA. (A) Pathogen richness; (B) Viral richness; (C) HLA-B genetic diversity as estimated with polymorphism informative content (PIC) as a proxy for HLA class I diversity; HLA-DRB1 genetic diversity as estimated with PIC as a proxy for HLA class II diversity.
Discussion
In this work, we used next generation sequencing to carry out high resolution typing of the HLA-A to HLA-DPB1 loci in a group of Lacandon Maya settled in the Lacandon Rainforest in the lowlands of Chiapas State in the southeast of Mexico. We determined the distribution of HLA alleles and CEHs and their possible ancestral origin, and assessed genetic diversity within the classical HLA genes. We also put together this Lacandon Maya population with other populations, both Native American and non-Native American, to asses not only genetic relationships but also their general tendency when correlating the genetic diversity of these populations with the geographic distance from Africa and both pathogen and viral richness.
The PCA plot (Fig. 2) showed that the Lacandon population is genetically similar to other Native American populations, such as Mayos28, Teenek58, Seri25, Maya30, Wayu33, and Quechua34. When we performed a phylogenetic analysis of the relationship between the Lacandon and other North and South American Native populations, we found that the Lacandon belonged to the same clade as Native North Americans from Oaxaca (Mixe, Mixtec and Zapotecs)29 and the very divergent Yucpa from Venezuela35. However, these analyses were performed using allelic frequencies, not haplotypic data. When it comes to HLA, allelic diversity in Mesoamerican-descent groups tends to group together most of the Native North American populations. Haplotypic diversity can distinguish finer scale relationships among Native American populations, reflecting that although a limited allelic diversity came into the continent when the first human settlers arrived, recombination played an important role in adaptation to new environments and population differentiation. This is exemplified by the fact that four out of the top ten CEH in the Lacandon were not previously reported (accounting for 30.58% of the total CEHs), although all four haplotypes contained alleles for both class I and class II that are commonly present in other Native American populations.
As it has been shown before49, Lacandon Mayans and other Mayan groups have a genetic component very distinct from other populations, even from those in the same (or at least related) linguistic families, or neighboring populations. This uniqueness can also be detected in the HLA genomic region. Distinctive alleles present in the Lacandon population include A*24:14 (A.F. = 0.0502; also found in the Mayan population from Guatemala with an A.F. = 0.004030, Buryat from Mongolia with an A.F. = 0.004059, and Saudi Arabia with and A.F. = 0.003260) and B*40:08 [A.F. = 0.0611; also found in Texas Hispanic from USA with an A.F. = 0.0170 (data collected by Cathi Murphey, available in61)], Polynesian from American Samoa with an A.F. = 0.010062 and northern India with an A.F. = 0.010063. Furthermore, we observed a high frequency of haplotypes containing the allele HLA-DRB1*04:11 in Lacandons, an allele that was found with a frequency of 0.3690, which is the highest frequency when compared to other neighboring indigenous groups29,30. Other authors37 have reported class II HLA alleles in the Lacandon (N = 162), and found an even higher frequency of HLA-DRB1*04:11 (Our study 0.3690 vs. ref. 37 0.5740, p = 0.0002). Only two out of nine HLA class II blocks present in previous reports37 and in our study were found to be at different frequencies: HLA-DRB1*04:11~DQB1*03:02 (0.3688 vs. 0.5670, p = 0.0001) and HLA-DRB1*04:07~DQB1*03:02 (0.2398 vs. 0.0960, p = 0.0003). Several studies have suggested that HLA-DRB1*04:11 allele is of Native American MPA and is frequent in several Native North and South American populations including Tarahumaras from Mexico, Wayúu from Venezuela, Mataco-Wichi, Toba Pilaga and Eastern Toba from Argentina, and Cayapa from Ecuador26,33,45,61,64,65. Recent reports have associated the HLA-DRB1*04:11 allele with an increased susceptibility to pulmonary tuberculosis in Amazon Brazilian population66. One SNP position in LD with non-DRB3, non-DRB5 haplotypes (i.e. the ones not linked with HLA-DRB1*04 allelic group, for instance) has been reported to be associated with a positive tuberculosis test in a recent study using genome-wide data paired with fine-mapping of the HLA region67. Although the significance is marginal, this finding further supports the involvement of the region in the susceptibility of tuberculosis. Also, the HLA-DRB1*04 allelic group alleles, highly prevalent in Lacandon Mayans, have been suggested to have a protective effect against hepatitis B virus infection68.
A high frequency of “rare alleles or haplotypes” may be present due to genetic isolation and small population number, both of which increase the effects of genetic drift. Class I (ABF: 0.4847), class II (ABF: 0.1659) and CEH (ABF: 0.5786) haplotypes previously unreported in Native American population account for an important part of haplotypic diversity in the Lacandon population, and they point to a distinctive Native American root that was overlooked until recent times49. For instance, haplotype HLA-A*31:01~B*40:02~C*03:04 (H.F: = 0.1310) was previously reported only in East Asian populations such as Japanese69,70, Chinese71 and Malaysia Peninsular Chinese (data collected by Sulaiman Salsabil, available in61), but it was also reported in mixed-ancestry populations such as Mexico City72 and “Hispanics” from USA73. Its frequency in Lacandon Maya is the highest frequency ever reported for this haplotype which would be indicative of a Native American MPA haplotype that can be traced back to its original East Asian ancestry thousands of years ago. HLA-B*35~C*07 associations, although uncommon, have been previously reported in Native North American populations such as Mixtec and Mixe29 but also in some Asian populations such as Rakhine from Myanmar (data collected by Thu ZinZin, available in61). Again, Lacandon Maya exhibit the highest frequency (H.F.: 0.1659) of this haplotype ever reported for any population. The reasons why these associations have never been previously found in Native American human groups include biased sampling procedures, but may also reflect genetic drift or pathogen selection having led to the extinction of specific haplotypes in other Native American groups, or the elevation of specific haplotypes in the Lacandon.
It is important to note that a European haplotype is present as part of the top ten most frequent haplotypes in our sample: A*02:01~B*18:01~C*07:01~DRB1*11:04~DQB1*03:01, with 2.40% (Table 6). That haplotype has been reported in frequencies ranging from 0.91% to 7.32% in populations from eastern and Mediterranean regions from Europe, such as Albania, Macedonia, Greece, Bosnia and Herzegovina, Romania and Italy71,74,75,76,77,78. The only African haplotype present at least twice in our sample was A*68:02~B*53:01~C*04:01~DRB1*13:03~DQB1*02:02 (HF = 0.44%; Table 6). This class I and class II haplotype can only be found in mixed ancestry populations such as Mexicans from Mexico City72 and African Americans73, but the class I block is present in populations from sub Saharan Africa such as the Bandiagara from Mali79, the Nandi and Luo from Kenya79,80 and the multicultural Worcester region in South Africa81. These two examples of non-Native American haplotypes give account of admixture events consistent with the ancestries brought into the Americas by conquerors during the conquest wars period and the colonial times82,83, even in what is considered to be one of the most isolated Native American human groups52,53.
HLA molecules have an important biological role as Killer cell Immunoglobulin-like Receptor (KIR) ligands. KIR2DL1, KIR2DL2/3 and KIR3DL1 bind HLA-C2, -C1 and -Bw4 ligands respectively, resulting in inhibition of natural killer (NK) cell-mediated cytolysis. C2, C1 and Bw4 are all found on HLA class I molecules: C1 and C2 are exclusive to HLA-C, while Bw4 can be found on some HLA-B and some HLA-A molecules. Several physiological functions of Natural Killer (NK) cells in human immunity and reproduction depend upon diverse interactions between KIRs and their HLA class I ligands84,85,86,87. In most populations, HLA-Bw4 alleles (i.e. those carrying asparagine, aspartic acid or serine in amino acid residue 77 and isoleucine or threonine in amino acid residue 80) are present in nearly 50% of the haplotypes, which means that around 75% of the individuals of any given population should express a ligand for the KIR3DL1 receptor88,89. Only 0.0503 of the HLA-B alleles present in Lacandon Maya are HLA-Bw4 alleles, and most of them (B*44:02, B*44:03, B*49:01, and B*13:02) are of European MPA. Nonetheless, Lacandon Maya have a relatively high frequency (A.F.: 0.0568) of the Bw4+ HLA-A allotype A*24:14, which is known to inhibit and to educate NK cells through interaction with KIR3DL189,90. In our Lacandon sample, therefore, approximately 10.7% of haplotypes carry a Bw4+ ligand. HLA-C2 alleles are also inhibitory KIR ligands91; HLA-C2 frequencies in humans range from <10% in certain East Asian populations to ~60% in Oceania native human groups and some African populations. In our sample, the C2 ligand is present on 0.2294 of the HLA-C alleles (Table 1). This level of HLA-C2 is consistent with other Native American populations in which HLA-C2 alleles are present in AF < 0.3 such as Barí (0.075) and Yucpa (0.214) from Venezuela’s tropical humid forests35,36, both of which share the same type of ecosystem with Lacandon Maya.
As expected, when PIC values observed in Lacandons (Table 8) are compared against those found in a mixed ancestry population72, diversity is lower in the Lacandon sample than in Mexico City. Nevertheless, Lacandon PIC values are higher than other Native American groups for the HLA-A locus (Lacandons: 0.8444; Guarani from Paraguay: 0.7810; Toba from Argentina: 0.7715; Terena from Brasil: 0.7540; Mixtec: 0.7525, Zapotec: 0.7400, and Mixe: 0.6800 from Oaxaca, Mexico) and HLA-B (Lacandons: 0.8227; Gila River Indian Community of Arizona: 0.8167; Tarahumara from Mexico: 0.8093; Toba from Argentina: 0.7895; Mixtec: 0.7592, Zapotec: 0.7742, and Mixe: 0.3750 from Oaxaca, Mexico). Lacandon Maya HLA-A diversity as measured by PIC is the highest among Native American groups, and Lacandon HLA-B diversity is the third highest, just below Pilaga from Argentina and Sioux from USA. Conversely, HLA-DRB1 shows relatively low diversity in Lacandon, below that found in Native South American groups (Lacandons: 0.7555; Toba: 0.8580, Pilaga: 0.8328, and Eastern Toba: 0.8326 from Argentina; Mapuche from Chile: 0.8333; Uro from Peru: 0.7879) and some Native North American groups (Zapotec from Oaxaca, Mexico: 0.8512; Sioux from USA: 0.8439).
In what could be called the “class I/class II diversity paradox”, the Lacandon population exhibits a relatively low diversity in HLA-DRB1 (globally one of the most variable regions in the human genome) compared to HLA-A and HLA-B. It is possible that the lower diversity of MHC class II genes in Native American populations (and, by extension, in mixed populations with high proportions of Native American ancestry) might result from the frequency increase of some alleles that provide efficient immune protection against highly prevalent extracellular pathogens in specific populations6,7,15,19,72.
Previous studies have demonstrated a positive correlation between HLA class I allele diversity and pathogen richness and a negative correlation between HLA class II diversity and pathogen richness, raising the possibility that HLA class I and class II genes undergo different types of evolutionary trajectory in response to pathogen selection5,6. However, Sanchez Mazas et al.6 found that the significance of both their observed positive correlation between HLA class I allele diversity and pathogen richness and the negative correlation between HLA class II diversity and pathogen richness disappeared when Native American and Taiwanese populations were removed from the dataset. Native American populations have low HLA class II diversity in a high pathogen environment (this in turn seems to have helped to generate the previously observed negative correlation between class II diversity and pathogen richness). Is it possible that low HLA class II diversity can be a form of adaptation to a high pathogen environment? The class II HLA evolutionary mechanisms proposed by Sanchez-Mazas et al.6 apply particularly to HLA-DQA1 and HLA-DQB1, whereas as noted previously the Lacandon also exhibit relatively low diversity in HLA-DRB1. Certain HLA alleles appear to be promiscuous and are capable of binding an exceptionally large set of epitope peptide segments15. Since the HLA class II alleles commonly found in Native Americans (except for HLA-DRB1*16:02), and specifically those reported here for Lacandon Mayans, do not fall within the category of “promiscuous alleles” established by Manczinger et al.15, we can in principle hypothesize that selection events happening in recent times may have driven this genetic structure. Other non-promiscuous alleles include those of the HLA-DRB1*04 allelic group (accounting altogether for 70.22% of the total HLA-DRB1 diversity observed in Lacandons), with some of them being associated with specific pathogens as discussed above. It is noteworthy that at least one allele of this allelic group has been implicated in resistance to the development of enteric fever caused by Salmonella enterica92 and that there is molecular evidence of this pathogen causing at least one outbreak after the conquest in a region not far away from Chiapas (i.e. the state of Oaxaca)93. New alleles, with a very specific peptide binding repertoire, might be the most efficient way to achieve resistance to specific pathogens, as high promiscuity may not be able to cope with the rise of novel pathogens15,94.
Sanchez Mazas et al.6 found a positive relationship between HLA-B genetic diversity and either pathogen score or viral score, once distance from Africa was taken into account. In our analysis, our fitted coefficients were consistent with a positive relationship, but we could not reject the null hypothesis of no relationship (Supplementary Tables 3 and 4). Sanchez Mazas et al. did not find evidence for a relationship between HLA-DRB1 genetic diversity and either pathogen score or viral score. Within our dataset, the strongest signal of any relationship (albeit one which was not robust to a Bonferroni correction) was for a negative relationship between HLA-DRB1 genetic diversity and viral score once distance from Africa is considered, in non-Native American populations. We found no evidence for a relationship between either distance from Africa or pathogen/viral scores and HLA diversity when the Native American populations were considered alone. This may be due to the small size of the dataset. A better picture may be drawn in the coming years when ancient DNA analyses are taken into account for studying the immunogenetic diversity of these and other populations before and after specific events that would have changed the immune challenges faced by Native Americans, especially after contact with so-called Old World populations.
It has been shown using a multilocus model of host–pathogen co-evolution with allele-specific adaptive immunity that if selection from a multi-epitope, strain-structured pathogen is maintaining associations between host recognition loci, alleles at those loci should not only be in linkage disequilibrium, but also exhibit non-overlapping associations24. Penman et al.24 showed in particular that pathogen selection has the potential to maintain non-overlapping associations between HLA loci despite the presence of recombination between those loci. They demonstrated that such long-range non-overlapping associations may be observed between HLA loci such as HLA-B and HLA-DRB1 in a dataset of HLA haplotypes from the Hutterite population of South Dakota. We examined non-overlapping associations within Lacandon HLA haplotypes in an effort to identify similar possible signatures of pathogen selection (Figs. 3, 4 and Supplementary Fig. 3). In our dataset, the highest levels of non-overlap between HLA loci are observed between the physically close HLA-B and -C pair and the similarly physically close HLA-DRB1 and HLA-DRB3/4/5. Such associations could be a result of pathogen selection, but given the likely low level of recombination between these loci we cannot rule out other population genetic processes. For HLA-DRB1 and HLA-DRB3/4/5, epistasis with the HLA-DRA locus with which the gene products of HLA-DRB1 or HLA-DRB3/4/5 must interact may contribute to particularly high levels of non-overlap. Recent work has also revealed that haplotypic associations between HLA-B and HLA-C in particular may be driven by the different mechanisms by which the proteins they encode are likely to be able to participate in NK cell education95.
There was no evidence for non-overlapping associations spanning class I and class II HLA loci in the Lacandon dataset, which may suggest that the most recent forms of pathogen selection in the Lacandon population have acted on the class I and/or class II loci separately. Furthermore, due to the particularly devastating infectious disease events the Lacandon, and other Native American populations have experienced – in which many people have died in large epidemics93,96,97,98,99– it is unlikely that the patterns seen in this group may be the result of the long-term pathogen selection simulated in the original Penman et al. model24. It may be that the Lacandon population maintained a set of haplotypes as a consequence of millennia of co-evolution with prevalent American pathogens, and the pattern seen today is the consequence of a short-term disruption of that state. It has been recently found100 that class II genes show a signature for selection when comparing Native Americans from the same region in the northwest coast of North America before and after the contact with Europeans and the European-borne pathogens during the conquest and colonial period98,99,101. Our results further add to the discussion on whether HLA class II region could have been under similar selective pressure in Native Americans.
In summary, the Lacandon Maya represent one of the most homozygous and least diverse populations regarding HLA class II genes among Native American populations. It is possible that a history of infections, genocides and inbreeding contributed to this limited diversity in the HLA system. The relationship between genetic diversity of the HLA system and both pathogen and viral diversity, as well as its correlation with the geographic distance from Africa, may become clearer if data from previous time periods are only considered. Future work using ancient DNA approaches to study populations before specific historic and prehistoric periods (e.g. the conquest of the Americas or great human migrations), but also identifying pathogens that caused outbreaks and epidemics, may shed light on the actual diversity of the HLA system in human populations before and after epidemic and pandemic events that may have shaped the immunogenetic diversity of our species.
Subjects and Methods
Subjects
A total of 218 first-degree non-related Lacandon individuals [146 ♀ (64.2%) and 82 ♂ (35.8%); average age = 31.7 ± 17.4 years] were studied for a final number of 436 chromosomes. All participants were inhabitants from small villages located in the Lacandon jungle in the southern State of Chiapas, Mexico including: Lacanjá, Bethel, E. Lacandón, Metzabok, Na Há, San Javier, and Tumbo, all of them belonging to the municipality of Ocosingo (Fig. 1). We confirmed the Lacandon ancestry (parents and grandparents born in the same region) of all included individuals by questionnaire. Collection of blood samples and demographic data was performed according to the requisites of the Helsinki Declaration (2008) and the General Health Law of Mexico and following the protocol approved jointly by the Ethics in Research Committee and the Research Committee from the National Institute for Medical Sciences and Nutrition “Salvador Zubirán” (INCMNSZ). All subjects provided written informed consent for these studies, and they authorized the storage of their DNA samples. Informed consent was obtained from all participants. When participants were under 18 years at the time of the sample collection, informed consent was obtained from a parent and/or legal guardian.
High resolution HLA typing by next generation sequencing
Genomic DNA was obtained from peripheral blood mononuclear cells using the QIAamp DNA mini kit (Qiagen®, Valencia, CA, USA). All samples were typed for 11 HLA loci utilizing low- and high-resolution methods. Low resolution typing was performed by a Luminex-based detection and typing method including PCR amplification and reverse oligonucleotide hybridization (LABType® SSO Typing Tests, One Lambda Inc., Canoga Park, CA, USA). High-resolution HLA class I and class II typing was performed by a recently developed high throughput sequencing technique involving long range PCR of each HLA locus, shearing of DNA and high throughput NGS typing as previously described102. In addition, these samples were typed with a commercial kit MIA FORATM NGS FLEX HLA Typing Kit (Immucor, Norcross, GA, USA) that utilizes a similar method which applies enzymatic fragmentation instead of Covaris sonication. HLA genotypes were assigned utilizing the ad hoc MIA FORA software HT9v1 (Immucor, Norcross, GA, USA), provided by the manufacturer with the reference sequence ver. 3.2.25 of the IMGT-HLA database103. The HLA typing system we used covers the class I genes in their full sequence. With exception of HLA-DPB1, the class II loci were covered in their full sequence. The sequence fragments (at least 300 bases long each) were put in phase. Given the number of SNPs, we could put in phase the sequences of almost all alleles. There were no ambiguities in the class I or class II loci with exception of HLA-DPB1. We had virtually no ambiguities in DPB1 because more than 65 percent of the subjects were homozygous in DPB1 resulting from the high frequency of DPB1*04:02 alleles (the frequency for DPB1*04:02:01:01 and DPB1*04:02:01:02 was ≥0.8). Among the few ambiguities observed, all of them resulted from genotypes that include alleles with identical sequences in exon-2 that differ in exon-3 and cannot be placed in phase because of lack of informative SNPs. Almost all ambiguous HLA-DPB1 genotypes (<2%) included DPB1*04:02; we assigned the likely genotype as the one including this allele. In summary, there were no ambiguities for all loci with exception of HLA-DPB1; the rate of ambiguity at HLA-DPB1 was low because of low allelic diversity at this locus in this population; the most likely genotype was assigned on the basis of allele distributions in unambiguous genotypes.
HLA blocks and haplotypes assignment
HLA allele and haplotype frequencies were obtained by gene counting; seventy of the 458 haplotypes were obtained by family segregation analysis, since they were obtained by HLA typing of 69 individuals from 36 known families (related individuals were excluded to avoid overrepresentation of alleles and haplotypes). In 160 individuals without available samples from close family members, frequencies for alleles, two-point, three-point, four-point and five-point associations were determined using direct counting using the computer program Arlequin ver. 3.5104 and further corroborated with Hapl-o-Mat105 using an Expectation-Maximization (EM) algorithm. Arlequin was also used to calculate HWE, OH and EH at a locus-by-locus level with 1 × 106 steps in the Markov chain; p-values ≤ 0.05 indicated statistical difference between OH and EH and thus a deviation from HWE and confirmed with an independent analysis done with PyPop106 ver. 0.7.0 using the Ewens-Watterson homozygosity (EWH) test of neutrality (tested by Slatkin’s implementation of the Monte-Carlo approximation of the Ewens-Watterson exact test). Listed HLA-B~C, HLA-DRB1~DQB1, HLA-DRB1~DRB3/4/5, HLA-DQB1~DQA1, HLA-DPB1~DPA1, Class I and Class II blocks, conserved extended haplotypes (HLA-B~C~DRB1~DQB1) and their extension to the HLA-A locus were estimated by maximum likelihood methods based on the standardized delta (Δ′) between alleles of two loci and between the two blocks and/or the extension to the HLA-A region, as previously described72,107. Estimation of delta (Δ) and standardized delta (Δ′) values to measure linkage disequilibrium (LD), nonrandom association of alleles at two or more loci, and their statistical significance, were calculated using previously described methods23,107. We used the statistic parameter t to validate all Δ′ data adjusted by sample size and number of times that each allele appeared in the sample72,108. Only t values ≥ 2.0 were considered significant. Most probable ancestry (MPA) for each haplotype was determined based on the relative frequencies of the haplotypes in well-defined continental populations. Based on previously published approaches109, haplotypes regarded as Native American MPA were defined as those found in significant frequencies (H.F. ≥ 1.0%) in Native American populations such as Argentina Gran Chaco Eastern Toba, Xavantes from Central Brazil64, South or Central America Native Americans70, Yucpa from Venezuela35, Aleut from Bering Island70,110, Penutian from British Columbia111, Tarahumaras from Northern Mexico26, Mixe, Mixtec and Zapotec from Oaxaca State, Southern Mexico29,32, North American Natives107 from USA, and Yup’ik112 from USA. European MPA haplotypes were defined as those present in significant frequencies (H.F. ≥ 1.0%) in European human groups such as Bosnia and Herzegovina [from the Deutsche Knochenmarkspenderdatei (DKMS), Germany], Greece (from the DKMS), Croatia (from the DKMS), Romania (from the DKMS), Spain (from the DKMS)71, Italy78, Gorski Kotar from Croatia113, Northern Ireland114 and Ireland115. Haplotypes of African MPA were defined as those found in significant frequencies (H.F. ≥ 1.0%) in African or African-descent human groups such as Azoreans from Terceira Islands (data collected by Jacome Bruges Armas, available in61), USA African American73, Bandiagara from Mali79,116, Kampala from Republic of Uganda79,117, Nandi from Kenya79 and Zambians79,118. Asian MPA haplotypes were found in H.F. above 1.0% in Asian human groups like Nganasan and Ket from Lower Yenisey River/Taimyr Peninsula region (Siberia)119, Chinese (from the DKMS)71, Taiwanese120, Japanese69, Ivatan from Bantanes, Republic of the Philippines121, and Koreans122. Mixed-ancestry populations included USA70,73 and Mexican72 well-defined mixed ancestry human groups. With these results, we calculated the aggregate block frequency (ABF)17,123,124 for each ancestry for class I, class II and CEHs.
Analysis of HLA genetic diversity and non-overlapping associations between HLA loci
The genetic diversity of each HLA locus was assessed by two previously described forensic parameters: PIC and PD125,126; computed using the PowerStat ver.1.2 spreadsheet (Promega Corporation, Fitchburg, WI, USA) as described previously72. PIC measures the strength of a genetic marker for linkage studies by indicating the degree of polymorphism of a locus. PIC > 0.5 is considered as highly polymorphic125. PD is defined as the probability of finding two random individuals with different genotypes for that locus in the studied population, and values higher than 0.8 indicate high polymorphism in the studied population context126.
Penman et al. have argued that certain types of non-overlapping association between HLA loci may be a signature of pathogen selection24. The previously developed \({f}_{adj}^{\ast }\) metric24,127 was calculated for each pairwise combination of HLA loci in the dataset, using 436 Lacandon haplotypes for which we had data for every HLA locus. Penman et al. used \({f}_{adj}^{\ast }\) to compare patterns of non-overlap between HLA-HLA pairs and HLA-non HLA pairs of loci. In the current study we have only data on Lacandon HLA loci. In order to provide a point of comparison for the \({f}_{adj}^{\ast }\) score of each locus pair we generated 5000 random permutations of the order of the 436 alleles at one of the loci in each pair, and re-calculated \({f}_{adj}^{\ast }\) for each set of randomized data We thus generated distributions of possible \({f}_{adj}^{\ast }\) scores for each pair of loci in the dataset that would be obtained if the alleles at those loci were associated entirely at random. We then calculated the difference between the \({f}_{adj}^{\ast }\) value calculated from the Lacandon dataset for each pair of loci and the mean of the \({f}_{adj}^{\ast }\) scores calculated from the randomized data for that pair of loci, then divided that difference by the standard deviation of \({f}_{adj}^{\ast }\) calculated from the randomized data. The resulting scores allowed us to rank the HLA pairs in order of how extreme a level of non-overlap they displayed relative to entirely random associations between the same alleles. There are many reasons why associations between HLA loci should not be entirely random, so we do not claim that a departure from randomness necessarily means selection has occurred. However, ranking the pairs of HLA loci by how much they each depart from a state of random association allows for a more meaningful assessment of which pairs of HLA loci are most strongly non-overlapping, since it accounts for the differing allelic diversity at each locus.
Analyzing genetic relationships between populations
A PCA plot and a population phylogenetic tree were constructed for 180 populations (including the Lacandon group studied in this work) with HLA-A, HLA-B and HLA-DRB1 low-resolution data from a worldwide population dataset (Supplementary Table 1) using the IBM SPSS Statistics 19 software (IBM Corporation, Armonk, NY, USA) for the PCA and POPTREEW128 to analyze the distribution of 115 human groups with high resolution HLA typing, including our Lacandon sample. We used DA distance129 and a NJ clustering method to construct a population phylogenetic tree with bootstrapping (1200 replications). References for each population group included in the PCA and phylogenetic analysis are listed given in Supplementary Table 1.
Assessment of the correlation between pathogen richness and HLA diversity
We used PIC as an estimator for genetic diversity for three HLA loci (HLA-A, HLA-B and HLA-DRB1) and conducted an analysis of HLA genetic diversity and pathogen species richness similar to that of Sanchez-Mazas et al.6. First, we tested the correlation between genetic diversity for each gene and the geographic distance from Africa. We maintained the five key geographic points suggested by the authors and modeled the distance (in Km) from Addis Ababa (as a suggested point of departure) to every one of the locations where the 122 populations analyzed in the previous point (with available high-resolution data) live today. In our dataset the same populations were used for the three genes analyzed. Distance was calculated with the Measure distance function of Google Maps130. Information on pathogen and viral richness was extracted from the GIDEON database57. In order to relate the level of HLA polymorphism within a population and the pathogen environment of this population, we compiled the number of infectious diseases present in all countries for which we had information on HLA genetic diversity (N = 115). To assess the effect of genetic drift/recent bottlenecks/selection on small sized, isolated populations, we ran the statistical analyses after excluding Native American populations, and also ran tests on Native American populations separately. The final datasets include, thus, the worldwide populations’ dataset, the Native American populations’ dataset and the worldwide non-Native American populations’ dataset.
We used general linear models to determine how the distance from Africa and either pathogen or viral richness explained the level of genetic diversity (as estimated with PIC) at each of the three loci, for each of the datasets previously listed. We logit-transformed the PIC values before carrying out the analyses in order to improve normality. Coefficients for the relationships between distance, pathogen score or virus score; their 95% confidence intervals and associated p values, and the R2 value for each model are shown in Supplementary Tables 3 and 4. We performed the analyses with and without outliers. Outliers were defined as populations for which the logit transformed PIC value at any of the three loci was further than 2 standard deviations away from the mean logit(PIC) value for that locus.
Data availability
All data from our sample sets, both frequencies and anonymized individual genotypes, can be found at The Allele Frequency Net Database website (www.allelefrequencies.net)131 (AFDN Population ID: 3666). Sequence data can be found at the European Nucleotide Archive under accession numbers: KU319387, KU233196, KU319386, KU319291, KU319274, KU319273, KU319277, KU319263, KU319282, KX825916, KU319262, KU319256, KU319255, KU319285, KU319287, KU319303, KU319304, KU319325, KU319272, and KX825915. Further information can be requested to J.G. and/or M.F.V.
References
Horton, R. et al. Gene map of the extended human MHC. Nat. Rev. Genet. 5, 889–899 (2004).
de Vries, R. R. P., Meera Khan, P., Bernini, L., van Loghem, E. & van Rood, J. Genetic control of survival in epidemics. J. Immunogenet. 6, 271–287 (1979).
Gilbert, S. C., Plebanski, M. & Hill, A. V. S. Association of malaria parasite population structure, HLA, and immunological antagonism. Science (80-.). 279, 1173–7 (1998).
Jeffery, K. J. M. & Bangham, C. R. M. Do infectious diseases drive MHC diversity? Microbes Infect. 2, 1335–1341 (2000).
Prugnolle, F. et al. Pathogen-driven selection and worldwide HLA class I diversity. Curr. Biol. 15, 1022–1027 (2005).
Sanchez-Mazas, A., Lemaître, J. F. & Currat, M. Distinct evolutionary strategies of human leucocyte antigen loci in pathogen-rich environments. Philos. Trans. R. Soc. B Biol. Sci. 367, 830–839 (2012).
Penn, D. J., Damjanovich, K. & Potts, W. K. MHC heterozygosity confers a selective advantage against multiple-strain infections. Proc. Natl. Acad. Sci. USA 99, 11260–11264 (2002).
Osborne, A. J. et al. Heterozygote advantage at MHC DRB may influence response to infectious disease epizootics. Mol. Ecol. 24, 1419–1432 (2015).
Hedrick, P. W. Balancing selection and MHC. Genetica 104, 207–214 (1998).
Hedrick, P. W. Pathogen Resistance and Genetic Variation at MHC Loci. Evolution (N. Y). 56, 1902–1908 (2002).
De Boer, R. J., Borghans, J. A. M., Van Boven, M., Keşmir, C. & Weissing, F. J. Heterozygote advantage fails to explain the high degree of polymorphism of the MHC. Immunogenetics 55, 725–731 (2004).
Borghans, J. A. M., Beltman, J. B. & De Boer, R. J. MHC polymorphism under host-pathogen coevolution. Immunogenetics 55, 732–739 (2004).
Vallender, E. J. & Lahn, B. T. Positive selection on the human genome. Hum. Mol. Genet. 13, 245–254 (2004).
Ejsmond, M. J. & Radwan, J. MHC diversity in bottlenecked populations: A simulation model. Conserv. Genet. 12, 129–137 (2011).
Müller, V. et al. Pathogen diversity drives the evolution of generalist MHC-II alleles in human populations. PLOS Biol. 1–21, https://doi.org/10.1371/journal.pbio.3000131 (2019).
Buhler, S. & Sanchez-Mazas, A. HLA DNA sequence variation among human populations: Molecular signatures of demographic and selective events. PLoS One 6, (2011).
Yunis, E. J. et al. Inheritable variable sizes of DNA stretches in the human MHC: Conserved extended haplotypes and their fragments or blocks. Tissue Antigens 62, 1–20 (2003).
Arrieta-Bolaños, E., Madrigal, J. A. & Shaw, B. E. Human Leukocyte Antigen Profiles of Latin American Populations: Differential Admixture and Its Potential Impact on Hematopoietic Stem Cell Transplantation. Bone Marrow Res. 2012, 1–13 (2012).
Doherty, P. C. & Zinkernagel, R. M. A biological role for the major histocompatibility antigens. Lancet 1, 1406–9 (1975).
Barquera-Lozano, R. El papel de la genética de poblaciones en la inmunología del trasplante en México. Gac. Med. Mex. 148, 52–67 (2012).
Ceppellini, R. et al. Genetics of leukocyte antigens: A family study of segregation and linkage. in Histocompatibility Testing 1967 (eds. Curtoni, E., Mattiuz, P. & Tosi, R.) 149–87 (Munksgaard, 1967).
de Bakker, P. I. W. et al. A high resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat. Genet. 38, 1166–1172 (2006).
Yunis, E. J. et al. Single Nucleotide Polymorphism Blocks and Haplotypes: Human MHC Block Diversity. Encyclopedia of Molecular Cell Biology and Molecular Medicine 13, 192–215 (2006).
Penman, B. S., Ashby, B., Buckee, C. O. & Gupta, S. Pathogen selection drives nonoverlapping associations between HLA loci. Proc. Natl. Acad. Sci. 110, 19645–19650 (2013).
Infante, E. et al. Molecular analysis of HLA class I alleles in the Mexican Seri Indians: Implications for their origin. Tissue Antigens 54, 35–42 (1999).
García-Ortiz, J. E. et al. High-resolution molecular characterization of the HLA class I and class II in the Tarahumara Amerindian population. Tissue Antigens 68, 135–146 (2006).
Arnaiz-Villena, A. et al. HLA genes in Mexican Mazatecans, the peopling of the Americas and the uniqueness of Amerindians. Tissue Antigens 56, 405–416 (2000).
Arnaiz-Villena, A. et al. HLA Genes in Mayos Population from Northeast Mexico. Curr Genomics 8, 466–475 (2007).
Hollenbach, J. A. et al. HLA diversity, differentiation, and haplotype evolution in mesoamerican natives. Hum. Immunol. 62, 378–390 (2001).
Gómez-Casado, E. et al. Origin of Mayans according to HLA genes and the uniqueness of Amerindians. Tissue Antigens 61, 425–436 (2003).
Vargas-Alarcon, G. et al. Origin of Mexican Nahuas (Aztecs) according to HLA genes and their relationships with worldwide populations. Mol. Immunol. 44, 747–755 (2007).
Arnaiz-Villena, A. et al. Mixtec Mexican Amerindians: An HLA Alleles study for America peopling, pharmacogenomics and transplantation. Immunol. Invest. 43, 738–755 (2014).
Silvera, C. et al. HLA genes in Wayu Amerindians from Colombia. Immunol. Invest. 40, 92–100 (2011).
Martinez-Laso, J. et al. Origin of Bolivian Quechua Amerindians: Their relationship with other American Indians and Asians according to HLA genes. Eur. J. Med. Genet. 49, 169–185 (2006).
Layrisse, Z. et al. Extended HLA haplotypes in a Carib Amerindian population: The Yucpa of the Perija Range. Hum. Immunol. 62, 992–1000 (2001).
Layrisse, Z., Montagnani, S. & Balbas, O. Barí from Perija Mountain Range, Venezuela. Anthropology/human genetic diversity population reports. in Immunobiology of the Human MHC: Proceedings of the 13th International Histocompatibility Workshop and Conference (ed. Hansen, J.) 640–642 (IHWG Press, 2007).
Aláez, C., Vazquez-García, M., Olivo, A. & Gorodezky, C. Lacandon Mayan Indians from Mexico. Anthropology/human genetic diversity population reports. in Immunobiology of the Human MHC: Proceedings of the 13th International Histocompatibility Workshop and Conference (ed. Hansen, J.) 632–633 (IHWG Press, 2007).
Cagliani, R. et al. Balancing selection in the extended MHC region maintains a subset of alleles with opposite risk profile for different autoimmune diseases. Genome Biol. 11, P38 (2010).
Pittman, K. J., Glover, L. C., Wang, L. & Ko, D. C. The Legacy of Past Pandemics: Common Human Mutations That Protect against Infectious Disease. Plos Pathog. 12, e1005680 (2016).
Matzaraki, V., Kumar, V., Wijmenga, C. & Zhernakova, A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 18, (2017).
Peschken, C. A. & Esdaile, J. M. Rheumatic diseases in North America’s indigenous peoples. Semin. Arthritis Rheum. 28, 368–391 (1999).
Molokhia, M. & McKeigue, P. Risk for rheumatic disease in relation to ethnicity and admixture. Arthritis Res. Ther. 2, 115 (2000).
Ghio, A. J. Particle Exposure and the Historical Loss of Native American Lives to Infections. Am. J. Respir. Crit. Care Med. 195, 1673 (2017).
Vargas-Alarcón, G. et al. Description of HLA-A*6803 and A*68N in Mazatecan Indians from Mexico. Immunogenetics 46, 446–447 (1997).
Marcos, C. Y., Moraes, M. E. & Moraes, J. R. Novel HLA-A and HLA-B alleles in South American Indians. 476–485 (1999).
Ramon, D. et al. HLA-A*6817, identified in the Kolla Amerindians of North-West Argentina possesses a novel nucleotide substitution. Tissue Antigens 55, 453–454 (2000).
Matson, G. A. & Swanson, J. Distribution of hereditary blood antigens among the Maya and non-Maya Indians in Mexico and Guatemala. Am. J. Phys. Anthropol. 17, 49–74 (1959).
López-López, M. et al. CYP2D6 genetic polymorphisms in Southern Mexican Mayan Lacandones and Mestizos from Chiapas. Pharmacogenomics 15, 1859–1865 (2014).
Moreno-Estrada, A. et al. The genetics of Mexico recapitulates Native American substructure and affects biomedical traits. Science (80-.). 344, 1280–1285 (2014).
Sharer, R. The ancient Maya. (Stanford University Press, 1994).
Kepecs, S. & Alexander, R. The Post-Classic to Spanish Era Transition in Mesoamerica. Archeological Perspectives. (The University of New Mexico Press, 2005).
Nations, J. D. & Nigh, R. B. The Evolutionary Potential of Lacandon Maya Sustained-Yield Tropical Forest Agriculture. J. Anthropol. Res. 36, 1–30 (1980).
Stewart, A., Martin, F. & Diemont, W. Lacandon Maya ecosistem management: sustainable design for subsistence and environmental restoration. Ecol. Appl. 19, 254–266 (2009).
Rojas, A. V. Kinship and Nagualism in a Tzeltal Community, Southeastern Mexico. Am. Anthropol. 49, 578–587 (1947).
Matson, G. A. & Swanson, J. Distribution of Hereditary Blood Antigens among American Indians in Middle America: Lacandón and Other Maya. Am. Anthropol. 63, 1292–1322 (1961).
Rodriguez-Reyna, T. S. et al. HLA Class I and II Blocks Are Associated to Susceptibility, Clinical Subtypes and Autoantibodies in Mexican Systemic Sclerosis (SSc) Patients. PLoS One 10, e0126727 (2015).
GIDEON Informatics, I. Infectious Diseases - Diseases. GIDEON - Global Infectious Diseases and Epidemiology Online Network (2019). Available at: https://web.gideononline.com/web/epidemiology/ (Accessed: 6th July 2019).
Vargas-Alarcón, G. et al. HLA genes in Mexican Teeneks: HLA genetic relationship with other worldwide populations. Mol. Immunol. 43, 790–799 (2006).
Akesaka, T. et al. Buryat from Angarsk, Russia. Anthropology/human genetic diversity population reports. in Immunobiology of the Human MHC: Proceedings of the 13th International Histocompatibility Workshop and Conference (ed. Hansen, J. A.) 627 (IHWG Press, 2007).
Hajeer, A. H. et al. HLA-A, -B, -C, -DRB1 and -DQB1 allele and haplotype frequencies in Saudis using next generation sequencing technique. Tissue Antigens 82, 252–258 (2013).
González-Galarza, F. F. et al. Allele frequency net 2015 update: New features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 43, D784–D788 (2015).
Severson, L., Crews, D. & Lang, R. Samoans from American Samoan. Anthropology/human genetic diversity population reports. in Immunobiology of the Human MHC: Proceedings of the 13th International Histocompatibility Workshop and Conference (ed. Hansen, J. A.) 623–4 (IHWG Press, 2007).
Rajalingam, R. et al. Distinctive KIR and HLA diversity in a panel of north Indian Hindus. Immunogenetics 53, 1009–1019 (2002).
Cerna, M. et al. Differences in HLA class II alleles of isolated South American Indian populations from Brazil and Argentina. Hum Immunol 37, 213–220 (1993).
Trachtenberg, E. A., Erlich, H. A., Rickards, O., DeStefano, G. F. & Klitz, W. HLA class II linkage disequilibrium and haplotype evolution in the Cayapa Indians of Ecuador. Am J Hum Genet 57, 415–424 (1995).
Souza de Lima, D. et al. Alleles of HLA-DRB1*04 associated with pulmonary tuberculosis in Amazon Brazilian population. PLoS One 11, 1–13 (2016).
Tian, C. et al. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat. Commun. 8, 599 (2017).
Yan, Z.-H. et al. Relationship between HLA-DR gene polymorphisms and outcomes of hepatitis B viral infections: a meta-analysis. World J. Gastroenterol. 18, 3119–3128 (2012).
Saito, S., Ota, S., Yamada, E., Inoko, H. & Ota, M. Allele frequencies and haplotypic associations defined by allelic DNA typing at HLA class I and class II loci in the Japanese population. Tissue Antigens 56, 522–529 (2000).
Gragert, L., Madbouly, A., Freeman, J. & Maiers, M. Six-locus high resolution HLA haplotype frequencies derived from mixed-resolution DNA typing for the entire US donor registry. Hum. Immunol. 74, 1313–1320 (2013).
Pingel, J. et al. High-resolution HLA haplotype frequencies of stem cell donors in Germany with foreign parentage: How can they be used to improve unrelated donor searches? Hum. Immunol. 74, 330–340 (2013).
Zúñiga, J. et al. HLA Class I and Class II Conserved Extended Haplotypes and Their Fragments or Blocks in Mexicans: Implications for the Study of Genetic Diversity in Admixed Populations. PLoS One 8, (2013).
Maiers, M., Gragert, L. & Klitz, W. High-resolution HLA alleles and haplotypes in the United States population. Hum. Immunol. 68, 779–788 (2007).
Sulcebe, G. et al. HLA allele and haplotype frequencies in the Albanian population and their relationship with the other European populations. Int. J. Immunogenet. 36, 337–343 (2009).
Sulcebe, G. et al. Human leukocyte antigen-A, -B, -C, -DRB1 and -DQB1 allele and haplotype frequencies in an Albanian population from Kosovo. Int. J. Immunogenet. 40, 104–107 (2013).
Sulcebe, G. & Shyti, E. HLA-A, -B, -C, -DRB1 and -DQB1 allele and haplotype frequencies in a population of 432 healthy unrelated individuals from Albania. Hum. Immunol. 77, 620–621 (2016).
Kirijas, M., Genadieva Stavrik, S., Senev, A., Efinska Mladenovska, O. & Petlichkovski, A. HLA-A, -B, -C and -DRB1 allele and haplotype frequencies in the Macedonian population based on a family study. Hum. Immunol. 79, 145–153 (2017).
Rendine, S. et al. Estimation of human leukocyte antigen class I and class II high-resolution allele and haplotype frequencies in the Italian population and comparison with other European populations. Hum. Immunol. 73, 399–404 (2012).
Cao, K. et al. Differentiation between African populations is evidenced by the diversity of alleles and haplotypes of HLA class I loci. Tissue Antigens 63, 293–325 (2004).
Arlehamn, C. S. L. et al. Sequence-based HLA-A, B, C, DP, DQ, and DR typing of 100 Luo infants from the Boro area of Nyanza Province, Kenya. Hum. Immunol. 78, 325–326 (2017).
Grifoni, A. et al. Sequence-based HLA-A, B, C, DP, DQ, and DR typing of 159 individuals from the Worcester region of the Western Cape province of South Africa. Hum. Immunol. 79, 143–144 (2018).
Axtell, J. The columbian mosaic in colonial America. Humanities 12, 12–18 (1991).
Grunberg, B. El universo de los conquistadores: resultado de una investigación prosopográfica. Signos Históricos 12, 94–118 (2004).
Moffett, A. & Loke, C. Immunology of placentation in eutherian mammals. Nat. Rev. Immunol. 6, 584–594 (2006).
Pyo, C. W. et al. Different patterns of evolution in the centromeric and telomeric regions of group A and B haplotypes of the human killer cell Ig-like receptor locus. PLoS One 5, (2010).
Parham, P. MHC class I molecules and KIRS in human history, health and survival. Nat. Rev. Immunol. 5, 201–214 (2005).
Rajagopalan, S. & Long, E. O. Understanding how combinations of HLA and KIR genes influence disease. J. Exp. Med. 201, 1025–1029 (2005).
Norman, P. J. et al. Unusual selection on the KIR3DL1/S1 natural killer cell receptor in Africans. Nat. Genet. 39, 1092–1099 (2007).
Falco, M., Moretta, L., Moretta, A. & Bottino, C. KIR and KIR ligand polymorphism: A new area for clinical applications? Tissue Antigens 82, 363–373 (2013).
Stern, M., Ruggeri, L., Capanni, M., Mancusi, A. & Velardi, A. Human leukocyte antigens A23, A24, and A32 but not A25 are ligands for KIR3DL1. Blood 112, 708–710 (2008).
Ahlenstiel, G., Martin, M. P., Gao, X., Carrington, M. & Rehermann, B. Distinct KIR/HLA compound genotypes affect the kinetics of human antiviral natural killer cell responses. J. Clin. Investig. 118, 1017–1026 (2008).
Dunstan, S. J. et al. Variation at HLA-DRB1 is associated with resistance to enteric fever. Nat. Genet. 46, 1333–1336 (2014).
Vågene, Å. J. et al. Salmonella enterica genomes from victims of a major sixteenth-century epidemic in Mexico. Nat. Ecol. Evol. 2, 520–528 (2018).
Kaufman, J. Generalists and Specialists: A New View of How MHC Class I Molecules Fight Infectious Pathogens. Trends Immunol. 39, 367–379 (2018).
Horowitz, A. et al. Class I HLA haplotypes form two schools that educate NK cells in different ways. Sci. Immunol. 1, eaag1672–eaag1672 (2016).
Lovell, W. G. & Lutz, C. H. The Historical Demography of Colonial Central America. Conf. Lat. Am. Geogr. 17/18, 127–138 (1992).
Patch, R. W. Sacraments and Disease in Mérida, Yucatán, Mexico, 1648–1727. Hist. 58, 731–743 (1996).
Marr, J. S. & Kiracofe, J. B. Was the Huey Cocoliztli a Haemorrhagic Fever? Med. Hist. 44, 341–362 (2000).
Acuña-Soto, R. Megadrought and Megadeath in 16th-Century Mexico. Emerg. Infect. Dis. 8, 360–62 (2002).
Lindo, J. et al. A time transect of exomes from a Native American population before and after European contact. Nat. Commun. 7, (2016).
Acuña-Soto, R., Stahle, D. W., Therrell, M. D., Griffin, R. D. & Cleaveland, M. K. When half of the population died: The epidemic of hemorrhagic fevers of 1576 in Mexico. FEMS Microbiol. Lett. 240, 1–5 (2004).
Wang, C. et al. High-throughput, high-fidelity HLA genotyping with deep sequencing. Proc. Natl. Acad. Sci. 109, 8676–8681 (2012).
Robinson, J. et al. The IPD and IMGT/HLA database: Allele variant databases. Nucleic Acids Res. 43, D423–D431 (2015).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Schäfer, C., Schmidt, A. H. & Sauter, J. Hapl-o-Mat: Open-source software for HLA haplotype frequency estimation from ambiguous and heterogeneous data. BMC Bioinformatics 18, 1–10 (2017).
Lancaster, A. K., Single, R. M., Solberg, O. D., Nelson, M. P. & Thomson, G. PyPop update - A software pipeline for large-scale multilocus population genomics. Tissue Antigens 69, 192–197 (2007).
Cao, K. et al. Analysis of the frequencies of HLA-A, B, and C alleles and haplotypes in the five major ethnic groups of the United States reveals high levels of diversity in these loci and contrasting distribution patterns in these populations. Hum. Immunol. 62, 1009–1030 (2001).
Haseman, J. K. & Elston, R. C. The investigation of linkage between a quantitative trait and a marker locus. Behav. Genet. 2, 3–19 (1972).
Arrieta-Bolaños, E. et al. High-resolution HLA allele and haplotype frequencies in majority and minority populations of Costa Rica and Nicaragua: Differential admixture proportions in neighboring countries. HLA 91, 514–529 (2018).
Moscoso, J. et al. HLA genes of Aleutian Islanders living between Alaska (USA) and Kamchatka (Russia) suggest a possible southern Siberia origin. Mol. Immunol. 45, 1018–1026 (2008).
Monsalve, M. V., Edin, G. & Devine, D. V. Analysis of HLA class I and class II of Na-Dene and Amerindian populations from British Columbia, Canada. Hum. Immunol. 59, 48–55 (1998).
Leffell, M. S. et al. HLA antigens, alleles and haplotypes among the Yup’ik Alaska natives: Report of the ASHI Minority Workshops, part II. Hum. Immunol. 63, 614–625 (2002).
Crnić-Martinović, M. et al. HLA class II polymorphism in autochthonous population of Gorski kotar, Croatia. Coll. Antropol. 31, 315–319 (2007).
Middleton, D., Williams, F., Hamill, M. A. & Meenagh, A. Frequency of HLA-B alleles in a caucasoid population determined by a two-stage PCR-SSOP typing strategy. Hum. Immunol. 61, 1285–1297 (2000).
Dunne, C., Crowley, J., Hagan, R., Rooney, G. & Lawlor, E. HLA-A, B, Cw, DRB1, DQB1 and DPB1 alleles and haplotypes in the genetically homogenous Irish population. Int. J. Immunogenet. 35, 295–302 (2008).
Lazaro, A. M. et al. HLA-B-Cw-DRB1-DQA1-DQB1 haplotypes diversity in Mali population. Hum. Immunol. 64, S135 (2003).
Okello, E. et al. Rheumatic heart disease in Uganda: The association between MHC class II HLA DR alleles and disease: A case control study. BMC Cardiovasc. Disord. 14, 2–6 (2014).
Tang, J. et al. HLA-DRB1 and -DQB1 alleles and haplotypes in Zambian couples and their associations with heterosexual transmission of HIV type 1. J. Infect. Dis. 189, 1696–704 (2004).
Uinuk-ool, T. S., Takezaki, N., Derbeneva, O., Volod’ko, N. & Sukernik, R. Variation of HLA class II genes in Nganasans and Kets, two indigenous Siberian populations. Tissue Antigens (2003).
Yang, K. L., Chen, S. P., Shyr, M. H. & Lin, P. Y. High-resolution human leukocyte antigen (HLA) haplotypes and linkage disequilibrium of HLA-B and -C and HLA-DRB1 and -DQB1 alleles in a Taiwanese population. Hum. Immunol. 70, 269–276 (2009).
Chu, C., Trejaut, J., Lee, H., Chang, S. & Lin, M. Ivatan from Bantanes, Philippines. Anthropology/human genetic diversity population reports. in Immunobiology of the Human MHC: Proceedings of the 13th International Histocompatibility Workshop and Conference (ed. Hansen, J. A.) 611–5 (IHWG Press, 2007).
Lee, K. W., Oh, D. H., Lee, C. & Yang, S. Y. Allelic and haplotypic diversity of HLA-A, -B, -C, -DRB1, and -DQB1 genes in the Korean population. Tissue Antigens 65, 437–447 (2005).
Romero, V. et al. Genetic fixity in the human major histocompatibility complex and block size diversity in the class I region including HLA-E. BMC Genet. 8, 1–12 (2007).
Dorak, M. T. et al. Conserved extended haplotypes of the major histocompatibility complex: Further characterization. Genes Immun. 7, 450–467 (2006).
Zhu, B. et al. Distributions of HLA-A and -B alleles and haplotypes in the Yi ethnic minority of Yunnan, China: relationship to other populations. J. Zhejiang Univ. Sci. B 11, 127–135 (2010).
Yasuda, N. HLA polymorphism information content (PIC). Jinrui Idengaku Zasshi 33, 385–7 (1988).
Buckee, C. O., Gupta, S., Kriz, P., Maiden, M. C. J. & Jolley, K. A. Long-term evolution of antigen repertoires among carried meningococci. Proc. R. Soc. B Biol. Sci. 277, 1635–1641 (2010).
Takezaki, N., Nei, M. & Tamura, K. POPTREEW: Web version of POPTREE for constructing population trees from allele frequency data and computing some other quantities. Mol. Biol. Evol. 31, 1622–1624 (2014).
Nei, M. & Chesser, R. Estimation of fixation indices and gene diversities. Ann. Hum. Genet. 47, 253–259 (1983).
Google. Google Earth Pro ©. (2019).
dos Santos, E. J. M., McCabe, A., Gonzalez-Galarza, F. F., Jones, A. R. & Middleton, D. Allele Frequencies Net Database: Improvements for storage of individual genotypes and analysis of existing data. Hum. Immunol. 77, 238–248 (2016).
Gumperz, J. E. et al. Conserved and variable residues within the Bw4 motif of HLA-B make separable contributions to recognition by the NKB1 killer cell-inhibitory receptor. J. Immunol. 158, 5237 LP–5241 (1997).
Acknowledgements
The authors are grateful to all the participants of this study. Jach nib óolal! We are thankful to Evelyn Collen for her analytical support. The authors want to express their gratitude to Prof. Derek Middleton, who kindly shared his time and expertise and helped us improving our work. Also, we want to thank the anonymous reviewers which with their time and suggestions contributed to improve our manuscript. The authors are grateful to Adriana Gómez Bustamante, head of the Public Health State Laboratory of Chiapas for all her valuable support. The experimental methods here described were approved jointly by the Ethics in Research Committee and the Research Committee from the National Institute for Medical Sciences and Nutrition “Salvador Zubirán” (Protocol number: 1738; Off. Letter Number: Mcontrol-506/2019; last re-approval: April 17th, 2019). All expenses for this project were covered by each of the participants, except for the lab work (including HLA typing and allele confirmation) which was carried out at Stanford Genome Technology Center, Palo Alto, CA, USA. Part of this work was funded by the Max Planck Society.
Author information
Authors and Affiliations
Contributions
R.B., J.Z., J.J.F.R., T.C., L.J.G., B.P., D.I.H.Z., J.A.A.V., J.G., M.F.V and E.Y. conceived the experiments. J.J.F.R., T.C., L.J.G., P.Y., A.O.M. and J.A.A.V. performed the collection of blood samples, did medical consultations for all participants and performed neurological assessments as part of the field work. R.B., J.J.F.R., T.C., M.S., D.I.H.Z., L.J.G., P.Y., A.O.M., J.A.A.V., M.G.L., L.M.H., E.O., J.G. and E.Y. conducted ethnographic work, including genealogy reconstructions for the population. K.C.M., K.B., M.M. and M.F.V. performed the sample processing and assigned the allele calls. R.B., J.Z., D.I.H.Z., B.P., K.C.M., K.B., M.M., M.Y., L.M.H. J.G., M.F.V. and E.Y. performed the statistical analyses. All authors contributed to the results discussion. R.B., J.Z., J.J.F.R., T.C., B.P., D.I.H.Z., M.S., J.G., M.F.V. and E.Y. wrote the paper with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barquera, R., Zuniga, J., Flores-Rivera, J. et al. Diversity of HLA Class I and Class II blocks and conserved extended haplotypes in Lacandon Mayans. Sci Rep 10, 3248 (2020). https://doi.org/10.1038/s41598-020-58897-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-58897-5
This article is cited by
-
Characterizing the diversity of MHC conserved extended haplotypes using families from the United Arab Emirates
Scientific Reports (2022)
-
Mayan alleles of the HLA-DRB1 major histocompatibility complex might contribute to the genetic susceptibility to systemic lupus erythematosus in Mexican patients from Tapachula, Chiapas
Clinical Rheumatology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
