
Abstract
The diagnostic specificity of prostate specific antigen (PSA) is limited. We aimed to characterize eight anti-PSA monoclonal antibodies (mAbs) to assess the prostate cancer (PCa) diagnostic utility of different PSA molecular forms, total (t) and free (f) PSA and PSA complexed to α1-antichymotrypsin (complexed PSA). MAbs were obtained by immunization with PSA and characterized by competition studies, ELISAs and immunoblotting. With them, we developed sensitive and specific ELISAs for these PSA molecular forms and measured them in 301 PCa patients and 764 patients with benign prostate hyperplasia, and analyzed their effectiveness to discriminate both groups using ROC curves. The free-to-total (FPR) and the complexed-to-total PSA (CPR) ratios significantly increased the diagnostic yield of tPSA. Moreover, based on model selection, we constructed a multivariable logistic regression model to predictive PCa that includes tPSA, fPSA, and age as predictors, which reached an optimism-corrected area under the ROC curve (AUC) of 0.86. Our model outperforms the predictive ability of tPSA (AUC 0.71), used in clinical practice. In conclusion, The FPR and CPR showed better diagnostic yield than tPSA. In addition, the PCa predictive model including age, fPSA and complexed PSA, outperformed tPSA detection efficacy. Our model may avoid unnecessary biopsies, preventing harmful side effects and reducing health expenses.
Similar content being viewed by others

Introduction
In Europe, prostate cancer (PCa) is the most common solid neoplasm in men, with an incidence rate of 25% of all newly diagnosed cancers and shows the highest death rate after lung and bronchus cancer1. Men surviving PCa are the largest population of male cancer survivors and comprise approximately 40% of all. Significant controversy concerning PCa overdetection and overtreatment has led to a search for better markers. Overdetection is a minor problem compared to underdetection. Overtreatment is an ethical problem that could be solved when effective tools to differentiate clinically significant from indolent tumours are adopted. Approximately 1.3 million of prostate biopsies are performed every year in the USA. Most of them are negative but 43% will require a new diagnostic biopsy in 3 years2. This situation involves side effects and unnecessary expenses.
Prostate specific antigen (PSA), also known as human glandular kallikrein 3, is a member of the kallikrein family which also includes tissue kallikrein and human glandular kallikrein 2 (hK2). PSA is secreted by prostate epithelial cells3 and is present in serum from patients with prostate disease4. High levels of PSA are a useful marker for PCa detection4, for monitoring follow-up and progression after radical prostatectomy5, and for monitoring local or systemic therapy6,7, However, levels of PSA are also increased in some patients with benign prostatic hyperplasia, acute prostatitis8 or prostate manipulations, leading to unnecessary negative biopsies or to over detection of non-significant cancers. Nonetheless, PSA screening has saved the lives of many men around the world, is an independent variable for PCa, and is a better predictor of cancer than digital rectal examination (DRE) or transrectal ultrasound diagnosis9. So, the suggestions to abandon PSA screening are unjustified. Instead, we should refine the diagnosis with better screening and better biopsy performing.
Different new biomarkers, cancer metabolism markers or microRNAs are being investigated to improve diagnosis of PCa in different types of samples, such as serum, urine, semen or cell cultures10,11,12,13. In addition, different strategies using several PSA molecular forms or ratios, PSA density or velocity14, proPSA forms, PSA glycoforms15 or PSA in combination with platelet volume and distribution16 have been developed in order to improve the specificity of PSA as a biomarker. Moreover, novel score tests to provide the risk of PCa derived from a mathematical algorithm for different kallikrein biomarkers as well as other clinical information, have been developed17,18. They are known as: 4 K Score [total PSA (tPSA), free PSA (fPSA) and intact PSA and human kallikrein 2 hK2)]19,20, Prostate Health Index (PHI) (tPSA, fPSA ratio and [−2]proPSA)21,22 or Stockholm-3 test23,24 (tPSA, fPSA and intact PSA, hK2, MSMB, MIC1, genetic polymorphisms, age, family history, previous prostate biopsy, DRE and prostate volume). Another approach, derived from these scores and estimated in large populations, is the risk calculators (RCs) prediction models, developed to assess patient’s individual PCa risk, which show a moderate to well discriminatory ability to predict PCa25. There are other prognostic scores based on genetic approaches, but their results are controversial3,26. In addition, mutations in kallikrein genes are associated with the risk of PCa and tumour aggressiveness27,28.
One of the most promising approaches to improve the specificity of the PSA test to better distinguish between PCa and non-PCa, is the development of assays for measuring different molecular forms of PSA in plasma or serum. PSA is present in circulation in different molecular forms, including fPSA and PSA complexed with α1-antichymotrypsin (PSA-α1ACT) and with α2-macroglobulin (PSA-α2M)29, although the most relevant forms are fPSA and PSA-α1ACT30,31. TPSA, fPSA and PSA-α1ACT complex levels vary with age, race and ethnicity, body mass index values or the assay kits used32. Nevertheless, these variations are mainly associated with prostate disease. The free-to-total (FPR) and complexed-to-total PSA (CPR) ratios have been shown to provide a better discrimination between PCA and non-PCa33,34. Additionally, hK2 has about 80% homology to PSA and some anti-PSA antibodies may cross-react with this protein. Therefore, the presence of different molecular forms of PSA and kK2 in serum, illustrates the need to develop new anti-PSA antibodies that do not cross-react with hK2 and may distinguish between fPSA and PSA-α1ACT more precisely.
Here, we report the preparation and characterization of eight anti-PSA monoclonal antibodies (mAbs) and their usefulness in specific sandwich ELISAs for tPSA, fPSA and PSA-α1ACT complex. We also evaluated the clinical usefulness to discriminate between PCa and non-PCa for these molecular forms, using ROC curves as well as combination of several biomarkers in a predictive model that may enhance sensitivity and specificity of PSA alone. All the assays were performed in citrated plasma samples as we have reported that citrated plasma samples provide higher specificity than serum samples when using FPR and CPR as markers35.
Results
Characterization of monoclonal anti-PSA antibodies
In our study, eight murine anti-PSA mAbs were generated and characterized: M1, M15, M21, M29, M40, M50, M63, and M73. All of them were of the IgG1 with kappa chain type and none exhibited cross-reactivity with female sera, determined by incubation with different relative concentration of antibodies and PSA (see Supplementary Fig. S1), showing that female serum does not contain any component that competes with PSA for antibody binding. The apparent dissociation constant (Kd) of each mAb for immobilized PSA or PSA in solution is shown in Supplementary Table S1, with the M40 mAb showing the highest affinity.
Only two of our eight mAbs showed cross-reactivity with hK2, especially M73, so it could only be used as a secondary antibody (see Supplementary Fig. S2). Four combinations, M1/M21*, M21/M40*, M40/M50* and M40/M73*, detected fPSA and PSA-α1ACT with the same efficiency (see Supplementary Fig. S3). Hence, we selected the pair M40/M73* to set up an ELISA for tPSA. This assay does not recognize hK2, is equimolar (detects all PSA molecular forms in equal molar ratios), and has a detection limit of 0.1 μg/L (see Supplementary Figs. S2–S4).
Immunoassays for the PSA molecular forms
All combinations with M63 were specific for fPSA (see Supplementary Fig. S4). We selected the pair M63/M50* to set up an assay specific for fPSA. The assay does not detect complexed PSA, has a detection limit of 0.04 μg/L (see Supplementary Fig. S4), and gave no signal with hK2 concentrations up to 20 μg/L.
There were three combinations (M1/M40*, M1/M50* and M1/M73*) that reacted slightly more efficiently with PSA-α1ACT than with fPSA (see Supplementary Fig. S4), suggesting that M1 may not be useful to measure PSA-α1ACT complex. Therefore, we selected the combination M40/polyclonal anti-α1ACT* pair to measure PSA-α1ACT in plasma. It gave a good dose-response curve with purified PSA-α1ACT diluted both in buffer and in plasma and no signal was obtained either with fPSA up to 2000 μg/L or with purified α1ACT up to 500 mg/L. The assay has a detection limit of 0.05 μg/L of complexed PSA (see Supplementary Fig. S4), and gave no signal with hK2 concentrations up to 20 μg/L.
Clinical usefulness of the PSA molecular forms
We studied 764 patients with benign biopsy (BB) and 301 patients with PCa. Table 1 shows median with the first and third quartiles in brackets, or n with % in parenthesis for tPSA, fPSA, PSA-α1ACT, PSA-α2M, PSA-hk2, as well as FPR, CPR and FPR/CPR ratio, as markers to discriminate between PCa and BB. The concentration of tPSA, fPSA, PSA-α1ACT, PSA-α2M, PSA-hK2 and CPR was significantly higher in patients with PCa than in those with BB (P < 0.001, P = 0.012, P < 0.001, P < 0.001 and P < 0.001, respectively), whereas prostate volume, FPR and FPR/CPR ratio were significantly higher in BB than in PCa (P < 0.001) (Table 1). Using tPSA levels, we also estimated the PSA density (tPSA/prostate volume) for all patients, which was significantly higher in PCa patients than in BB (P < 0.001).
We used two types of analyses to assess the clinical performance of the parameters studied, one through ROC curves grouping patients into subgroups according to their tPSA level, and the other through multivariable logistic regression models with continuous variables, for generalizable results.
Table 2 shows the sensitivity, specificity, and AUC values for the group of patients with tPSA between ≥4 and <10 μg/L. For this subgroup of patients, FPR (AUC = 0.81), CPR (AUC = 0.79) and FPR/CPR ratio (AUC = 0.79) gave a better discrimination than tPSA (AUC = 0.56).
Supplementary Tables S4–S7 show the sensitivity, specificity, and AUC values for the different subgroups analysed: whole cohort of patients, patients with tPSA between ≥2.5 and ≤4 μg/L, patients with tPSA between ≥4 and <10 μg/L and patients with tPSA between 10 and <20 μg/L. For the whole cohort of analysed patients, the FPR/CPR ratio (0.82) and FPR (0.78) gave a better discrimination than tPSA (0.69) (Supplementary Table S4). For the group of patients with tPSA between ≥2.5 and <4 ng/ml, the FPR/CPR ratio (0.77) and FPR (0.76) again gave better discrimination than tPSA (0.53) (Supplementary Table S5). For the tPSA range between ≥10 and <20 μg/L, FPR (0.80) and CPR (0.79) gave the best discrimination compared to tPSA (0.55) (Supplementary Table S7). In this range, using a cut-off point of 4.4 for the FPR/CPR ratio we would have avoided 30% biopsies without losing any PCa patient.
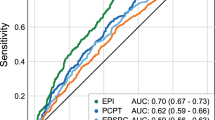
Clinical variables, age, PSA density, tPSA, fPSA, PSA-α1ACT, PSA-α2M, FPR, CPR and FPR/CPR were also analyzed in multivariable logistic regression models. Table 3 shows the different models used for discriminating between PCa and BB using the akaike information criterion (AIC). The best model, according to the AIC criterion, included only the variables: age (OR = 1.75, 95% CI: 1.31–3.36 (per 10 years), P < 0.001,), tPSA and fPSA values (OR = 22.55, 95% CI: 13.1–40.5, P < 0.001; and OR = 0.05, 95% CI: 0.025–0.095, P < 0.001, respectively), as well as their interaction (OR = 1.31, 95% CI: 1.08–1.6, P = 0.007) (Table 4). Due to their skewed distribution, tPSA, fPSA and their interaction were log-transformed prior to modelling. With a likelihood ratio test we compared the performance of our elected model and the other models proposed. As depicted in Table 3, our model outperformed all others. Our model substantially improves the predictive capacity of PCa compared to that of tPSA. It achieved an apparent AUC of 0.86 (95% CI: 0.83–0.89) and an optimism-corrected AUC of 0.86, compared to AUC of 0.71 (95% CI: 0.67–0.75) for tPSA (Fig. 1). The formula for predicting the probability (Pr) of PCa would be:

ROC curves for the predictive model (age, tPSA, fPSA and tPSA*fPSA) compared to that obtained for tPSA using mAbs. The area under the curve (AUC) and interquartile range in parenthesis are shown.
In order to assess whether our selected model was really better than by biopsy all, we performed a decision curve analysis comparing our selected model to total PSA and biopsy all (Supplementary Fig. S5). The results show that our model improves the standardized net benefit over all the range of thresholds compared to biopsy all and over most of the threshold values compared to total PSA values. Thus, standardized net benefit values are higher in our model compared to biopsy all starting at a probability of 4%. This difference is statistically significant starting at a probability of 13%.
In order to ease the interpretation of the results of the model, we represented its sensitivity and specificity profile plot (Supplementary Fig. S6). Using this profile plot we may select a sensitivity (for example 90%), obtaining a specificity of 58%.
We also generated an effect plot depicting the relationship between tPSA, fPSA and the probability of cancer (Supplementary Fig. S7). It shows that increasing levels of tPSA are associated with higher probabilities of cancer, and increasing levels of fPSA are associated with lower probabilities of cancer. The effect of one biomarker can mask the effect of the other at extreme values (i.e., low values of tPSA always result in low probabilities of cancer no matter what the fPSA values are, and high values of fPSA always result in low probabilities of cancer no matter how high the tPSA values are). We also provided as supplementary material a spreadsheet (see supplementary Dataset) for performing straightforward predictions. For example, for an 80 years old patient, if tPSA is 9 ng/mL and fPSA is 1.5 ng/mL, the calculated risk of PCa is 44.5% (see supplementary Dataset).
Furthermore, we estimated the improvement of our new predictive model versus the classical markers (tPSA levels) using the net reclassification improvement (NRI) and the integrated discrimination improvement (IDI). We observed that our predictive model had an NRI value of 0.870, splitted as NRI for the event (PCa) of 0.374 and NRI for the non-event (BB) of 0.496 (Supplementary Table S7). The IDI value was 0.201. So, the NRI and IDI scores indicate that our combination of several biomarkers in a predictive model may enhance sensitivities [increase for PCa (sensitivity) = 0.142] and specificities [decrease for BB (specificity) = 0.0587] of PSA alone, improving the ability to estimate the risk of PCa.
Additionally, we have analyzed the correlation between the Gleason score and the risk of PCa, calculated with our predictive model. For these analyses involving the Gleason score we only included samples from patients with reliable Gleason scores. In many cases, we couldn’t find the data, or the grading was not reliable because of the sample specimen analyzed. In the 164 PCa patients for whom we had the Gleason score, the coefficient of correlation was r = 0.56 (95% CI: 0.45–0.66, P < 0.0001) and the number of patients with tumours with Gleason scores ≥ 7 increased as the PCa risk increased (Fig. 2). Moreover, the AUC estimated by introducing the PCa risk, calculated with our model, and the dichotomized Gleason score (<7 and ≥ 7), was 0.83 (95% CI: 0.77–0.89, P < 0.0001). Using a cut-off value of PCa risk > 50%, the specificity was 39% for a sensitivity of 95%. This means that, using a sensitivity of 95%, we could have avoided about 39% of biopsies to PCa patients with a Gleason score <7.

Percentage of PCa patients with Gleason score ≥7 in relation to PCa risk.
Discussion
Over the past decade, PSA has been shown to be the most valuable diagnostic and prognostic marker in oncology. However, its reliability as screening tool for PCa remains controversial due to its lack of specificity. Several non-malignant conditions of the prostate, such as BB, are associated with an increased PSA levels35. Alternative biomarkers for PCa have emerged with the aim of increasing the diagnostic specificity, prognosis and staging of this cancer3,14,36,37,38,39,40,41. Some of these new diagnostic tools are related to PSA, such as PHI21,40, 4 K Score19,20 and the STHLM3 test23,24, or RCs to assess the PCa risk of a patient25. However, their clinical applicability is controversial and remain a matter of personal choice whether to use it in daily clinical practice.
PSA circulates in two forms, fPSA (30%) or PSA complexed to α1ACT (70%). The FPR or the CPR have become important markers to improve the specificity of tPSA and the differential diagnosis of BB and PCa33,42,43,44,45. There are different immunoassays for detection of these PSA molecular forms with different assay manufacturers. In systematic review, Roddam et al.34 described the diagnostic ability of the FPR and CPR in men with tPSA levels between 2 and 10 ng/mL, and its impact on clinical practice. They concluded that their use in this segment of patients could reduce the number of unnecessary biopsies whilst maintaining a high cancer detection rate.
Accordingly, we proposed the use of specific homemade ELISAs for tPSA, fPSA and PSA-α1ACT complex by using different pairs of antibodies that do not recognize hK2. These assays are equimolar and show detection limits and variation coefficients that are in all cases adequate, and have been validated in a large cohort of patients with PCa and BB (more information is shown in Supplementary material).
Our results show that PSA-α1ACT, FPR, CPR and FPR/CPR ratio significantly increase the diagnostic yield of tPSA when patients are classified according to different tPSA ranges (see Supplementary Tables S4–S6). In the diagnostic gray zone of 4 to 10 μg/l tPSA, using a cut-off value of 27 for FPR, 69 for CPR or 6 for FPR/CPR ratio (95% specificity), we could avoid about 35% of biopsies compared to 6% of biopsies avoided when using a cut-off value of 4.2 μg/l tPSA.
Our results compare well with those obtained with the PHI and [−2]proPSA markers, showing similar or better clinical performance for PCa detection46. There, the PHI was significantly higher in PCa patients than in patients without PCa, with an AUC of 0.70. Our results show AUCs for FPR, CPR and FPR/CPR, ranging from 0.79 to 0.81.
In order to improve the discrimination between PCa and BB, we compared our new predictive model to the classical marker (tPSA levels) using the NRI and the IDI. Our results, compared to those obtained with the PHI, with an AUC ranging from 0.70 to 0.7745,46, showed that our predictive model including age, tPSA, fPSA and the interaction of tPSA and fPSA, rendered a superior AUC (0.85; 95% CI = 0.83–0.89), demonstrating a better clinical performance for PCa detection. Additionally, our model shows another advantage, the independence from prostate volume. Only age is required as a valuable clinical factor, combined with the PSA molecular forms. Similar results are found when comparing our predictive model with the results described for the 4 K Score panel20, which could distinguish PCa and BB with good accuracy, with an AUC from 0.81 to 0.84, respectively, similar to that estimated with our predictive model (0.85). When we compared our predictive model with the seven well known RCs, that shown an AUC range between 0.64 and 0.72, we observed again how our predictive model exhibits a better discriminatory ability to predict PCa. And if we select only patients with clinically significant PCa, the RCs study with the highest AUC is 0.77, lower than the AUC obtained in our predictive model for all patients25.
Finally, Vickers et al.19, described a predictive model similar to ours, based on how additional kallikreins (fPSA, intact PSA, and hK2) could enhance discrimination of PCa diagnosis compared to a classical laboratory model (including age and tPSA) or a classical clinical model (including age, tPSA and DRE). They described a homemade ELISA to identify intact PSA and hK2 kallilkreins, based on modified mAbs with less nonspecific assay interference. This study obtained an AUC of 0.64 for tPSA levels, 0.70 including DRE, and 0.76 including age and additional kallikreins. We propose an alternative mAb design to quantify other markers (fPSA and PSA-α1ACT), based on equimolar tests that show no unspecific interactions with others kallikreins. Our strategy has allowed us to develop a predictive model with highest AUC, simple and easy to introduce in daily clinical practice.
Although our study had as main objective to identify a predictive model of PCa, given the current interest in identifying patients with more aggressive tumours, we decided to also analyze whether the predictive model of PCa obtained was also able to discriminate between aggressive and indolent phenotypes of PCa, for which we included in the study the 164 PCa patients for whom we had a reliable Gleason score. We observed a significant correlation between the Gleason score and PCa risk obtained with our model. Furthermore, the number of patients with a score ≥7 increased with the increase in the risk of PCa (Fig. 2), and the risk of PCa was significantly higher in patients with Gleason score ≥7 tumours (70%) compared with Gleason score 6 tumours (39%) (P < 0.0001). A similar result was reported by Stephan et al.45 using the PHI index. From the AUC performed introducing the PCa risk, calculated with our model, and the dichotomized Gleason score (<7 and ≥7), using a cut-off point of PCa risk >50%, the specificity was 39% for a sensitivity of 95%, indicating that with our model we could have avoided about 39% of biopsies to PCa patients with a Gleason score <7.
A limitation of this study is that our predictive model needs to be validated in a prospective study before prostate biopsy has been conducted, and needs an external validation using a multicenter study population. However, prospective population-based screening strategies are difficult to implement in daily clinical practice and require approval by local authorities. Another limitation is the unfeasibility to gather the variables needed for the estimation of proPSA and 4K-panel, so no head-to-head comparison of the different methods could be made. Finally, of the 301 patients with PCa we only had 164 patients with the available Gleason score. It would be necessary to confirm our results with a higher number of PCa patients.
In conclusion, we have developed and validated specific and sensitive immunoassays to quantify different PSA molecular forms. Our results using ROC curves for different tPSA ranges show that the FPR and CPR significantly increase the discriminating power of tPSA, fPSA and PSA-α1ACT.
In addition, the combination of tPSA, fPSA and age in a predictive model, increases the diagnostic power of tPSA, widely used in clinical practice, and may identify patients with a more aggressive tumours. Thus, the use of our predictive model may avoid unnecessary biopsies while high sensitivity is maintained, thus reducing unnecessary side effects in hundreds of thousands of patients every year and consequently unnecessary health expenses. A diagnosis in time and in early stages is very advisable and valuable, since it represents the critical stage regarding treatment and survival of the patient.
Material and Methods
Study subjects
This case-finding study included 1,065 patients with at least one prostate biopsy, 764 with BB and 301 with PCa, selected for having a positive DRE and/or a tPSA ≥4 μg/L. Samples were collected between 1997 and 2001.
PCa was objectively diagnosed by transrectal ultrasound-guided prostate biopsy, or by examination of tissue specimens following transurethral prostatectomy (incidental diagnosis). According to clinical situation, only 17 of the 301 PCa patients studied (5.7%) underwent transurethral resection for prostatic obstruction, a small percentage that would not cause deviation in the study population.
A diagnosis of BB was considered when patients had a negative biopsy and also when patients had an increase in prostate volume (>40 cm3) along with the following symptoms: obstructive symptoms, hesitant or intermittent micturition, decreased strength and thinning of the urinary stream gauge.
All participants were enrolled after giving written informed consent according to protocols approved by the ethics review board at La Fe University Hospital (reference no. 3009/0085). The procedures followed were in accordance with the Helsinki Declaration of 1975 as revised in 2008.
Blood collection
Blood was collected into Vacuette© sodium citrate tubes, centrifuged at 1,800 x g for 30 minutes at 4 °C and plasma samples were aliquoted and frozen at −70 °C until analysis.
Reagents
Purified hK2 and a monoclonal anti-hK2 antibody (HK1G86.1) that does not cross-react with PSA were provided by Hybritech Inc. (San Diego, CA). Biotin-NHS, human α1ACT and α2M, and rabbit anti-human α2M (IgG fraction) were obtained from Calbiochem (La Jolla, CA). Rabbit anti-human α1ACT (IgG fraction) was purchased from Dako A/S (Glostrup, Denmark). Aprotinin-Sepharose, casein, bovine serum albumin, horseradish peroxidase (HRP, type VI, RZ = 3.2), anti-mouse IgG (goat)-HRP, anti-mouse IgG (rabbit)-HRP whole molecule, Tween 20, hydrogen peroxide (H2O2), O-phenylenediamine (OPD), benzamidine chloride, dimethyl sulfoxide and dithiothreitol were from Sigma Chem. Co. (St. Louis, MO). Glutaraldehyde and 1,10-phenanthrolinium chloride were from E. Merck (Darmstadt, Germany). Streptavidin-HRP (SAHRP), biotinylated horse polyclonal anti-mouse IgG antibody and NBT/BCIP were from Pierce (Thermo Scientific, Waltham, MA USA). Streptavidin-alkaline phosphatase was from Bio-Rad. CM-Sephadex, Sephacryl S-200 and CNBr-activated Sepharose 4B were obtained from Pharmacia (Uppsala, Sweden).
Production and characterization of monoclonal anti-PSA antibodies
See supplementary results for the production, purification, isotyping, calculation of the apparent Kd (Supplementary Table S1), biotinylation, serum competition (Supplementary Fig. S1), SDS-PAGE and Western blots, reactivity of anti-PSA mAbs on immunoblots towards fPSA and PSA-α1ACT complex (Supplementary Table S2), competition between unlabelled and labelled mAbs for binding to PSA (Supplementary Table S3), cross-reactivity with hK2 (Supplementary Fig. S2) and reactivity of several pairs of anti-PSA mAbs towards fPSA and PSA-α1ACT complex (Supplementary Fig. S3).
Immunoassay for total PSA
The assay for tPSA was performed with M40 as capturing antibody and biotinylated (*) M73 as detecting antibody. Plates were coated with 5 mg/L of M40. After washing and blocking with blocking buffer, 50 μL/well of duplicated samples or calibrators were added and incubated for 1 h at room temperature (RT). After washing, 50 μL/well of M73* at a dilution of 1/8000 in 0.01 mol/L Tris-HCl, pH 7.4, 0.14 mol/L NaCl, 0.5 g Thimerosal per liter, 0.5 mL Tween 20 per liter, was added and incubated for 1 h at RT. After washing, SAHRP at 1/4000 was applied as described above. Colour was developed with OPD substrate and the reaction was stopped after 5 min with 35 μL/well of 4 mol/L H2SO4.
Immunoassay for PSA -α1ACT complex
The assay for PSA-α1ACT was performed with M40 as capturing antibody and a polyclonal HRP-labelled anti-α1ACT antibody as detecting antibody. Plates were coated with 5 mg/L of M40. After washing and blocking, 50 μL/well of duplicated samples or calibrators were added and incubated for 1 h at RT. After washing, 50 μL/well of HRP-labelled anti-α1ACT antibody at a dilution of 1/2000 was added and incubated for 1 h at RT. After washing, colour was developed with OPD substrate and the reaction was stopped after 5 min with 35 μL/well of 4 mol/L H2SO4.
Immunoassay for fPSA
The assay for fPSA was performed with M63 as capturing antibody and M50* as detecting antibody. Plates were coated with 5 mg/L of M63. After washing and blocking, 50 μL/well of duplicated samples or calibrators were added and incubated for 1 h at RT. After washing, 50 μL/well of M50* at a dilution of 1/10000 was added and incubated for 1 h at RT. After washing, SAHRP at 1/4000 was applied as described above. Colour was developed with OPD substrate and the reaction was stopped after 5 min with 35 μL/well of 4 mol/L H2SO4.
Immunoassay for PSA -α2M complex
The assay for PSA-α2M complex was performed as reported before for the activated protein C-α2M complex47. Briefly, plasma samples were pre-treated with dithiothreitol and then with iodoacetamide, in order to expose PSA epitopes. The M40 anti-PSA antibody was used as coating antibody and biotinylated anti-IgG antibody was used as detecting antibody. The detection range of the PSA-α2M assay was 0.1 to 4.0 ng/ml of complex.
Statistical analysis
Data were summarized using median, standard deviation and 1st and 3rd quartile in the case of continuous variables and relative and absolute frequencies in the case of categorical variables. Multivariable logistic regression models were constructed with different combinations of the PSA molecular forms and clinical variables. Selection of the best model for discriminating between PCa and BB was performed using the AIC48. Calibration of the final model was assessed using a scatter plot of the predicted versus observed probabilities using 500 bootstrap replicates of the sample. Although the bootstrap is not a substitute of a large sample external validation, it is the best method for assessing calibration when no external validation is feasible. The predictive ability of the model was assessed by estimating an optimism-corrected area under the curve (AUC) for the receiver operator characteristic (ROC) analysis, and using 1000 bootstrap replicates. All these statistical analyses were performed using R (version 3.4.2) and R-packages pROC (1.10.0) and rms (5.1–1). Moreover, the improvement of the new (multivariable logistic regression model) versus the classical marker, tPSA, were estimated by using the NRI and the IDI. For that, the R-package Hmisc (4.1–1) was used for the NRI and IDI of logistic regression models49. NRI and IDI values were provided as a complement of the AUC values for the different models, following specific guidelines49, and providing only 95% CI for NRI and IDI. The AUC to assess the diagnostic accuracy of each parameter was calculated using a commercially available computer program for medical statistics50. Analysis of ROC curves using clinical groups was performed on patients according to tPSA levels: a) tPSA between 0.76 and 975 µg/L (whole cohort), b) ≥ 2.5 and <4.0 μg/L, c) ≥ 4 and <10 μg/L and d) ≥ 10 and <20 μg/L. Using the same statistical program, we estimated the AUC introducing the PCa risk, calculated with our model, and the dichotomized Gleason score (<7 and ≥7).
References
Jemal, A. et al. Cancer statistics, 2008. CA Cancer J Clin. 58, 71–96 (2008).
Pinsky, P. F. et al. Repeat prostate biopsy in the prostate, lung, colorectal and ovarian cancer screening trial. BJU Int. 99, 775–779 (2007).
Filella, X. & Foj, L. Emerging biomarkers in the detection and prognosis of prostate cancer. Clin. Chem. Lab. Med. 53, 963–973 (2015).
Piironen, T. et al. Immunofluorometric assay for sensitive and specific measurement of human prostatic glandular kallikrein (hK2) in serum. Clin. Chem. 42, 1034–1041 (1996).
Stamey, T. A. et al. Prostate-specific antigen as a serum marker for adenocarcinoma of the prostate. N. Engl. J. Med. 317, 909–916 (1987).
Heidenreich, A. et al. EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur. Urol. 65, 467–479 (2014).
Roach, M. et al. Defining biochemical failure following radiotherapy with or without hormonal therapy in men with clinically localized prostate cancer: recommendations of the RTOG-ASTRO Phoenix Consensus Conference. Int. J. Radiat. Oncol. Biol. Phys. 65, 965–974 (2006).
Christensson, A. et al. Serum prostate specific antigen complexed to alpha 1-antichymotrypsin as an indicator of prostate cancer. J. Urol. 150, 100–105 (1993).
Catalona, W. J. et al. Comparison of digital rectal examination and serum prostate specific antigen in the early detection of prostate cancer: results of a multicenter clinical trial of 6,630 men. J. Urol. 151, 1283–1290 (1994).
Park, J. et al. Low testosterone level is an independent risk factor for high-grade prostate cancer detection at biopsy. B.J.U. Int. 118, 230–235 (2015).
De Luca, S. et al. High prostate cancer gene 3 (PCA3) scores are associated with elevated Prostate Imaging Reporting and Data System (PI-RADS) grade and biopsy Gleason score, at magnetic resonance imaging/ultrasonography fusion software-based targeted prostate biopsy after a previous negative standard biopsy. B.J.U. Int. 118, 723–730 (2016).
Jablonka, F. et al. Serum crosslinked-N-terminal telopeptide of type I collagen (NTx) has prognostic implications for patients with initial prostate carcinoma (PCa): a pilot study. Clin. Chim. Acta 436, 316–318 (2014).
Matin, F. et al. A Plasma Biomarker Panel of Four MicroRNAs for the Diagnosis of Prostate Cancer. Sci. Rep. 8, 6653, https://doi.org/10.1038/s41598-018-24424-w (2018).
Shariat, S. F., Karam, J. A., Margulis, V. & Karakiewicz, P. I. New blood-based biomarkers for the diagnosis, staging and prognosis of prostate cancer. B.J.U. Int. 101, 675–683 (2008).
Ferrer-Batallé, M. et al. Comparative Study of Blood-Based Biomarkers, α2,3-Sialic Acid PSA and PHI, for High-Risk Prostate Cancer Detection. Int. J. Mol. Sci. 18, https://doi.org/10.3390/ijms18040845 (2017).
Fu, S., Zhang, X., Niu, Y. & Wang, R. T. Prostate Specific Antigen, Mean Platelet Volume, and Platelet Distribution Width in Combination to Discriminate Prostate Cancer from Benign Prostate Hyperplasia. Asian Pac J Cancer Prev 19, 699–702 (2018).
Vickers, A. J. et al. A panel of kallikrein markers can reduce unnecessary biopsy for prostate cancer: data from the European Randomized Study of Prostate Cancer Screening in Göteborg, Sweden. B.M.C. Med. 6, 19 (2008).
Foj, L. et al. Real-time PCR PCA3 assay is a useful test measured in urine to improve prostate cancer detection. Clin. Chim. Acta 435, 53–58 (2014).
Vickers, A. et al. Reducing unnecessary biopsy during prostate cancer screening using a four-kallikrein panel: an independent replication. J. Clin. Oncol. 28, 2493–2498 (2010).
Carlsson, S. et al. Predictive value of four kallikrein markers for pathologically insignificant compared with aggressive prostate cancer in radical prostatectomy specimens: results from the European Randomized Study of Screening for Prostate Cancer section Rotterdam. Eur. Urol. 64, 693–699 (2013).
Boegemann, M. et al. The percentage of prostate-specific antigen (PSA) isoform [-2]proPSA and the Prostate Health Index improve the diagnostic accuracy for clinically relevant prostate cancer at initial and repeat biopsy compared with total PSA and percentage free PSA in men aged ≤65 years. B.J.U. Int. 117, 72–79 (2016).
Filella, X. & Gimenez, N. Evaluation of [−2] proPSA and Prostate Health Index (phi) for the detection of prostate cancer: a systematic review and meta-analysis. Clin. Chem. Lab. Med. 51, 729–739 (2013).
Loeb, S., Roehl, K. A., Thaxton, C. S. & Catalona, W. J. Combined prostate-specific antigen density and biopsy features to predict “clinically insignificant” prostate cancer. Urology 72, 143–147 (2008).
Ström, P., Nordström, T., Grönbergk, H. & Eklund, M. The Stockholm-3 Model for Prostate Cancer Detection: Algorithm Update, Biomarker Contribution, and Reflex Test Potential. Eur. Urol. 74, 204–210 (2018).
Pereira-Azevedo, N., Verbeek, J. F. M., Nieboer, D., Bangma, C. H. & Roobol, M. J. Head-to-head comparison of prostate cancer risk calculators predicting biopsy outcome. Transl. Androl. Urol. 7, 18–26 (2018).
Liss, M. A., Xu, J., Chen, H. & Kader, A. K. Prostate genetic score (PGS-33) is independently associated with risk of prostate cancer in the PLCO trial. Prostate 75, 1322–1328 (2015).
Lose, F. et al. Common variation in Kallikrein genes KLK5, KLK6, KLK12, and KLK13 and risk of prostate cancer and tumor aggressiveness. Urol. Oncol. 31, 635–643 (2013).
Chunhua, L. et al. Clinical Significance of Peripheral Blood PCA3 Gene Expression in Early Diagnosis of Prostate Cancer. Transl. Oncol. 11, 628–632 (2018).
España, F. et al. Quantitative immunoassay for complexes of prostate-specific antigen with alpha2-macroglobulin. Clin. Chem. 42, 545–550 (1996).
McCormack, R. T. et al. Molecular forms of prostate-specific antigen and the human kallikrein gene family: a new era. Urology 45, 729–744 (1995).
Zhu, L. et al. PSA forms complexes with α1-antichymotrypsin in prostate. Prostate 73, 219–226 (2013).
Zhu, Y. et al. Effect of body mass index on the performance characteristics of PSA-related markers to detect prostate cancer. Sci. Rep. 6, 19034, https://doi.org/10.1038/srep19034 (2016).
Catalona, W. J. et al. Evaluation of percentage of free serum prostate-specific antigen to improve specificity of prostate cancer screening. J.A.M.A. 274, 1214–1220 (1995).
Roddam, A. W. et al. Use of prostate-specific antigen (PSA) isoforms for the detection of prostate cancer in men with a PSA level of 2-10 ng/ml: systematic review and meta-analysis. Eur. Urol. 48, 386–399 (2005).
España, F. et al. Free and complexed prostate-specific antigen in the differentiation between benign prostatic hyperplasia and prostate cancer. Studies in serum and plasma samples. J. Urol. 160, 2081–2088 (1998).
Catalona, W. J. et al. Measurement of prostate-specific antigen in serum as a screening test for prostate cancer. N. Engl. J. Med. 324, 1156–61 (1991).
Richardsen, E. et al. MicroRNA 141 is associated to outcome and aggressive tumor characteristics in prostate cancer. Sci. Rep. 9, 386, https://doi.org/10.1038/s41598-018-36854-7 (2019).
Bravaccini, S. et al. PSMA expression: a potential ally for the pathologist in prostate cancer diagnosis. Sci. Rep. 8, 4254, https://doi.org/10.1038/s41598-018-22594-1 (2018).
Bansal, N. et al. Relevance of MIC-1 in the Era of PSA as a Serum Based Predictor of Prostate Cancer: A Critical Evaluation. Sci. Rep. 7, 16824, https://doi.org/10.1038/s41598-017-17207-2 (2017).
Catalona, W. J. et al. A multicenter study of [−2]pro-prostate specific antigen combined with prostate specific antigen and free prostate specific antigen for prostate cancer detection in the 2.0 to 10.0 ng/ml prostate specific antigen range. J. Urol. 185, 1650–1655 (2011).
Nordström, T., Akre, O., Aly, M., Grönberg, H. & Eklund, M. Prostate-specific antigen (PSA) density in the diagnostic algorithm of prostate cancer. Prostate Cancer Prostatic Dis. 21, 57–63 (2018).
España, F. et al. Longitudinal evaluation of the complexed-to-total prostate specific antigen ratio in men with prostate disease. Effect of treatment. Eur. J. Cancer 34, 1375–1380 (1998).
Martínez, M. et al. Prostate-specific antigen complexed to alpha(1)-antichymotrypsin in the early detection of prostate cancer. Eur. Urol. 38, 85–90 (2000).
Vashi, A. R. & Oesterling, J. E. Percent free prostate-specific antigen: entering a new era in the detection of prostate cancer. Mayo Clin. Proc. 72, 337–344 (1997).
Stephan, C. et al. Multicenter evaluation of [−2]proprostate-specific antigen and the prostate health index for detecting prostate cancer. Clin. Chem. 59, 306–314 (2013).
Na, R. et al. Performance of serum prostate-specific antigen isoform [−2]proPSA (p2PSA) and the prostate health index (PHI) in a Chinese hospital-based biopsy population. Prostate 74, 1569–1575 (2014).
Martos, L. et al. α2-macroglobulin is a significant in vivo inhibitor of activated protein C and low APC:α2M levels are associated with venous thromboembolism. Thromb. Haemost. 118, 630–638 (2018).
Burnham, K. P. & Anderson, D. R. Multimodel inference: understanding AIC and BIC in Model Selection. Sociol. Methods Res. 33, 261–304 (2004).
Leening, M. J., Vedder, M. M., Witteman, J. C., Pencina, M. J. & Steyerberg, E. W. Net reclassification improvement: computation, interpretation, and controversies: a literature review and clinician’s guide. Ann. Intern. Med. 160, 122–131 (2014).
Schoonjans, F., Zalata, A., Depuydt, C. E. & Comhaire, F. H. Medcalc: A new computer program for medical statistics. Comput. Methods Programs Biomed. 48, 257–262 (1995).
Acknowledgements
We would like to thank María José Solmoirago, Cristina Elías and Noelia Santillana for their excellent technical support and Arash Javadinejad for his assistance in the English editing. This research was supported by research grants from Instituto de Salud Carlos III (PI14/00079, PI14/00512, FI14/00269, CPII15/00002, PI17/00495), FEDER una manera de hacer Europa, Generalitat Valenciana (PrometeoII/2015/017, ACIF/2017/138).
Author information
Authors and Affiliations
Contributions
A.F.P., M.R., L.M. and J.O. performed the experiments, analyzed the data and revised the manuscript. M.M. and C.D.V.-D. recruited patients and revised the manuscript. D.H. performed the statistical analysis. M.J.H. participated in the study concept and design, analyzed the data and revised the manuscript. F.E., P.M. and S.N. performed the research, participated in the study concept and design, analyzed the data, wrote and revised the manuscript. All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oto, J., Fernández-Pardo, Á., Royo, M. et al. A predictive model for prostate cancer incorporating PSA molecular forms and age. Sci Rep 10, 2463 (2020). https://doi.org/10.1038/s41598-020-58836-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-58836-4
This article is cited by
-
Alpha-1-antichymotrypsin as a novel biomarker for diagnosis, prognosis, and therapy prediction in human diseases
Cancer Cell International (2022)
-
Applying a genetic risk score for prostate cancer to men with lower urinary tract symptoms in primary care to predict prostate cancer diagnosis: a cohort study in the UK Biobank
British Journal of Cancer (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
