
Abstract
The PROteKT study tested the hypothesis that rosuvastatin can inhibit aminoglycoside-induced nephrotoxicity in children with Cystic Fibrosis (CF). This open label, parallel group, randomised controlled trial recruited children and young people aged 6 to 18 years with CF at 13 paediatric CF treatment centres in the UK. Participants were randomised equally to either receive oral rosuvastatin (10 mg once daily) or no intervention (control) throughout clinically indicated treatment with intravenous tobramycin. The primary outcome was the difference between the groups in mean fold-change in urinary Kidney Injury Molecule-1 (KIM-1). Fifty (rosuvastatin n = 23, control n = 27) participants were recruited between May 2015 and January 2017. Primary outcome data was available for 88% (rosuvastatin n = 20, control n = 24). The estimated mean treatment difference in the geometric mean-fold change of normalised KIM-1 was 1.08 (95% CI 0.87–1.35, p = 0.48). In total there were 12 adverse reactions, all mild, reported by five participants randomised to rosuvastatin, and one serious adverse event in each group. Whilst no protective effect of rosuvastatin was seen, there was a lower than expected level of nephrotoxicity in the cohort. Therefore, we can neither confirm nor refute the hypothesis that rosuvastatin protects against aminoglycoside nephrotoxicity.
Similar content being viewed by others

Introduction
In Cystic fibrosis (CF) the resistant pathogen Pseudomonas aeruginosa is commonly implicated in secondary bacterial lung infections and colonisation. Aminoglycoside antibiotics, usually combined with a beta-lactam, are frequently used to treat respiratory exacerbations in CF1, as they are effective against P. aeruginosa. Whilst this approach generally leads to improved patient outcomes, aminoglycoside use is also associated with an increased risk of nephrotoxicity.
The incidence of acute kidney injury (AKI) in children with CF is increased with current or recent exposure to aminoglycosides2,3,4, with rates of 20% reported using daily monitoring of serum creatinine5. The risk of AKI is higher with longer duration of aminoglycoside use and with recent previous aminoglycoside exposure6. In one cohort of adults with CF, between 31% and 42% had evidence of chronic renal impairment which was significantly associated with cumulative aminoglycoside exposure7. However, this has not been replicated in other cohorts8,9. Strategies such as therapeutic drug monitoring and extended-interval dosing10 are only partially effective in preventing aminoglycoside-induced nephrotoxicity. It is therefore important to develop further, more effective, strategies.
Aminoglycosides cause targeted toxicity to renal proximal tubule epithelial cells. Megalin, a multi-ligand receptor, facilitates the endocytosis and accumulation of aminoglycosides in these cells11. This pathway is activated by intermediates derived from mevalonate, which is formed from 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) via the enzyme HMG-CoA reductase12. We hypothesised that inhibiting HMG-CoA reductase with a statin would lead to a reduction in toxicity. Statins are inhibitors of megalin-mediated endocytosis in vitro13,14, and are therefore likely to reduce uptake of aminoglycosides in the proximal tubule. Indeed, in vivo studies of aminoglycoside-induced nephrotoxicity in rats have consistently demonstrated a protective effect of statins15,16,17,18,19,20.
In this paper, we describe the first clinical trial to evaluate the repurposing of a statin for the prevention of aminoglycoside-induced kidney toxicity. Rosuvastatin was chosen as it is hydrophilic, has minimal hepatic metabolism, and has some renal elimination of the parent drug21 (and therefore theoretically preferable in preventing megalin-mediated uptake rather than a hepatically metabolised statin). In addition, it is highly potent, and licensed for use in children. This trial was designed to test the hypothesis that rosuvastatin can inhibit aminoglycoside-induced nephrotoxicity in children with CF.
Results
Study participants
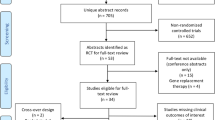
A total of 258 patients were assessed for eligibility across 13 CF centres in the UK (May 2015-January 2017); 208 were not randomised as they failed to meet the inclusion/exclusion criteria or did not provide consent. The planned sample size was met with twenty-three participants randomised to the intervention and 27 to the control (Fig. 1). Baseline characteristics are shown in Table 1. The two arms were well balanced and no clinical differences were deemed to be important.

CONSORT Flow Diagram.
Five (22%) and four (15%) participants randomised to the intervention and control groups, respectively, discontinued tobramycin treatment early. Median duration of treatment was 14 days in both the intervention and control groups (Supplemental Table 1). Four participants in the intervention group discontinued tobramycin treatment due to a change in their condition and one due to line failure. One control group participant discontinued tobramycin treatment early due to a change in their condition, two were a clinical decision to cease and, in one case, no reason was given.
Assessment of outcome measures
In terms of the primary outcome, the estimated geometric mean fold-change of normalised Kidney Injury Molecule-1 (KIM-1) was 1.85 and 2.00 in the control and intervention groups respectively (Table 2). The estimated mean treatment difference was 1.08 (95% CI: 0.87,1.35; p = 0.48). Four participants (intervention n = 1 and control n = 3) did not have baseline urine samples and were excluded from the primary analysis. Five sensitivity analyses confirmed the conclusion found in the primary analysis (Supplemental Table 2). The estimated mean treatment difference in the area under the curve (AUC) of normalised KIM-1 between the two treatment groups was 12.41 ng/mgCr (95% CI: −0.89,25.70; p = 0.07) (Supplemental Table 3). There were no statistically significant differences between the treatment groups and the treatment by time interactions were not significant for the secondary outcomes (Table 3). Mean profile plots for each outcome measure are included in Fig. 2.

Mean profile plots. Mean profile plots for control and intervention (rosuvastatin) groups during tobramycin exposure for creatinine (A), KIM-1 normalised to urinary creatinine (B), Schwartz eGFR (C), and NGAL normalised to urinary creatinine (D).
The estimated geometric mean fold-change of normalised Neutrophil Gelatinase-Associated Lipocalin (NGAL) was 8.90 and 4.99 in the control and intervention groups respectively. The estimated mean treatment difference was 0.56 (95% CI: 0.27,1.15; p = 0.11) (Supplemental Table 4). The estimated mean treatment difference in the AUC of normalised NGAL between the two arms was 557.8 ng/mgCr (95% CI: 46.5,1069.2; p = 0.03) (Supplemental Table 3). The tobramycin and rosuvastatin data are reported in Supplemental Tables 5 and 6, respectively.
An analysis of serum creatinine data using the Kidney disease: Improving global outcomes (KDIGO) AKI criteria22 demonstrated that a total of four participants fulfilled the criteria for AKI during the trial: two patients in the control group both reached AKI stage 3, and two patients in the intervention group both reached AKI stage 1 (Supplemental Table 7).
Safety and adverse events
Two participants randomised to the intervention group withdrew consent prior to receiving the intervention and were therefore not included in the safety analysis. Twelve adverse reactions were reported by 5 (25%) of the 21 participants in the intervention group (Table 4). All events were reported as mild in severity.
Recurrence of pulmonary exacerbation during follow-up, requiring hospitalisation, was the only serious adverse event in the control group, reported for one (4%) participant. Abnormal blood results requiring prolonged hospitalisation were reported as a serious adverse event for one (5%) participant in the intervention group but this was not considered to be related to the intervention.
Discussion
This is the first randomised controlled trial in man to assess whether rosuvastatin has a protective effect against tobramycin-induced nephrotoxicity. It was designed as an early phase trial using KIM-1 as a surrogate outcome measure. We found little evidence of a protective effect of rosuvastatin in this trial as determined by both primary and secondary outcomes, but this should be interpreted with caution as the control group only demonstrated a mean fold-change of KIM-1 of 1.85. The sample size calculation for this study was, however, based upon an expected mean fold-change in KIM-1 of 3.03 during exposure to tobramycin derived from an early analysis of samples in the URBAN CF study23. The trial was powered to detect a difference in fold-change between the groups of 2. The PROteKT and URBAN CF populations were similar, although PROteKT contained a greater proportion of female participants, and had higher mean age, height and weight (due to differences in the inclusion criteria for age). There were no differences in aminoglycoside prescribing practices between the two studies. We do not believe that the differences between the populations used in PROteKT and URBAN CF account for lower than expected levels of nephrotoxicity observed in our randomised controlled trial. Therefore, it is unclear whether this reflects lower than usual levels of toxicity in the PROteKT cohort, or higher than usual levels of toxicity in the cohort upon which the sample size calculation was based. This makes it difficult to definitively exclude the possibility of a protective effect of rosuvastatin. Indeed, some of our secondary outcome measures suggest a trend towards a protective effect. Importantly, the use of rosuvastatin was generally well tolerated.
Urine KIM-1 is a cell membrane glycoprotein upregulated by proximal tubule epithelial cells in response to toxicity24. It was chosen as the primary outcome measure as it has previously shown potential for the identification of aminoglycoside-induced nephrotoxicity in preterm neonates25, and in children with CF23,26, as well as outperforming other biomarkers of aminoglycoside-induced nephrotoxicity in pre-clinical models27. In clinical practice, changes in serum creatinine meeting agreed criteria for AKI22 are more widely accepted for defining nephrotoxicity. However, changes in serum creatinine are a relatively late event in the process of nephrotoxicity, and for the purposes of a phase IIa trial, it was felt that KIM-1 was a good surrogate measure. Indeed, we identified no significant treatment effect upon serum creatinine in this study, consistent with our previous experience in the URBAN CF study23. The use of a change in serum creatinine as an outcome measure may be relevant to use in future trials, but given that this is estimated to occur in 14–20%5,6, a much larger sample size will be required.
Urine NGAL is a 25 kDa protein expressed by kidney epithelial cells (as well as other tissues, including neutrophils)28. It is a sensitive predictor for AKI29, and has previously been shown to be elevated during aminoglycoside exposure in neonates25, and in children with CF23. Patients in the intervention group had a smaller estimated geometric mean fold-change of normalised NGAL (p = 0.11), and a significantly lower AUC of normalised NGAL. The difference in AUC between the groups must be interpreted in the light of a higher baseline urinary NGAL in the control compared to the intervention group (61.08 ng/mgCr and 22.46 ng/mgCr respectively). This may, in part, be explained by the higher proportion of females in the control versus intervention groups as females have been consistently shown to have higher baseline NGAL concentrations than males30. Whilst our results may be interpreted to suggest a protective effect of rosuvastatin, we feel that these should be interpreted cautiously.
Published studies in rat models of aminoglycoside-induced nephrotoxicity have consistently demonstrated a protective effect with a range of statins, including atorvastatin17,18,19, simvastatin15,16 and rosuvastatin20. We felt that rosuvastatin was the most promising candidate based upon its pharmacology. It is a hydrophilic statin, and has greater renal excretion than the more lipophilic atorvastatin and simvastatin. It causes more proteinuria than other statins, probably because it is secreted in the proximal tubules31 and inhibits megalin-mediated endocytosis. We considered whether we should have given the statin for a few days in advance of commencing the tobramycin in order to achieve steady-state concentrations. However, published pre-clinical studies demonstrated a protective effect of statins without the need for this15,17,18,19,20.
The recruitment of children with CF from thirteen different sites in the UK to participate in this trial ensures that differences in practice between centres should be accounted for in the randomisation. The trial was designed to cause the least disruption possible to standard daily care, and for the intervention to be minimally burdensome to patients (requiring only one additional medicine per day for 2 weeks). This was reflected in the good adherence to the randomised intervention, and by a low number of reported adverse reactions.
In conclusion, given the lower than expected level of nephrotoxicity seen in this study we feel that we can neither confirm nor refute the hypothesis that statins protect against aminoglycoside nephrotoxicity. Given the large amount of pre-clinical data which shows that statins can be protective15,16,17,18,19,20, we feel further studies in humans are warranted. These would need to be powered for a clinically acceptable primary outcome measure such as AKI (rise in creatinine). There are of course other interventions which have been attempted to reduce the risk of aminoglycoside nephrotoxicity, including extended-interval dosing and drug trough level monitoring10, and morning (versus evening) administration of the aminoglycoside32. All these interventions are not mutually exclusive, and it may be that combinations of interventions may be more successful in preventing renal injury when compared with individual interventions, and such a possibility should be investigated in an appropriately designed clinical trial in the future.
Methods
Protocol and trial population
This multi-centre, randomised, open-labelled, parallel group, phase IIa trial was registered in duplicate on both the EU Clinical Trials Register (EudraCT number 2014-002387-32, 27/06/2014) and the ISRCTN Registry (ISRCTN26104255, 05/09/2014). It received ethical approval from the National Research Ethics Service Committee North West – Greater Manchester Central, and was conducted in accordance with the Declaration of Helsinki. Participants were recruited at 13 paediatric CF treatment centres in the UK. Each parent or guardian gave written informed consent, and each child gave assent where appropriate.
Children and young people with CF, aged 6 to 18 years, who had a planned, clinically indicated (usually for a respiratory exacerbation of CF), course of treatment with IV tobramycin were eligible for randomisation. Key exclusion criteria included participants of Asian ancestry (specifically Japanese, Chinese, Filipino, Vietnamese and Korean subjects, as they have clinically significant increased systemic exposure to rosuvastatin33), previous adverse reaction to a statin, and existing treatment with a statin. Participants with renal disease, elevation of either transaminases and/or creatine kinase were also excluded. The full inclusion and exclusion criteria are described in the trial protocol (Appendix 1).
There were amendments to the eligibility criteria during the trial. The age inclusion criteria were changed from 10–18 to 6–18 years following a change in the licence for rosuvastatin34. A review of published pharmacokinetic data suggested that concomitant itraconazole35 or Asian-Indian ethnicity33 would result in clinically insignificant increases in rosuvastatin exposure and therefore these were removed from the exclusion criteria.
Randomisation and trial procedures
Randomisation was performed, by the Principal Investigator or delegated other at site, using a secure web-based system with random permuted blocks of 2 and 4 stratified by trial site. Randomisation lists were generated using Stata v9.0 by an independent statistician. Emergency back-up randomisation envelopes were available when there was a problem with this system. At enrolment eligible participants were randomised 1:1 to receive either rosuvastatin or no intervention.
Each participant randomised to the intervention arm received a daily oral dose of 10 mg rosuvastatin (Crestor®; AstraZeneca UK Ltd) for the duration of a treatment course of IV tobramycin (usually 14 days). Tobramycin was the first choice aminoglycoside in CF for all centres, and dosing and monitoring of drug concentrations followed the British National Formulary36 (standard dosing is 10 mg/kg once daily) and local protocols. The first dose of rosuvastatin was given on the same calendar day and prior to the first dose of IV tobramycin. Subsequent daily doses of rosuvastatin were given prior to that day’s dose of tobramycin. Treatment with oral rosuvastatin continued for the duration of the course of IV tobramycin. If the tobramycin course was shorter than 14 days, rosuvastatin was discontinued after the final tobramycin dose. If the tobramycin course was longer than 14 days, rosuvastatin was continued until the final day of the tobramycin course. Participants randomised to the no intervention arm received usual care during their IV tobramycin treatment course. No placebo was provided.
Sample collection
Baseline assessment before tobramycin therapy (T0) included collection of urine and blood samples for confirmation of eligibility criteria, and spirometry. During treatment with IV tobramycin urine samples were collected daily from each child. Participants had assessments on days T + 1, T + 8 and T + 13. These included adverse event assessment, a symptom-directed physical examination if required, and collection of blood and urine samples. Spirometry was performed at the T + 8 and T + 13 visits. A final follow-up visit, with collection of blood/urine samples and spirometry, was conducted 3–5 weeks following completion of tobramycin treatment.
Urine samples were normally collected by clean catch into a sterile container. If the participant was an inpatient, the daily samples were sent directly to the local laboratory. If the participant received IV tobramycin at home, samples were stored in the home refrigerator in a sealed container until the next scheduled study visit. All samples were stored at fridge temperature (4 °C) for a total of one week (including time spent at the patient’s home) and then aliquoted and stored at −80 °C (or −20 °C for a maximum of 6 months) in the local laboratory. Stability of KIM-1 has previously been reported in urine stored at 4 °C for periods of one week37,38.
Wherever possible, study bloods were collected by venepuncture at the same time as any clinically indicated bloods in order to minimise the burden to the patient. A minimum of two 1.2 ml samples in Lithium/Heparin tubes, and one 1.2 ml sample in a serum tube were collected at each time point. Plasma and serum were extracted locally at each site, aliquoted and stored at −80 °C.
Batched samples were couriered on dry ice to the Good Clinical Practice Laboratory (GCPLab) facility in Liverpool for subsequent storage and analysis (here samples were stored at −80 °C). All laboratory analyses were undertaken in a standardised manner, being blinded to allocation, time of sampling and clinical outcomes of patients.
Outcome measures
The primary outcome was the difference in mean fold-change in urinary KIM-1 from baseline to ‘highest value’ concentration during exposure to tobramycin between the intervention and control arms.
The secondary outcome measures were the difference in serum creatinine, estimated glomerular filtration rate (eGFR) and urinary NGAL between the intervention and control arms during exposure to tobramycin. We assessed for pharmacokinetic interaction between rosuvastatin and tobramycin by comparing tobramycin concentrations between the rosuvastatin treated arm and the control arm. We assessed for pharmacodynamic interaction between rosuvastatin and tobramycin, by comparing change in percent of predicted Forced Expiratory Volume in 1 second (FEV1), between the rosuvastatin treated and control arms. This is a widely used indirect measure of aminoglycoside treatment outcome in children with CF39, and was measured locally during study visits. We also compared change in C-Reactive Protein (CRP), as a marker of inflammation/infection, between the two groups. Rosuvastatin concentrations were measured in the intervention arm participants to describe the pharmacokinetic profile of rosuvastatin, to assess compliance, and to relate rosuvastatin concentrations to change in urinary KIM-1.
Safety
All the participants who received at least one dose of rosuvastatin were included in the safety analysis. Adverse reactions and serious adverse events were recorded from the point of informed consent and throughout the trial treatment period up until the final assessment (3–5 weeks after the participant had taken the final dose of rosuvastatin).
Laboratory analyses
Urinary KIM-1 and NGAL were measured using validated electrochemiluminescent assays (Meso Scale Discovery (MSD), US) as previously described23,30. Biomarker values were normalised to urinary creatinine which was determined spectrophotometically as previously described40. Serum creatinine, tobramycin concentrations and CRP were measured in the laboratory serving the local hospital site.
Rosuvastatin concentrations were measured centrally at the University of Liverpool. A high performance liquid chromatography mass spectrometric method for the estimation of rosuvastatin in plasma, was developed and validated using rosuvastatin-D6 as internal standard. Sample preparation was accomplished by protein precipitation. The samples were chromatographed on a C-18 Halo column using mobile phases consisting of 0.1% formic acid in water and 0.1% formic acid in acetonitrile. The method was validated over a concentration range of 0.5–100 ng/mL for rosuvastatin.
Statistical analysis
The statistical analysis plan (Appendix 2) was written prior to any formal analysis. The trial was designed with a 92% power to detect a difference in fold-change in KIM-1 between the 2 groups at a 5% significance level (two sided). A standard deviation of 1.84 was derived from an early analysis of samples in the URBAN CF study23 from 10 participants receiving a single course of treatment with tobramycin. The same data was also inspected to assess that the assumption of normality is reasonable. Using these assumptions and utilizing a 2-sample t-test, a sample size of 20 in each arm would be sufficient. We planned to include 50 patients in order to compensate for 10% loss to follow up.
The principle of intention-to-treat was the main strategy of the analysis adopted for the primary outcome and all the secondary outcomes. These analyses were conducted on all randomised participants, in the group to which they were allocated, and for whom the outcome(s) of interest have been observed/measured.
The primary outcome was analysed using the method of analysis of covariance (ANCOVA). The outcome measure was the mean log-transformed fold-change of normalised KIM-1 (ng/mgCr) calculated by dividing the peak value, corresponding to the ‘highest value’ of normalised KIM-1 during exposure to tobramycin, by the baseline normalised KIM-1 value for each participant. The explanatory variables were treatment group and baseline normalised KIM-1 value. 95% confidence intervals will be reported and a p-value of <0.05 considered statistically significant. The secondary outcomes were analysed longitudinally using random intercept models with unstructured covariance matrices, including an interaction between treatment group and visit.
In an additional analysis of normalised NGAL (ng/mgCr), an ANCOVA model was used, as for KIM-1 above, comparing log-transformed mean fold-change from baseline to peak NGAL between the treatment groups, controlling for the baseline normalised NGAL. The model estimates were exponentiated to be interpretable on the normal scale.
In an additional descriptive analysis of serum creatinine data, change from baseline serum creatinine during tobramycin exposure was defined according to the KDIGO AKI criteria (Stage 1, increase ≥1.5 and <2 times from baseline; Stage 2, increase ≥2 and <3 times from baseline; Stage 3, increase ≥3 times from baseline)22.
Pre-specified sensitivity analyses tested impact of missing data; the statistical analysis plan includes full details of sensitivity analyses and analyses of secondary outcomes. Analyses were performed using SAS® version 9.4 (SAS Inc., USA).
Data availability
Data may be requested by submitting a written request to the corresponding author outlining the purposes for which the data would be used.
Change history
10 March 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Robinson, C. A. & Kuhn, R. J. Management of pulmonary disease in patients with cystic fibrosis. J. Pharm. Pract. 14, 207–227 (2001).
Bertenshaw, C., Watson, A. R., Lewis, S. & Smyth, A. Survey of acute renal failure in patients with cystic fibrosis in the UK. Thorax 62, 541–545 (2007).
Saban, J. A., Pizzi, M., Caldwell, J., Palijan, A. & Zappitelli, M. Previous aminoglycoside use and acute kidney injury risk in non-critically ill children. Pediatr. Nephrol. 32, 173–179 (2017).
Smyth, A. et al. Case-control study of acute renal failure in patients with cystic fibrosis in the UK. Thorax 63, 532–535 (2008).
Downes, K. J. et al. Daily serum creatinine monitoring promotes earlier detection of acute kidney injury in children and adolescents with cystic fibrosis. J. Cyst. Fibros. 13, 435–441 (2014).
Downes, K. J. et al. Risk factors for acute kidney injury during aminoglycoside therapy in patients with cystic fibrosis. Pediatr. Nephrol. 30, 1879–1888 (2015).
Al-Aloul, M. et al. Renal impairment in cystic fibrosis patients due to repeated intravenous aminoglycoside use. Pediatr. Pulmonol. 39, 15–20 (2005).
Pedersen, S. S., Jensen, T., Osterhammel, D. & Osterhammel, P. Cumulative and acute toxicity of repeated high-dose tobramycin treatment in cystic fibrosis. Antimicrob. Agents Chemother. 31, 594–599 (1987).
Stehling, F., Büscher, R., Grosse-Onnebrink, J., Hoyer, P. F. & Mellies, U. Glomerular and Tubular Renal Function after Repeated Once-Daily Tobramycin Courses in Cystic Fibrosis Patients. Pulm. Med. 2017 (2017).
McWilliam, S. J., Antoine, D. J., Smyth, R. L. & Pirmohamed, M. Aminoglycoside-induced nephrotoxicity in children. Pediatr. Nephrol. 32, 2015–2025 (2017).
Schmitz, C. et al. Megalin deficiency offers protection from renal aminoglycoside accumulation. J. Biol. Chem. 277, 618–622 (2002).
Khwaja, A., Connolly, J. O. & Hendry, B. M. Prenylation inhibitors in renal disease. Lancet 355, 741–744 (2000).
Sidaway, J. E. et al. Inhibitors of 3-hydroxy-3-methylglutaryl-CoA reductase reduce receptor-mediated endocytosis in opossum kidney cells. J. Am. Soc. Nephrol. 15, 2258–2265 (2004).
Verhulst, A., D’Haese, P. C. & De Broe, M. E. Inhibitors of HMG-CoA reductase reduce receptor-mediated endocytosis in human kidney proximal tubular cells. J. Am. Soc. Nephrol. 15, 2249–2257 (2004).
Chinnapa Reddy, V. et al. Effect of simvastatin in gentamicin induced nephrotoxicity in albino rats. Asian J. Pharm. Clin. Res. 5, 36–40 (2012).
Jabbari, M., Rostami, Z., Jenabi, A., Bahrami, A. & Mooraki, A. Simvastatin ameliorates gentamicin-induced renal injury in rats. Saudi J. Kidney Dis. Transpl. 22, 1181–1186 (2011).
Jaikumkao, K. et al. Atorvastatin improves renal organic anion transporter 3 and renal function in gentamicin-induced nephrotoxicity in rats. Exp. Physiol. 101, 743–753 (2016).
Jaikumkao, K. et al. Amelioration of renal inflammation, endoplasmic reticulum stress and apoptosis underlies the protective effect of low dosage of atorvastatin in gentamicin-induced nephrotoxicity. PLoS One 11 (2016).
Ozbek, E. et al. Atorvastatin prevents gentamicin-induced renal damage in rats through the inhibition of p38-MAPK and NF-kB pathways. Ren. Fail. 31, 382–392 (2009).
Selim, A., khalaf, M. M., Gad, A. M. & Abd El-Raouf, O. M. Evaluation of the possible nephroprotective effects of vitamin E and rosuvastatin in amikacin-induced renal injury in rats. J. Biochem. Mol. Toxicol. 31 (2017).
AstraZeneca UK Limited. Crestor 10 mg film-coated tablets - Summary of Product Characteristics. Available at, www.medicines.org.uk/emc/product/7559/smpc, (Accessed: 4th December 2019) (2019).
Kellum, J. A. et al. Kidney disease: Improving global outcomes (KDIGO) acute kidney injury work group. KDIGO clinical practice guideline for acute kidney injury. Kidney International Supplements 2, 1–138 (2012).
McWilliam, S. J., Antoine, D. J., Jorgensen, A. L., Smyth, R. L. & Pirmohamed, M. Urinary Biomarkers of Aminoglycoside-Induced Nephrotoxicity in Cystic Fibrosis: Kidney Injury Molecule-1 and Neutrophil Gelatinase-Associated Lipocalin. Sci. Rep. 8 (2018).
Zhang, J. et al. Immunolocalization of Kim-1, RPA-1, and RPA-2 in Kidney of Gentamicin-, Mercury-, or Chromium-Treated Rats: Relationship to Renal Distributions of iNOS and Nitrotyrosine. Toxicol. Pathol. 36, 397–409 (2008).
McWilliam, S. J. et al. Mechanism-based urinary biomarkers to identify the potential for aminoglycoside-induced nephrotoxicity in premature neonates: A proof-of-concept study. PLoS One 7 (2012).
Lahiri, T., Guillet, A., Diehl, S. & Ferguson, M. High-dose ibuprofen is not associated with increased biomarkers of kidney injury in patients with cystic fibrosis. Pediatr. Pulmonol. 49, 148–153 (2014).
Vaidya, V. S. et al. Kidney injury molecule-1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat. Biotechnol. 28, 478–485 (2010).
Kjeldsen, L., Johnsen, A. H., Sengelov, H. & Borregaard, N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J. Biol. Chem. 268, 10425–10432 (1993).
Haase, M. et al. Accuracy of Neutrophil Gelatinase-Associated Lipocalin (NGAL) in Diagnosis and Prognosis in Acute Kidney Injury: A Systematic Review and Meta-analysis. Am. J. Kidney Dis. 54, 1012–1024 (2009).
Mcwilliam, S. J. et al. Reference intervals for urinary renal injury biomarkers KIM-1 and NGAL in healthy children. Biomark. Med. 8, 1189–1197 (2014).
Agarwal, R. Statin induced proteinuria: Renal injury or renoprotection? J. Am. Soc. Nephrol. 15, 2502–2503 (2004).
Prayle, A. P. et al. The pharmacokinetics and toxicity of morning vs. evening tobramycin dosing for pulmonary exacerbations of cystic fibrosis: A randomised comparison. J. Cyst. Fibros. 15, 510–517 (2016).
Birmingham, B. K. et al. Rosuvastatin pharmacokinetics and pharmacogenetics in Caucasian and Asian subjects residing in the United States. Eur. J. Clin. Pharmacol. 71, 329–340 (2015).
Braamskamp, M. J. A. M. et al. Efficacy and safety of rosuvastatin therapy in children and adolescents with familial hypercholesterolemia: Results from the CHARON study. J. Clin. Lipidol. 9, 741–750 (2015).
Cooper, K. J. et al. Effect of itraconazole on the pharmacokinetics of rosuvastatin. Clin. Pharmacol. Ther. 73, 322–329 (2003).
Paediatric Formulary Committee. BNF for children. MeReC Extra Available at, www.medicinescomplete.com, (Accessed: 4th December 2019) (2005).
Seo, M. S. et al. Effect of treatment on urinary kidney injury molecule-1 in IgA nephropathy. BMC Nephrol. 14 (2013).
De Carvalho, J. A. M. et al. Evaluation of the diagnostic characteristics of urinary kidney injury molecule 1 (uKIM-1) and uKIM-1/creatinine ratio in the assessment of incipient diabetic kidney disease. Clin. Chem. Lab. Med. 53, e51–e54 (2015).
Szczesniak, R., Heltshe, S. L., Stanojevic, S. & Mayer-Hamblett, N. Use of FEV1 in cystic fibrosis epidemiologic studies and clinical trials: A statistical perspective for the clinical researcher. Journal of Cystic Fibrosis 16, 318–326 (2017).
Waikar, S. S., Sabbisetti, V. S. & Bonventre, J. V. Normalization of urinary biomarkers to creatinine during changes in glomerular filtration rate. Kidney Int. 78, 486–494 (2010).
Acknowledgements
The study was funded by The JP Moulton Charitable Foundation. The funder had no role in design and conduct of the study or the analysis and interpretation of the results. The study was conducted with the support of the MRC Centre for Drug Safety Science, University of Liverpool. SJM is funded by a National Institute for Health Research (NIHR) Academic Clinical Lectureship for this research project. Both MP and RLS are NIHR Emeritus Senior Investigators. This publication presents independent research funded by the National Institute for Health Research (NIHR) and The JP Moulton Charitable Foundation. The views expressed are those of the authors and not necessarily those of the JP Moulton Charitable Foundation, the NHS, the NIHR or the Department of Health and Social Care. The PROTEKT Trial Management Group would like to acknowledge and thank all participating centres, the multi-disciplinary teams supporting the trial, and all children, young people and families who took part in this trial. Alder Hey Children’s Hospital: Dr. Kevin Southern, Naomi Rogers. Birmingham Children’s Hospital: Dr. Maya Desai, Rehana Bi, Helen Williamson. Bristol Royal Hospital for Children: Dr. Simon Langton-Hewer, Hope Lacy, Anja Keating, Lisa Tucker. Countess of Chester Hospital: Dr. Ravi Jayaram, Caroline Burchett. Great Ormond Street Hospital: Dr. Helen Spencer, Laura McCarthy, Sahra Shah. King’s College Hospital: Dr. Gary Ruiz, Hannah Fletcher, Eniola Nsirim. Norfolk and Norwich University Hospital: Dr. Caroline Kavanagh, Louisa Fear, Louise Coke. Nottingham Children’s Hospital: Prof Alan Smyth, Dr. Andrew Prayle, Lindsay Crate. Royal Alexandra Children’s Hospital, Brighton: Dr. Akshat Kapur, Catherine Olden, Sebastien Martin. Royal Devon & Exeter Hospital: Dr. Patrick Oades, Suzanne Wilkins. Sheffield Children’s Hospital: Dr. Sonal Kansra, Stuart Gormley. University Hospitals of Leicester NHS Trust: Dr. Erol Gaillard, Judy Maynard-Mills. University Hospitals of North Midlands NHS Trust: Dr. Francis Gilchrist, Rachel Pringle, Victoria Riches. The PROTEKT Trial Management Group would like to thank the help and support of the independent committees that had oversight of the trial. The Trial Steering Committee comprised: Professor Deborah Ashby, Professor Stuart Elborn and Dr. Malcolm Brodie; and the Independent Data and Safety Monitoring Committee: Professor Jane Davies, Dr. Edward Simmons and Dr James Wason.
Author information
Authors and Affiliations
Contributions
S.J.M., A.R.-H., A.P.J., V.S., W.G., T.J., A.S., R.L.S. and M.P. wrote the manuscript. S.J.M., T.J., R.L.S. and M.P. designed the research. S.J.M., A.R.-H., A.P.J., V.S., W.G., A.S., R.L.S. and M.P. performed the research. S.J.M., A.R.-H. and A.P.J. analyzed the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
McWilliam, S.J., Rosala-Hallas, A., Jones, A.P. et al. A randomised controlled trial of rosuvastatin for the prevention of aminoglycoside-induced kidney toxicity in children with cystic fibrosis. Sci Rep 10, 1796 (2020). https://doi.org/10.1038/s41598-020-58790-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-58790-1
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
