
Abstract
Lack of routine surveillance in countries endemic for bovine tuberculosis (TB) and limited laboratory support contributes to the inability to differentiate the Mycobacterium tuberculosis Complex species, leading to an underestimated burden of the disease. Here, Whole-Genome Sequencing of Mycobacterium bovis isolated from tissues with TB-like lesions obtained from cattle and buffalos at Marajó Island, Brazil, demonstrates that recent transmission of M. bovis is ongoing at distinct sites. Moreover, the M. bovis epidemiology in this setting is herein found to be dominated by an endemic and unique clade composed of strains evolved from a common ancestor that are now genetically differentiated from other M. bovis clades. Additionally, envisioning a rapid strain differentiation and tracing across multiple settings, 28 globally validated strain-specific SNPs were identified, three of which considered as robust markers for the M. bovis Marajó strain. In conclusion, this study contributes with data regarding the identification of a novel M. bovis phylogenetic clade responsible for ongoing transmission events in both cattle and buffalo species in Brazil, provides a framework to investigate the dissemination of this highly prevalent strain and, holds the potential to inform TB control strategies that may help to prevent the spread of bovine and zoonotic TB.
Similar content being viewed by others


Introduction
Tuberculosis (TB) is a worldwide important infectious disease in humans and other animals resulting in substantial morbidity and mortality caused by the Mycobacterium tuberculosis Complex (MTBC) pathogens1,2. Among these, Mycobacterium bovis is the main etiological agent of bovine TB (bTB) in herds and is associated with a decreased livestock productivity due to early disposal of animals of high zootechnical value with subsequent economic impact3. The impact of bTB and M. bovis is not only restricted to economic aspects of livestock production. Although the economic losses can ascend to, e.g., US$18 200 in a single region as reported for Makurdi in Nigeria due to a prevalence 1.9% among 61 654 slaughtered cattle4, zoonotic TB is still a major public health problem as it is estimated to cause 140 000 new cases and more than 12 000 deaths in humans worldwide5.
An important factor for the control of zoonotic TB is that human TB caused by M. bovis is possibly underestimated6. In fact, combating zoonotic TB is a goal of WHO’s End TB Strategy since the lack of scientific attention to this problem warrants further studies, especially in areas where bTB remains endemic. Besides lack of routine surveillance data to differentiate M. bovis from M. tuberculosis sensu stricto in humans, the burden of the disease in humans is unknown, often showing uncommon clinical presentations, and a possible cause of treatment failure due to M. bovis intrinsic resistance to pyrazinamide (PZA), an important first-line drug5. The latter is of special concern as it may pose a stepping-stone to the already reported human M. bovis infections by multidrug-resistant strains7.
The mere presence of M. bovis among cattle warns of the zoonotic risk to humans, especially those living at the human-animal interface. In the northern region of Brazil, the Marajó Island in the State of Pará, an unusual insular system, harbours the largest buffalo herd at a nationwide scale (over 520,000 head)8. No data presently exists regarding notification rates of bTB in this island, but previous evidence supports the presence of M. bovis in cattle and buffalo with biopsy specimens suggestive of TB9.
While the body of knowledge concerning the distribution of M. tuberculosis sensu stricto strains is already well understood in most settings, molecular data and phylogeny of the M. bovis ecotype are scarce10. Envisioning strain differentiation, genotyping schemes and molecular methods were developed to discriminate MTBC clinical samples, such as spoligotyping and MIRU-VNTR (Mycobacterial Interspersed Repetitive Units - Variable Number of Tandem Repeats). However, these methods have limitations for phylogenetic studies due to a low discriminatory power or, e.g., an existing propensity of molecular markers for convergent evolution, leading to identical or similar patterns in strains that are phylogenetically and epidemiologically unrelated11,12. In this aspect, the advances made over the last decade on high-throughput sequencing technology now enable a comprehensive access to the entire genomic information of any given strain13.
Herein, to elucidate the genetic clonality and transmission dynamics of M. bovis, we have employed classical genotyping methods, along with state-of-the-art genome-wide phylogenetic reconstruction to evaluate the genomic clustering among cattle and buffalo from an abattoir in the Marajó Island while simultaneously providing a global phylogenetic context for these strains.
Results
bTB in the Marajo Island, Brazil, and M. bovis genotypic diversity
The initial approach to investigate the clonality and population structure of M. bovis in this setting relied on the use of classical typing methods (spoligotyping and MIRU-VNTR) in the characterization of 22 M. bovis isolates from nine cattle and nine buffalos from October 2014 to December 2015. The genotypic characterization by spoligotyping yielded two distinct profiles (Table 1): the predominant spoligotype was SB0822/SIT997, detected in 21 isolates, which is characterized by the absence of spacers 3, 4, 9, 16, and 39 to 43; the remaining isolate corresponded to the spoligotype SB0885/SIT986, which lacks spacers 3, 4, 5, 6, 9, 16 and 39 to 43.
Comparing both profiles, one can speculate that the SB0822/SIT997 profile is the parental strain with the isolate belonging to SB0855/SIT986 type being a derived strain that underwent diversification at the DR locus by losing the 5th and 6th spacer regions as this is the only difference between these isolates and is coherent with the unidirectional evolution observed at the DR locus12.
To further support this notion, and contrary to spoligotype diversity, 24-loci MIRU-VNTR of 20 M. bovis isolates recovered from 17 animals (two isolates/animals were excluded as these failed to amplify more than 10 MIRU-VNTR loci after multiple attempts) revealed a more diverse scenario composed of five profiles. Only VNTR loci 2165 (ETRA), 2461 (ETRB), 1644 (MIRU 16), and 2347 (Mtub29) presented allelic diversity amongst the isolates with a total of three clusters herein detected encompassing a total of 18 isolates.
The most common MIRU-VNTR profile was shared by 14 isolates, the second and third most common MIRU-VNTR profiles were shared by two isolates and, two isolates exhibited unique profiles and were therefore classified as non-clustered strains (Table 1). Noteworthy, under the 24-loci MIRU-VNTR typing method, the single SB0855/SIT986 clinical isolate was clustered in the largest MIRU-VNTR cluster, mostly comprised by SB0822/SIT997 isolates (Table 1 and Fig. 1).

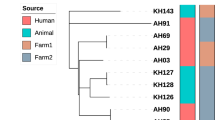
Geographical distribution of genomic clusters, maximum-likelihood phylogenetic and minimum spanning trees (MSTs) of the M. bovis isolates from the Marajó Island, Brazil. The geographic distribution of the three genomic clusters found in the Marajó Island shows that M. bovis transmission is ongoing at multiple cities in the island (A) as a result of clonal expansion of a unique M. bovis clade that has disseminated across both cattle and buffalo species (B). MSTs with node coloring according to host species (C), city of origin (D) and genomic cluster (E) also support the dissemination of this strain across multiple cities and host species with moderate diversification observed mostly between genomic clusters. The maximum-likelihood phylogenetic tree based on 1773 high-quality genome-wide SNPs (B) is shown annotated with the genomic clusters (see legend), host species, MIRU-VNTR and spoligotyping profiles and city of origin of the animal. MIRU-VNTR profiles are represented in a 24-digit numeric string where each-digit represents the number of repeats at a particular locus according to the following order of the loci: 154, 424, 577, 580, 802, 960, 1644, 1955, 2059, 2163b, 2165, 2347, 2401, 2461, 2531, 2687, 2996, 3007, 3171, 3192, 3690, 4052, 4156, 4384 and 4348. Numbers annotated in links between the nodes of the MSTs (C–E) represent the number of segregating SNP sites between nodes. Figure generated using the Interactive Tree of Life v5 online tool (available at https://itol.embl.de), Microsoft PowerPoint 2016 (Version 1707) and Microsoft Excel 2016 (Version 1707), incl. Microsoft Power Map 3D Data Visualization Tool (https://products.office.com/en-us/business/office) and Phyloviz v2.0 (available at https://online2.phyloviz.net).
The latter lends support to the previous notion that SB0855/SIT986 is a likely divergent strain from the predominant SB0822/SIT997 strains detected in the Marajo Island (Fig. 2). Overall, both MIRU-VNTR and spoligotyping suggest that a highly clonal M. bovis population structure exists in the Marajó Island albeit slightly more diverse under a MIRU-VNTR perspective as would already be expected given its superior discriminatory power14,15.

Frequency histogram of SNP pairwise distances across the Marajó Island M. bovis isolates (B) and across the global M. bovis dataset (B). Pairwise distance distribution across the Marajó M. bovis isolates (A) show a restricted distribution when compared with the global dataset (B) with the strains isolated from the Marajó Island showing a SNP pairwise distance of up to 64 SNPs. In panel A, two additional distance peaks around 450 and 1050 bp represent the distance of the Marajó strains towards M. bovis AF2122/97 and M. caprae EPDC01, which were used as reference for mapping and to root the phylogenetic trees, respectively.
Genomic diversity of M. bovis in the Marajó Island reveals the Marajó M. bovis strain as a unique monophyletic branch within M. bovis
Out of the 22 M. bovis isolates sequenced, 17 genomes were retained for downstream genome-wide phylogenetic analysis after genome quality control. These isolates originated from the four municipalities: Soure (n = 9), Chaves (n = 6), Cachoeira do Arari (n = 1) and Santa Cruz do Arari (n = 1) (Table 1).
Search for drug resistance-conferring mutations using TB-Profiler16 showed that all isolates were genotypically susceptible to all anti-TB drugs except PZA due to the M. bovis specific H57D missense mutation in the pncA gene of M. bovis17.
Upon phylogenetic analysis, the Marajó Island M. bovis isolates were found to comprise a monophyletic clade composed of isolates within a maximum pairwise distance of 64 SNPs and is herein referred as the “Marajó-strain” (Fig. 1). These findings are also corroborated by the analysis of the frequency distribution of pairwise SNP distances between isolates from the Marajó Island (0–64 SNPs) which, depending on the isolate are 445–488 and 995–1039 SNPs apart from M. bovis AF2122/97 and M. caprae EPDC01, respectively (Fig. 2). Also, three genomic clusters (C1–3) harboring isolates within a maximum pairwise distance of 5 SNPs were detected with each cluster being composed of isolates originating from the same city within the Marajó Island and isolated from the same animal species. Moreover, the topology of the reconstructed phylogenetic tree is suggestive of historical dissemination by this strain across both cattle and buffalo species and, at multiple geographical locations in the Marajó Island (Fig. 1).
Next, in order to provide an adequate evolutionary background for this strain, we compared the SNP pairwise distance between the isolates of our study and a global M. bovis dataset comprised of 3 402 M. bovis genomes publicly available, along with two genomes obtained from ANSES (Maisons-Alfort, France), isolated in France and belonging to the SB0822 and SB0855 types (Supplementary Table S1; Fig. 2). This global dataset showed an ample and continuous distribution of pairwise SNP distances, congruent with the M. bovis global phylogenetic representation of this dataset (Fig. 2). Across these, only eight genomes were found to be within a maximum pairwise distance of 150 SNPs from the Marajó strains. To further investigate the phylogenetic context of the the Marajó strains, a total of 240 M. bovis isolates, representative of 150 SB types (along with 18 spoligotyping patterns not found in the Mycobacterium bovis Spoligotyping Database) and originating from over 15 countries, were selected for maximum-likelihood phylogenetic analysis (Fig. 3). This sample of 240 M. bovis genomes included the SB0822 and SB0885 strains isolated in France with the same spoligotype pattern as the Marajó isolates and the M. bovis reference AF2122/97. The phylogenetic tree obtained confirmed the monophyletic nature of the Marajó M. bovis isolates in a global context, and, albeit unclassified as per the current rules and genetic markers associated with the different clonal complexes, these strains did form a parallel branch to the European 2 clonal complex18 and therefore shared a more recent common ancestor with this specific clade when compared with the other clonal complexes. The French strains, albeit sharing the same spoligotyping profiles, were positioned to distinct M. bovis evolutionary branches and were separated from the Marajó strains by 413–492 SNPs. Despite the structural similarity at the DR locus, the phylogenetic positioning and topological structure of the phylogenetic tree denotes genotypic convergence at the DR locus level by distinct and unrelated clades: the Marajó strain in Brazil and the A11 (SB0822) and C8 (SB0885) French isolates. Minimum spanning trees constructed using the goeBURST algorithm also position the Marajó strains as a separate branch close to strains from Germany and among unclassified isolates regarding the M. bovis clonal complexes (Supplementary Figures S1 and S2).

Global phylogenetic tree of 257 M. bovis isolates (including the 17 Marajó M. bovis isolates) highlighting the monophyletic nature of the M. bovis Marajó clade. The tree is shown annotated with the isolate ID or ENA run accession, clonal complex colored according to the legend in the bottom-left corner, country of isolation, year of isolation, host species, and spoligotyping profile (in this order). Figure generated using the Interactive Tree of Life v5 online tool (available at https://itol.embl.de).
The Marajó M. bovis strain comprises a genetically differentiated family
Given the monophyletic nature of the Marajó strains in a global evolutionary context, we next sough to investigate the genetic differentiation of these strains when compared with the remaining 240 M. bovis isolates by using a total of 11 544 core SNPs as variable genetic loci. Principal Component Analysis (PCA) across three main components shows a clear genetic distinctiveness between the Marajó strains and the remaining strains that were herein included for comparative purposes. The PCA also showed that all Marajó strains cluster together across the three main axes and appear to be positioned more closely to isolates from Germany and the United States of America (USA) which, is also compatible with the topology of the phylogenetic tree (Fig. 4). Also, Principal Coordinate Analysis (PCoA) encompassing country of isolation and clonal complex as population descriptives, also do demonstrate that the Marajó strains and the Marajó Island epidemiology is genetically differentiated from other geographical sites and genetic groupings (Fig. 4). This latter notion was also corroborated by pairwise FST distances calculated between the Marajó strains and the previously described clonal complexes (European 1–2 and African 1–2)18,19,20,21 which convey a notion of a higher level of genetic differentiation of the Marajó strain when compared with other sub-populations of M. bovis (Table 2). In fact, all populations were significantly differentiated although unclassified strains did show a lower level of genetic differentiation, probably owing to its polyphyletic nature (Fig. 5 and Table 2).

Principal Component Analysis (PCA, panel A) and Principal Coordinate Analysis (PCoA) showing the genetic differentiation of the M. bovis Marajó strains upon comparison with additional 240 M. bovis isolates (B and C). PCA demonstrates the clustering of the M. bovis Marajó strains across all three principal components while showing the genetic divergence from the remaining isolates (A). PCoA corroborates this analysis by demonstrating the unique differentiation of the Marajó M. bovis population when compared with other settings (B) or with the known and previously described clonal complexes (C). Points across the PCA and PCoA plots are coloured according to the study population (country of origin) or clonal complex (see legends).

Boxplots of pairwise FST distances between populations (herein defined as clade or clonal complex), revealing the higher level of genetic differentiation of the Marajó strain when compared with other sub-populations of M. bovis.
Defining a specific set of SNPs to trace and screen for the Marajó M. bovis strain
In order to facilitate rapid strain screening across publicly available genome data and to enable the implementation of laboratory fast tracking of this strain across different settings, we defined a set of specific SNPs that can be used as a variant signature set for this specific clade. Using ancestral reconstruction methods, we compared the most recent common ancestor (MRCA) of the Marajó clade with ERR2815574, herein used as the outgroup isolate (Fig. 3). Using this approach, we detected 28 SNPs occurring between the two MRCA nodes of the phylogenetic tree.
Ideal clade-specific SNPs were defined as synonymous variants occurring in essential genes, which simultaneously decreases the likelihood of homoplasy driven by convergent evolution while ensuring that the region associated with the variant is conserved. Eight out of the 28 SNPs were found to be intragenic and synonymous and, of these, 3 were found to occur at genes know to be essential in M. tuberculosis H37Rv22. To validate this specific set, the 3 402 M. bovis publicly available genomes were re-screened for the initially identified SNPs. All 28 SNPs were found to occur only among the M. bovis isolates from Marajó Island with SNPs 2718745 T, 2830430 C and 3858304 C (M. bovis AF2122/97) considered as robust clade-specific SNPs for the Marajó strain (Table 3).
Discussion
Livestock production is of the utmost economic importance in Brazil and is the mainstay of most inhabitants in the Marajó Island. The latter is the largest fluviomarine island in the world and its barriers to gene flow are highly relevant in the understanding of microbial biodiversity in socioeconomically relevant diseases, such as TB, and of the adaptation of local strains to specific ecological niches23. Over recent years, several aspects of the Marajó Island in the State of Pará has motivated special concern regarding bTB: the region is isolated from the mainland and notification of human TB by M. bovis is inexistent along with a very close contact of humans and animals (buffalo and cattle).
Two unusual spoligotypes (SB0822/SIT997 and SB0855/SIT986) of rare occurrence in Brazil were the only two profiles found among the 18 animals whose isolates were analyzed. This prompts us to a scenario of high strain endemicity and suggestive of a low, if any, strain flow from outside of this insular system. Moreover, 24-loci MIRU-VNTR typing yielded five profiles and three clusters were detected, yielding a clustering rate of 90%, and was unable to discriminate the single SB0855/SIT986 strain from most SB0822/SIT997 isolates. This preliminary data suggested that the single SB0855/SIT986 strain descends from the SB0822/SIT997 isolates and it also suggests that bTB epidemiology in this setting is dominated by a single strain undergoing clonal expansion across different herds of cattle and buffalos.
The SB0855/SIT986 profile is an uncommon one but it has been previously detected in European countries, such as France, Germany, Spain and Belgium. On the other hand, the SB0822/SIT986 profile is not as uncommon and, according to SITVIT2 and Mycobacterium bovis Spoligotyping Database (Mbovis.org), it shows widespread distribution across Europe, South America and North Africa with increased incidence in France. This latter profile has been in fact detected in Southeast Brazil (Minas Gerais and Espírito Santo states) but at a low prevalence when compared with other spoligotyping profiles24.
A possible link to France can be speculated since it has been proposed that bTB in South America has been introduced via cattle importation from Europe25,26. However, an alternative origin is also plausible since in the end of the 19th century a ship taking buffalos from India to the French Guyana sank near the Marajó coast with the surviving animals becoming well adapted to the island27. Before this event, no records of water buffalos exist in this setting.
A more resolved phylogenetic scenario was obtained using WGS of 17 isolates from animals. Upon examining the overall distribution of pairwise SNP distances, and considering the dominance of a single spoligotype profile, we have obtained a broad SNP distance distribution indicating many missing links across the transmission chains and a large timeframe for M. bovis evolution in the area.
The genomic findings do highlight the limitations of 24-loci MIRU-VNTR in this specific setting where this highly prevalent SB0822/SIT997 strain already underwent significant diversification on a genome-wide scale. A total of three genomic clusters were identified, two of which encompassing three isolates and the remaining genomic cluster being composed of two clinical isolates for a total of eight clustered isolates (47%). Each cluster was found to be restricted to the same city and animal species which, per se, suggests that transmission is occurring at multiple geographical points with unapparent dissemination to other animal herds. While the genomic clusters herein detected are restricted to animals of the same species (two involving cattle and one involving buffalos) the tree topology is consistent with an evolutionary history marked by cross-species strain dissemination at multiple points in time since a specific sub-lineage to cattle or buffalos is not observed. Though the present study failed to detect recent transmission between different species, we hypothesize that these transmission events may well occur but at a lower rate since the phylogenetic structure of the clade does support it. Moreover, comparison with a global genomic dataset confirmed the uniqueness and genomic distinctiveness of the M. bovis Marajó strain herein described. Although the Marajó strains comprise a monophyletic clade, these strains are not classifiable in a known clonal complex but do in fact form a parallel branch with the European 2 strains while showing a high genetic differentiation from these. M. bovis European 2 strains are more prevalent in the Iberian Peninsula, are present at low frequencies in France and Italy, and are absent from the British Isles18. The phylogenetic history and multivariate populational genetics analysis show that isolates detected in Germany and USA are genetically closer, arguing mostly in favor of the emergence of this strain due to cattle importation from Europe with later dissemination across Buffalo herds.
Although the samples included in the study originated from the only abattoir with veterinary inspection in the island and the animals included were sourced from multiple sites, a limitation of this study is its sample size which may have hampered the detection of rare inter-species transmission events. This limitation might be related with the fact that only samples from animals displaying anatomopathological abnormalities or lesions suggestive of bTB were included in the study. A study conducted in South Brazil (Santa Catarina State) revealed that in M. bovis cattle positive for tuberculin/PPD antigen, only 8% of the animals showed clinical signs28.
To enable a rapid molecular screening and tracing of this unusual strain across multiple settings, we sought to identify strain-specific SNPs that can inform rapid molecular assays. The genomic SNP distance between the Marajó isolates and other isolates bearing the same spoligotyping profile but isolated in France revealed that spoligotyping is not an adequate marker since the strains from France and Marajó were phylogenetically located on distinct branches of the M. bovis phylogenetic tree, more than 413 SNPs apart. Given the clonal population structure of the MTBC species, SNP markers pose as attractive candidates for strain differentiation29. We were able to identify 28 potential SNPs that can be used as a barcode for these strains. Moreover, a total of three SNPs comprised synonymous intragenic SNPs located in genes classified as essential in M. tuberculosis H37Rv by saturated Himar1 transposon libraries22, which decreases the probability that the associated regions are lost due to further genome downsizing. The latter is a major mode of genome diversification throughout the evolutionary history of the MTBC30.
This set of specific SNPs identified in this study therefore has the potential to be incorporated in molecular screening strategies aimed at detecting the Marajó M. bovis strain at a global level using in silico approaches or, at the state or national level in M. bovis strain biobanks to further evaluate the dissemination of this clade to other regions in Brazil and to assess the risk to human health. This type of approach, using allele-specific PCR amplification has been successfully used to evaluate the dissemination of specific strains outside its endemic region, understand the transmission dynamics at the cross-border level or enable the evaluation of TB transmission in settings without routine or universal molecular typing31.
Cultural aspects associated with living conditions in the Marajó such as drinking raw milk or eating dairy products produced from raw unpasteurized milk pose a specific threat to human health. So far, M. bovis infection in humans has not been reported or detected in the island population or in the state of Pará, however, this could be due to a combination of a low detection rate, inexistent laboratory confirmation of clinically suspected cases or identification at the species-level and, lack of routine strain typing at the state-level.
In conclusion, this study combines classical typing methods along with genome-wide sequence data in the identification and delineation of a unique M. bovis strain, that is herein designated as the Marajó strain. As the real magnitude and impact of bTB as a zoonotic disease in northern Brazil remains unknown and there is a considerable and underestimated bTB risk to humans in the Marajó Island, the study provides information on isolates from the livestock sectors in Brazil and on the origin of M. bovis strains. Such information is essential in the development and implementation of future bTB control strategies for the north of Brazil and, the molecular markers identified will aid in the development of rapid molecular assays that can be deployed at multiple settings and assess the specific risk posed by this strain to human health and food safety.
Materials and Methods
Data and sample collection
The present study includes a total of 20 M. bovis isolates from 18 different animals slaughtered at the Soure municipal abattoir, Marajó Island, in North of Brazil. These isolates ensued from the screening of 48 cattle and buffalo, slaughtered for meat production purpose, presenting TB-like lesions, from October 2014 to December 2015. Briefly, 13 isolates were obtained from nine cattle and the remaining nine isolates from buffalos. Different points of origin for these animals were detected within the island: six animals from Chaves, 10 from Soure, one from Santa Cruz do Arari and one from Cachoeira do Arari (Table 1). The samples were obtained during official post-mortem routine inspection performed by the veterinarian technical manager of the municipal abattoir and this study was submitted for ethical review by the Animal Use Ethics Committee of the State University of Pará, which issued an opinion waiver certificate in accordance with Brazilian Law 11794 (October 08, 2008).
Isolation and identification of mycobacterium bovis
Tissue sample was macerated and decontaminated with SDS(3%) and NaOH(1%), neutralized with bromothymol blue solution and spread evenly onto Löwenstein-Jensen media slants supplemented with pyruvate or with pyruvate and p-nitrobenzoic acid. Screening for the presence of acid-fast bacilli was performed by Ziehl-Neelsen staining and microscopy. Isolates displaying a slow-growth rate incapable of growing on media supplemented with p-nitrobenzoic acid were presumptively identified as MTBC isolates. Molecular confirmation was carried out by PCR amplification and partial sequencing of the hsp65 gene as previously described32. DNA extraction was carried out using a phenol-chloroform extraction method33.
Spoligotyping
Spoligotyping was performed as previously proposed by Kamerbeek et al.34 and implemented on a Luminex 200™ platform automated system (Luminex Corp, Austin, TX) using polystyrene microbeads35. The result was analyzed in the SITVIT2 (http://www.pasteur-guadeloupe.fr:8081/SITVIT2/) and Mycobacterium bovis Spoligotyping Database (https://www.mbovis.org/).
Mycobacterial interspersed repetitive units-variable number of tandem repeat (MIRU-VNTR) typing
PCR amplification of DNA for MIRU-VNTR typing was performed for a set of 24 tandem repeat loci as previously described36. PCR reactions were performed in using GoTaq® DNA Polymerase following manufacturer’s instructions (Promega Corporation, Wisconsin, USA). Amplicon sizing and allelic determination was performed by agarose gel electrophoresis on a 2% (w/v) agarose gel. A MIRU-VNTR cluster was defined as a group of two or more isolates sharing identical profiles.
Whole genome sequencing
DNA quantification was performed using the Qubit™ dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, USA) and the Agilent High Sensitivity DNA Kit (Agilent, California, USA). WGS of M. bovis isolates was carried out on a NextSeq instrument (Illumina, San Diego, CA) using a 2 × 150 paired-end chemistry and the Nextera XT library preparation kit (Illumina, San Diego, CA).
Phylogenomic analysis
Quality trimming and filtering of Illumina reads were performed using the Trimmomatic v0.36 by cutting reads whose average quality falls below an average PHRED score of 20 in a 4 bp sliding window and retaining reads with minimum length of 36 bp37. Filtered reads were mapped against the M. bovis AF2122/97 genome (GenBank Accession NC_002945.4)38 using the Burrows Wheeler Aligner tool (BWA-MEM algorithm)39. Mapping statistics were obtained using Qualimap40. SAMtools and GATK were used for variant calling and only concordant variants were retained for downstream analysis41,42.
High-quality SNPs were extracted and concatenated into a single DNA pseudo-molecule. A minimum site coverage of 20 reads were used and variant sites retained only if the majority nucleotide reached a relative coverage depth of 75%. A missing call was assigned if a base call did not met the previous criteria and samples or SNP sites having an excess of 10% missing calls were excluded as to remove heterogenic and low coverage sites43. SNP positions falling in mycobacterial PPE/PE genes and repeat regions were removed from the final alignment. The final dataset across 17 isolates and M. bovis AF2122/97 (reference) encompassed a total of 1 773 high-quality SNPs, 1 559 (87.9%) of which had no missing genotypes (core SNP sites). Pairwise SNP distance was evaluated using snp-dists (https://github.com/tseemann/snp-dists).
A maximum-likelihood phylogenetic tree rooted on Mycobacterium caprae EPDC0144 was created with SeaView v.4 software implementing PhyML using a Generalized Time Reversible (GTR) model without gamma variation and invariable sites45. Tree topology was optimized by searching the tree space using the Subtree Pruning and Regrafting (SPR) and, the Nearest Neighborhood Interchange (NNI) methods. Branch reliability was estimated using the approximate Likelihood Ratio Test (aLRT)46.
The resulting tree was annotated and rooted using the Interactive Tree of Life v5.3 (iTOL) online tool (available at https://itol.embl.de/)47. Ancestral sequence reconstruction was carried out using the package phangorn in R. The goeBURST/Phyloviz tool (available at https://online2.phyloviz.net) was used to identify genomic transmission clusters using a 5 SNP threshold and to generate minimum spanning trees.
Additionally, variants associated with drug resistance were detected using TB-Profiler v2.6.1 (https://github.com/jodyphelan/TBProfiler)16,48.
Comparison with a global M. bovis dataset
Genomic variant data was compared to a global collection of 3 402 M. bovis genomes publicly available on the European Nucleotide Archive (ENA; Supplementary Table S1) until December 2018 (initial: 3 590, 188 isolates excluded due to low coverage of sequence data or inability to generate a spoligotyping profile using SpoTyping). This genome collection is part of a M. bovis genomic variant dataset available at iMed.ULisboa and, is composed of variant call data and individual site mapping statistics obtained through the Snippy mapping pipeline using M. bovis AF2122/97 (GenBank Accession NC_002945.4) as reference genome49. Two additional M. bovis genomes from ANSES (Agence Nationale de Sécurité Sanitaire de l’Alimentation, de l’Environnement et du Travail), Maisons-Alfort, France, sharing similar spoligotyping profiles were included (ENA study accession SRP161870). SpoTyping was used to determine in silico spoligotyping profiles for all isolates50. If available, metadata on the year of isolation and country of origin was extracted from sample associated XML files available at ENA. Genome-wide high-quality SNPs (Total: 42 419 segregating sites) were extracted from VCF files and coverage-validated using the same parameters described above across the entire genomic dataset, including the Marajó isolates. A phylogenetic tree was constructed for a final dataset composed of 257 M. bovis isolates which included the 17 isolates recovered at the Marajó Island, the two additional M. bovis isolates from ANSES and 237 M. bovis genomes of isolates from more than 16 countries, representative of all spoligotypes (SB type, Mbovis.org) available across the 3 402 M. bovis isolates whose genome was publicly available. For this final dataset, high-quality SNPs were obtained as described for the 17 Marajó M. bovis isolates totalling 20 103 high-quality SNP sites, of which 11 544 (57.4%) had no missing genotypes (core SNPs).
Population genetics
Populational genetic multivariate analysis was carried out in R using the adegenet package. Principal Component Analysis (PCA) and Principal Coordinate Analysis (PCoA) was carried out for the 257 M. bovis dataset using the 11 544 coreSNPs identified across this sample and along three principal components. Inter-populational comparison and differentiation analysis was done by considering the previously described Clonal Complexes (European 1–2 and African 1–2) and assigning each isolate to one of these populations based on specific genomic markers18,20,21,22. Isolates deemed unclassifiable as per this scheme were assigned to the Unclassified population and isolates recovered at the Marajó Island were, given its monophyletic nature, assigned to the Marajo population. Pairwise FST distances between populations was computed as a metric of populational paiwise differentiation using R along with the dartR and StAMPP packages implementing the method described by Weir and Cockerham (1984) with 1 000 bootstraps51.
Accession numbers
Study accession ERP116404.
Data availability
Raw sequence data has been submitted to the ENA under the study accession ERP116404. Sample information along with run accession numbers for publicly available genomes used in this study can be found at the Supplementary Table S1.
References
Brites, D. & Gagneux, S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol. Rev. 264, 6–24 (2015).
Coscolla, M. & Gagneux, S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin. Immunol. 26, 431–44 (2014).
Zumárraga, M. J. et al. Understanding the relationship between Mycobacterium bovis spoligotypes from cattle in Latin American Countries. Res. Vet. Sci. 94, 9–21 (2013).
Ejeh, E. F. et al. Prevalence and direct economic losses from bovine tuberculosis in Makurdi, Nigeria. Vet. Med. Int.; 904861 (2014)
World Health Organization. Roadmap for zoonotic tuberculosis. World Health Organization (WHO), Food and Agriculture Organization of the United Nations (FAO) and World Organisation for Animal Health (OIE), 2017. Geneva. [cited 2019 Jul 16]. https://www.who.int/tb/publications/2017/zoonotic_TB/en/
Olea-popelka, F. et al. Zoonotic tuberculosis in human beings caused by Mycobacterium bovis-a call for action. Lancet Infect. Dis. 17, 21–5 (2017).
Vazquez-Chacon, C. A. et al. Human multidrug-resistant Mycobacterium bovis infection in Mexico. Tuberculosis . 95, 802–9 (2015).
Brazilian Institute of Geography and Statistics. Sistema IBGE de Recuperação Automática - SIDRA. Pesquisa da Pecuária Municipal. [cited 2019 Jul 11]. https://sidra.ibge.gov.br/tabela/3939
Da Conceição, M. L. et al. Outbreak of tuberculosis due to Mycobacterium bovis in cattle and buffalo in Marajó Island, Amazon Region. In: Abstracts of the 40th Annual Congress of The European Society of Mycobacteriology; Valencia; 2019. 30 June,30–July,6; Abstract P122.
Gagneux, S. Strain Variation in the Mycobacterium tuberculosis Complex: Its Role in Biology, Epidemiology and Control. Springer, Heidelberg, 2017.
Comas, I. et al. Genotyping of genetically monomorphic bacteria: DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologies. PLoS ONE. 12, 1–11 (2009).
Jagielski, T. et al. Current methods in the molecular typing of Mycobacterium tuberculosis and other Mycobacteria. Biomed. Res. Int.; 645802 (2014).
Meehan, C. J. et al. Whole genome sequencing of Mycobacterium tuberculosis: current standards and open issues. Nat Rev Microbiol. s41579-019-0214-5 (2019).
Perdigão, J. et al. Clonal expansion across the seas as seen through CPLP-TB database: a joint effort in cataloguing Mycobacterium tuberculosis genetic diversity in Portuguese-speaking countries. Infect. Genet. Evol. 72, 44–58 (2019).
Supply, P. et al. Proposal for Standardization of Optimized Mycobacterial Interspersed Repetitive Unit–Variable-Number Tandem Repeat Typing of Mycobacterium tuberculosis. J. Clin. Microbiol. 44, 4498–510 (2006).
van Beek, J., Haanpera, M., Smit, P. W., Mentula, S. & Soini, H. Evaluation of whole genome sequencing and software tools for drug susceptibility testing of Mycobacterium tuberculosis. Clin. Microbiol. Infect. 25, 82–6 (2019).
Espinosa de los Monteros, L. E. et al. Allele-specific PCR method based on pncA and oxyR sequences for distinguishing Mycobacterium bovis from Mycobacterium tuberculosis: intraspecific M. bovis pncA sequence polymorphism. J. Clin. Microbiol. 36, 239–42 (1998).
Rodriguez-Campos, S. et al. European 2–a clonal complex of Mycobacterium bovis dominant in the Iberian Peninsula. Infect. Genet. Evol. 12, 866–72 (2012).
Smith, N. H. et al. European 1: A globally important clonal complex of Mycobacterium bovis. Infect. Genet. Evol. 11, 1340–51 (2011).
Müller, B. et al. African 1, an epidemiologically important clonal complex of Mycobacterium bovis dominant in Mali, Nigeria, Cameroon, and Chad. J. Bacteriol. 191, 1951–60 (2009).
Berg, S. et al. African 2, a Clonal Complex of Mycobacterium bovis Epidemiologically Important in East Africa. J. Bacteriol. 193, 670–8 (2011).
DeJesus, M. A. et al. Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis. MBio 17, 02133–16 (2017).
Acevedo, P. et al. Tuberculosis epidemiology in islands: insularity, hosts and trade. PLoS One 8, 1–8 (2013).
Zanini, M. S. et al. Molecular typing of Mycobacterium bovis isolates from south-east Brazil by spoligotyping and RFLP. J. Vet. Med. B Infec Dis. Vet Public. Health. 52, 129–33 (2005).
Njanpop-Lafourcade, B. M. et al. Molecular typing of Mycobacterium bovis isolates from Cameroon. J. Clin. Microbiol. 39, 222–7 (2001).
Cataldi, A. A. et al. The genotype of the principal Mycobacterium bovis in Argentina is also that of the British Isles: did bovine tuberculosis come from Great Britain? [in Spanish]. Rev. Argent. Microbiol. 34, 1–6 (2002).
Sousa, G. S. M., Salvarani, F. M., Bomjardim, H. A., Brito, M. F. & Barbosa, J. D. Brucellosis in water buffaloes. Brucellosis in water buffaloes. Pesq. Vet. Bras. 37, 234–40 (2017).
Menin, Á. et al. Asymptomatic cattle naturally infected with Mycobacterium bovis present exacerbated tissue pathology and bacterial dissemination. PLoS One. 8, e53884 (2013).
Coll, F. et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 5, 4812 (2014).
Gagneux, S. et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl Acad. Sci. USA 103, 2869–73 (2006).
Domínguez, J. et al. Simplified Model to Survey Tuberculosis Transmission in Countries Without Systematic Molecular Epidemiology Programs. Emerg. Infect. Dis. 25, 507–514 (2019 Mar).
Kim, H. et al. Differentiation of Mycobacterium species by analysis of the heatshock protein 65 gene (hsp65). Int. J. Syst. Evol. Microbiol. 55, 1649–56 (2005).
Sambrook, J. Molecular Cloning: a Laboratory Manual. Cold Spring Harbor, N.Y.:Cold Spring Harbor Laboratory Press, 2001.
Kamerbeek, J. et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 35, 907–14 (1997).
Gomgnimbou, M. K. et al. Tuberculosis-spoligo-rifampin-isoniazid typing: an all-in-one assay technique for surveillance and control of multidrug-resistant tuberculosis on Luminex devices. J. Clin. Microbiol. 51, 3527–34 (2013).
Yasmin, M. et al. Quick and cheap MIRU-VNTR typing of Mycobacterium tuberculosis species complex using duplex PCR. Tuberculosis . 101, 160–3 (2016).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–20 (2014).
Malone, K. M. et al. Updated reference genome sequence and annotation of Mycobacterium bovis AF2122/97. Genome Announc. 5, e00157–17 (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–60 (2009).
Okonechnikov, K., Conesa, A. & García-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 32, 292–4 (2016).
Li, H. et al. 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 25, 2078–9 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–303 (2010).
Coll, F. et al. Genome-wide analysis of multi- and extensively drug-resistant Mycobacterium tuberculosis. Nat. Genet. 50, 307–16 (2018).
Yoshida, S. et al. Mycobacterium caprae Infection in Captive Borneo Elephant, Japan. Emerg. Infect. Dis. 24, 1937–40 (2018).
Gouy, M., Guindon, S. & Gascuel, O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evo. 27, 221–4 (2010).
Anisimova, M. & Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 55, 539–52 (2006).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–5 (2016).
Coll, F. et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 7, 51 (2015).
Seemann, T. (2015) snippy: fast bacterial variant calling from NGS reads https://github.com/tseemann/snippy
Xia, E., Teo, Y. Y. & Ong, R. T. SpoTyping: fast and accurate in silico Mycobacterium spoligotyping from sequence reads. Genome Med. 8, 19 (2016).
Weir, B. S. & Cockerham, C. C. Estimating F‐statistics for the analysis of population structure. Evolution 38, 1358–70 (1984).
Sassetti, C. M. & Rubin, E. J. Genetic requirements for mycobacterial survival during infection. Proc. Natl Acad. Sci. USA 100, 12989–94 (2003).
Griffin, J. E. et al. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7, e1002251 (2011).
Acknowledgements
We are grateful to Professor Márcio R. T. Nunes (Center for Technological Innovation, Evandro Chagas Institute), Maria L. Lopes and Jacira S. Nascimento (Mycobacteriology Group at the Evandro Chagas Institute) for technical assistance and to Professor Stefan Niemann, head of the Molecular and Experimental Mycobacteriology Group at the Research Center Borstel, for the comments and collaboration. This work was supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brazil (CAPES), Instituto Evandro Chagas/MS/SVS and Fundação para o desenvolvimento científico e tecnológico em saúde [PRES-012-FIO-16]. M.L.C. is supported by CAPES – Finance Code 001, [194–88881.187587/2018–01, 2018]. Parts of the work were funded by the German Center for Infection Research (DZIF). The phylogenetic and bioinformatic analysis work at iMed.ULisboa is supported by the European Society of Clinical Microbiology and Infectious Diseases, for which we would like to would like to acknowledge the Study Group for Mycobacterial Infections and Fundação para a Ciência e Tecnologia (UID/DTP/04138/2019). J.P. is supported by a research contract [CEECIND/00394/2017] from FCT.
Author information
Authors and Affiliations
Contributions
M.L.C., E.C.C., J.P., K.V.B.L. conceived the project. M.L.C. and J.P., undertook the experiments. The manuscript was primarily written by M.L.C., E.C.C., J.P. and K.V.B.L., with secondary contributions, input, and feedback from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Conceição, M.L.d., Conceição, E.C., Furlaneto, I.P. et al. Phylogenomic Perspective on a Unique Mycobacterium bovis Clade Dominating Bovine Tuberculosis Infections among Cattle and Buffalos in Northern Brazil. Sci Rep 10, 1747 (2020). https://doi.org/10.1038/s41598-020-58398-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-58398-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
