
Abstract
Host genetic susceptibility to leprosy has been intensively investigated over the last decades; however, there are no studies on the role of genetic variants in disease recurrence. A previous initiative identified three recurrent cases of leprosy for which none of the M. leprae strains, as obtained in the first and the second diagnosis, had any known genomic variants associated to resistance to Multidrug therapy; in addition, whole genome sequencing indicated that the same M. leprae was causing two out of the three recurrences. Thus, these individuals were suspected of being particularly susceptible to M. leprae infection, either as relapse or reinfection. To verify this hypothesis, 19 genetic markers distributed across 11 loci (14 genes) classically associated with leprosy were genotyped in the recurrent and in three matching non-recurrent leprosy cases. An enrichment of risk alleles was observed in the recurrent cases, suggesting the existence of a particularly high susceptibility genetic profile among leprosy patients predisposing to disease recurrence.
Similar content being viewed by others

Introduction
Leprosy is a chronic infectious disease caused by Mycobacterium leprae, an obligatory intracellular, slow growing, alcohol-acid resistant bacillus with tropism for skin macrophages and Schwann cells of the peripheral nervous system1. The disease affects more than 200.000 people every year globally, with high incidence and prevalence concentrating in India and Brazil2. Leprosy presents a large spectrum of clinical manifestations that varies depending on the host’s immune response, ranging from the localized tuberculoid to the systemic lepromatous forms as defined in the classic Ridley & Jopling classification system3. Alternative, the World Health Organization (WHO) classification protocol distributes leprosy cases as paucibacillary (PB) or multibacillary (MB) for treatment purposes4.
Leprosy is treated by a multidrug therapy (MDT) consisting of rifampicin, dapsone and clofazimine carried out for six monthly doses for PB patients and for 12 monthly doses for MB patients5; treatment is distributed for free in endemic countries since 19956. Currently, there is the proposal for a uniform multidrug therapy regimen for leprosy patients that consists of six doses of MDT prescribed regardless of any clinical classification criteria. A Clinical Trial for Uniform Multidrug Therapy Regimen for Leprosy Patients in Brazil, (U-MDT/CT-BR) was recently conducted to compare outcomes of regular 12 doses MDT (R-MDT) and the uniform six doses MDT (U-MDT). The study included results from laboratory tests (bacilloscopic index, serology and histopathology) and clinical evaluation during long term follow-up7. Among the recurrent cases of leprosy identified in the U-MDT/CT-BR trial; for three of them, M. leprae isolates obtained in the first and second diagnosis were submitted to whole genome sequencing (WGS); analyses did not show any mutations associated with drug resistance. Based on comparative analysis between M. leprae genomes obtained in the first and second infection of each patient, the authors concluded for two cases of relapses caused by same strain and one case of reinfection caused by a different M. leprae strain8.
Considering that (i) all patients were followed and completed the treatment correctly, (ii) U-MDT was efficient in treating the disease, and (iii) no mutation associated with drug resistance was detected in the genomes of any M. leprae strain recovered from the three reinvestigated recurrent cases, it is reasonable to assume that leprosy recurrence was not due to characteristics of the etiologic agent but yet, to a combination of continuous exposure to M. leprae associated with an increased host susceptibility genetic background.
Host genetics accounts for a great proportion of disease heritability in leprosy and many genes were already identified with substantial evidences in support of their association with leprosy9. Among them, genes HLA-DR-DQ (major histocompatibility complex, class II, DR – class II DQ)10, LTA (lymphotoxin alpha)11, IL10 (interleukin 10)12, PRKN/PACRG (parkin RBR E3 ubiquitin protein ligase/parkin coregulated gene)13, NOD2 (nucleotide binding oligomerization domain containing 2)14, LACC1/CCDC122 (laccase domain containing 1/coiled-coil domain containing 122)14, SOD2 (superoxide dismutase 2)15, NEBL (nebulette) (unpublished data), GATA3 (GATA binding protein 3)16, IFNG (interferon gamma)17 and TLR1 (toll like receptor 1)18 have been associated with leprosy in Brazilian population samples, the majority of them playing important roles related to the host immune system. However, to date, no attempt has been made to translate this genetic knowledge into the definition of an individual susceptibility profile that could help explain complex leprosy phenotypes such as recurrence/reinfection. Here we genotyped 19 Single Nucleotide Polymorphism (SNP) distributed across these 11 loci associated with leprosy per se on six leprosy cases from the U-MDT/CT-BR cohort: two cases previously reported as relapses and one as reinfection8, and three matching, successfully treated leprosy patients that did not relapse. Allele frequencies were compared across the two groups and with publicly available databases.
Results and Discussion
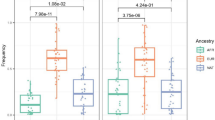
All 19 SNPs were successfully genotyped for all six samples of leprosy patients and results are summarized in Table 1. A global count including all genes/markers assessed revealed that risk alleles were more frequently observed in recurrent patients #3208 and #2188, in contrast to controls and recurrent patient #1126.
Comparison of the combined genotypic distribution of all variants tested between the two groups revealed an enrichment of homozygous genotypes for the risk alleles among the three recurrent patients (Table 2): 40% vs. 19% among controls (P = 0.02; OR = 2.83; 95% CI 1.22–6.58). The same comparison applied for the combined allelic distribution also revealed an enrichment of risk alleles among cases, with a suggestive difference that failed to reach statistical significance at the 0.05 level (57% vs. 45% among controls; P = 0.08). These results must be interpreted with caution, given the extremely low sample size and the moderate to high degree of linkage disequilibrium across markers located within gene loci PRKN/PACRG (r2 = 1 between rs1040079 and rs2276201 and r2 = 0.7 between rs1040079 or rs2276201 and rs9356058), LTA, (r2 = 0.5 between rs2239704 and rs909253), NOD2 (r2 = 0.6 between rs2111234 and rs8057341, and r2 = 0.66 between rs2111234 and rs3135499), HLA-DRB1/DQA1 (r2 = 0.46 between rs602875 and rs1071630) and CCDC122/LACC1 (r2 = 1 between rs2275252 and rs4942254). Importantly: it is possible that high LD is in fact reflecting the additive effect of two or more variants of distinct functional impact upon the net risk for that gene. This effect would be similar to what has been observed previously for gene TNFSF819: extensive LD has been detected between associated markers identified as independent expression quantitative trait loci (eQTL) for TNFSF8; the authors argue that this combined functional impact could reflect the high correlation observed between the SNP alleles.
It is interesting to note that recurrent patient #1126 presented allelic and genotypic global counts similar to the controls and in contrast with the two other recurrent individuals. Patient #1126 has been described previously as the only case of recurrence caused by distinct M. leprae strains isolated in the first and the second diagnosis8. Also, patient #1126 presented the shortest time to relapse and the latest age of diagnosis – 32 years – similar to the controls (Table 3). One cannot disregard the possibility that recurrent patients #3208 and #2188 have also suffered reinfection – and not relapse – caused by the same M. leprae strain. This hypothesis is reinforced by the fact that none of the M. leprae strains have the classically recognized resistance mutations8, thus relapse caused by treatment failure is unlikely. Thus, the most striking host-related, non-genetic difference between recurrent patients #1126 and #3208 and #2188 is the age of diagnosis. In leprosy, the presence of the age-dependent association has already been shown for PRKN-PACRG20, LTA11, miR-146a21 (leprosy per se) and TNFSF15-TNFSF822 (leprosy reaction). The enrichment of risk alleles observed for recurrent patients #3208 and #2188 is compatible with the hypothesis that early onset leprosy is more heavily dependent on genetic mechanisms, whereas late onset disease is a balanced combination of genetic and environmental factors.
A higher frequency of the risk allele is observed for markers of several well-known leprosy susceptibility genes – IL10, PACRG, NOD2, HLA-DRB1/DQA1, LTA, GATA3, IFNG and TLR1 – when relapse cases are compared with controls and the frequency available on public databases (Table 1). This observation, although interesting, has only qualitative value in the context of a description of a series of cases and should be interpreted with extreme caution.
Materials and Methods
Ethics approval
This study was performed following the Declaration of Helsinki and Brazilian research regulations and was approved by the National Ethics Commission of Research (CONEP) of the Ministry of Health, protocol number 12949/2007, as previously described7. Informed consent was obtained from all individual participants included in the study.
Subjects
Six male leprosy patients were selected from the previous U-MDT/CT-BR study; three did not relapse during an eight-year follow-up; two were diagnosed as relapse caused by the same M. leprae strain and one was considered a reinfection caused by a distinct M. leprae isolate8. The three non-recurrent patients were matched with the recurrent cases by leprosy clinical form, ethnicity (estimated based on phenotypical features and skin color and distributed as Caucasian, African, Asian, Native American or Mixed), bacilloscopic index, treatment protocol, occurrence of leprosy reaction and place of residence (all six patients were from a leprosy hyperendemic area of Fortaleza, Brazil). A summary of the demographic and diagnostic information is presented in Table 3.
Gene selection, genotyping and analysis
Genes were selected for genotypic analysis based on previous evidence for validated/replicated association with leprosy in Brazilian population samples. SNP markers have been selected based on previous published, association with leprosy in one or more Brazilian population samples and/or in previous genome-wide association studies (GWAS) (Supplementary Table S1). Genomic DNA was obtained from peripheral blood by salting-out23; the DNA was quantified spectrophotometrically using NanoDrop 2000/2000c and the concentration of genomic DNA was normalized to 20 ng/µL. Genotyping was performed by fluorescence-based allelic discrimination using TaqMan, as implemented in the Applied Biosystems® 7500 Real-Time PCR platform. The reactions mix was composed by ultrapure water (1.9 μl per sample), TaqMan® SNP Genotyping Assays (40×, 0.1 μl per sample), TaqMan™ Genotyping Master Mix (2×, 3 μl per sample) and template DNA (3 μl). Reaction was conducted using a denaturing temperature of 95 °C for 15 seconds and an oligonucleotide hybridization and extension temperature of 60 °C for 2 minutes on each of a total of 50 cycles.
Linkage disequilibrium (LD) was estimated using Haploview v.4.2 using the combined dataset of all six patients. A descriptive analysis was performed using the Statistical Package for the Social Sciences (SPSS) v.22. Genetic profiles of recurrent and non-recurent cases were described against the frequencies of risk/protection alleles obtained from previous studies as available on Ensembl Genome Browser with data of 1000 genomes, phase 3, combined population. The Fisher’s exact test for small sample sizes was used to compare combined allelic and genotypic frequencies across the two groups.
Conclusion
Our study presents, for the first time, suggestive evidence that genes associated with susceptibility to leprosy may also play an important role on disease recurrence. Our data indicates that a combination of alleles in different genes may confer hyper-susceptibility to leprosy detectable even among leprosy susceptible individuals, increasing the chances of disease recurrence. In addition, one could speculate, based on the allelic profile of patient #1126 – similar to the non-recurrent and distinct of the other two recurrent individuals – on the possible existence of distinct genetic mechanisms controlling disease relapse and reinfection. We are fully aware of the limitations of our design, particularly related to the small sample size; thus, this mainly descriptive, qualitative report of a series of cases must be perceived as a hypothesis-generating initiative, opening the possibility for additional studies of larger sample sizes of non-recurrent and recurrent leprosy affected individuals, aiming to identify biomarkers of risk of reinfection that may be used to monitor treated patients continuously exposed to the bacilli in endemic areas, with potential positive effects over leprosy control programs.
Data availability
The authors state that all data produced in the study is available.
References
Britton, W. J. & Lockwood, D. N. Leprosy. Lancet 363, 1209–1219, https://doi.org/10.1016/S0140-6736(04)15952-7 (2004).
WHO, W. H. O. Weekly epidemiological record, Global leprosy update, 2017: reducing the disease burden due to leprosy (2018).
Ridley, D. S. & Jopling, W. H. Classification of leprosy according to immunity. A five-group system. International journal of leprosy and other mycobacterial diseases: official organ of the International Leprosy Association 34, 255–273 (1966).
World Health Organization. A guide for surveillance of antimicrobial resistance in leprosy: 2017 update. (2017).
WHO/Department of Communicable Disease Prevention, C. a. E. 38 (2000).
WHO. Multidrug therapy (MDT), http://www.who.int/lep/mdt/en/ (2018).
Penna, G. O. et al. Uniform multidrug therapy for leprosy patients in Brazil (U-MDT/CT-BR): Results of an open label, randomized and controlled clinical trial, among multibacillary patients. PLoS neglected tropical diseases 11, e0005725, https://doi.org/10.1371/journal.pntd.0005725 (2017).
Stefani, M. M. A. et al. Whole genome sequencing distinguishes between relapse and reinfection in recurrent leprosy cases. PLoS neglected tropical diseases 11, doi:ARTN e000559810.1371/journal.pntd.0005598 (2017).
Sauer, M. E. et al. Genetics of leprosy: expected and unexpected developments and perspectives. Clinics in dermatology 33, 99–107, https://doi.org/10.1016/j.clindermatol.2014.10.001 (2015).
da Silva, S. A. et al. HLA-DR and HLA-DQ alleles in patients from the south of Brazil: markers for leprosy susceptibility and resistance. BMC infectious diseases 9, 134, https://doi.org/10.1186/1471-2334-9-134 (2009).
Alcais, A. et al. Stepwise replication identifies a low-producing lymphotoxin-alpha allele as a major risk factor for early-onset leprosy. Nature genetics 39, 517–522, https://doi.org/10.1038/ng2000 (2007).
Alvarado-Arnez, L. E. et al. Association of IL10 Polymorphisms and Leprosy: A Meta-Analysis. PloS one 10, e0136282, https://doi.org/10.1371/journal.pone.0136282 (2015).
Mira, M. T. et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 427, 636–640, https://doi.org/10.1038/nature02326 (2004).
Sales-Marques, C. et al. NOD2 and CCDC122-LACC1 genes are associated with leprosy susceptibility in Brazilians. Human genetics 133, 1525–1532, https://doi.org/10.1007/s00439-014-1502-9 (2014).
Ramos, G. B. et al. Association Analysis Suggests SOD2 as a Newly Identified Candidate Gene Associated With Leprosy Susceptibility. The Journal of infectious diseases 214, 475–478, https://doi.org/10.1093/infdis/jiw170 (2016).
Medeiros, P. et al. The GATA3 gene is involved in leprosy susceptibility in Brazilian patients. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases 39, 194–200, https://doi.org/10.1016/j.meegid.2016.01.015 (2016).
Cardoso, C. C. et al. IFNG +874T > A single nucleotide polymorphism is associated with leprosy among Brazilians. Human genetics 128, 481–490, https://doi.org/10.1007/s00439-010-0872-x (2010).
Marques Cde, S. et al. Toll-like receptor 1 N248S single-nucleotide polymorphism is associated with leprosy risk and regulates immune activation during mycobacterial infection. The Journal of infectious diseases 208, 120–129, https://doi.org/10.1093/infdis/jit133 (2013).
Fava, V. M. et al. Association of TNFSF8 regulatory variants with excessive inflammatory responses but not leprosy per se. The Journal of infectious diseases 211, 968–977, https://doi.org/10.1093/infdis/jiu566 (2015).
Alter, A. et al. Linkage disequilibrium pattern and age-at-diagnosis are critical for replicating genetic associations across ethnic groups in leprosy. Human genetics 132, 107–116, https://doi.org/10.1007/s00439-012-1227-6 (2013).
Cezar-de-Mello, P. F. et al. Pre-miR-146a (rs2910164 G > C) single nucleotide polymorphism is genetically and functionally associated with leprosy. PLoS neglected tropical diseases 8, e3099, https://doi.org/10.1371/journal.pntd.0003099 (2014).
Fava, V. M., Sales-Marques, C., Alcais, A., Moraes, M. O. & Schurr, E. Age-Dependent Association of TNFSF15/TNFSF8 Variants and Leprosy Type 1 Reaction. Frontiers in immunology 8, 155, https://doi.org/10.3389/fimmu.2017.00155 (2017).
John, S. W., Weitzner, G., Rozen, R. & Scriver, C. R. A rapid procedure for extracting genomic DNA from leukocytes. Nucleic acids research 19, 408, https://doi.org/10.1093/nar/19.2.408 (1991).
Acknowledgements
This study was funding from UMDT Clinical Trial supported by Department of Science and Technology and National Council for Scientific and Technological Development (CNPq Number 403293/2005-7). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
P.V.U.S.: molecular analysis, statistical analysis, manuscript preparation; G.O.P.: clinical assessment, study design, financial support, manuscript preparation; S.B.S.: clinical assessment, study design; M.A.A.P.: clinical assessment, study design; H.S.G.: clinical assessment; R.C.: clinical assessment; M.C.L.V.: clinical assessment; I.M.F.D.B.: molecular analysis. P.S.R.: molecular analysis. M.L.F.P.: clinical assessment, statistical analysis; V.M.F.: statistical analysis, manuscript preparation; M.M.A.S.: study design, senior supervisor, manuscript preparation; M.T.M.: study design, senior supervisor, manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Uaska Sartori, P.V., Penna, G.O., Bührer-Sékula, S. et al. Human Genetic Susceptibility of Leprosy Recurrence. Sci Rep 10, 1284 (2020). https://doi.org/10.1038/s41598-020-58079-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-58079-3
This article is cited by
-
Leprosy Transmission in Amazonian Countries: Current Status and Future Trends
Current Tropical Medicine Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
