
Abstract
The modern-day distribution of freshwater fishes throughout multiple rivers is likely the result of past migration during times when currently separate drainages were once connected. Here, we used mitochondrial and microsatellite analyses for 248 individuals of Rhodeus notatus collected from seven different rivers to obtain better understand historical gene flow of freshwater fish on the Korean Peninsula. Based on our phylogenetic analyses, this Korean species originated through the paleo-Yellow River from China and first colonized near the west coast. These genetic data also provided evidence of estuary coalescences among the rivers flowing to the west and southwest coast on well-developed continental shelf. In addition, the pattern of population structure revealed the biogeodispersal route from the west coast to the south coast. It could be inferred that massive migration was not involved in the formation of southern populations, since the signature of historical genetic drift was clearly observed. Our study is the first genetic attempt to confirm hypotheses describing the migration of freshwater species towards the end of East Asia, which have previously been developed using only geological reasoning.
Similar content being viewed by others

Introduction
Primary freshwater fish species living in separate river systems are not able to come into contact naturally. Even within a single river system, populations can be isolated by landscape structures1,2,3. The high degree of interpopulation genetic differentiation normally found in freshwater fish species has often been attributed to the fragmented nature of freshwater environments4,5,6,7. However, the distribution of freshwater fish species throughout multiple drainage basins is likely the result of past migration during times when currently separate drainages were once connected8. Insight into historical changes in past drainage systems and gene flow in a given region can be obtained by characterizing the pattern of genetic diversity and structure among populations that are currently spatially separated9,10.
The geography of the Korean Peninsula provides an excellent opportunity to explore the connections among the historical formation of freshwater systems and the colonization of freshwater fish9,11,12,13. This peninsula is a small but complex mountainous terrain that was created in the course of large-scale tectonic events from the early Triassic to early Miocene eras14. The sea between the western part of the Korean Peninsula and China is known to have been a lake into which the Yellow River once flowed; this lake was connected to the estuaries of many rivers flowing to the west coast on the Korean Peninsula15. Previous studies11,15,16 have claimed that many fish species found in these western-flowing rivers on the Korean Peninsula originated from the paleo-Yellow River system. Since the estuaries of these western-flowing rivers are connected or located in proximity on the huge continental shelf that is present around the west coast, it is presumed that freshwater fish species could move to other rivers whenever the sea levels became much lower than at present during the Pleistocene17,18. These rivers may have been isolated from the rivers that flow to the south coast where the continental shelf is not as well developed9. The coastal rivers on the east of the Korean Peninsula, which are surrounded by mountain ranges and are thought to have historically been influenced by the Amur River system from the north (east Russia), can also be regarded as an isolated region13,15,19. In this way, the freshwater system on the Korean Peninsula can be seen as having three independent historical subdistricts15.
Rhodeus notatus is a small freshwater bitterling species (Acheilognathidae; Cyprinoidea) native to China and the Korean Peninsula15,20. Before Kim revised the taxonomy of Rhodeus15, R. notatus of the Korean Peninsula was called R. atremius or R. suigensis in Japan or treated as a subspecies of R. atremius (i.e., R. a. atremius and R. a. suigensis)21. In fact, R. notatus has often been recognized as a member of the species complex including these two Japanese (sub)species and R. fangi in China, because these species share both the same number of chromosomes (i.e., 2n = 46), which is one pair less than other congeners, and other morphological features22,23,24. The Korean species was eventually synonymized with R. notatus after it was demonstrated that it had more morphological traits in common with Chinese R. notatus than other species within this species complex15. On the Korean Peninsula, R. notatus inhabits rivers flowing into the west and south coasts25. The distribution of this species preserves the historical imprints of migration through the paleo-Yellow River and the process of spreading to the freshwater system on the Korean Peninsula. The species could have migrated between rivers by sharing estuaries on the west coastal continental shelf11. Given that the continental shelf is not well-developed around the south coastal region, however, estuary coalescence might not be a major way to colonize the rivers flowing into the south coast, but instead it is more likely that gene flow occurred through the sharing of spatially proximal watersheds due to topographical erosion13. If so, large-scale population migration could not have been possible.
In this study, six mitochondrial and eight microsatellite loci were analyzed to estimate the level of intrapopulation genetic diversity, genetic structure among all major populations found on the Korean Peninsula and phylogenetic placement of R. notatus (Fig. 1). The results of these analyses were used to test four specific hypotheses about the geological and biogeographical history of freshwater ecosystem formation on the Korean Peninsula: i) R. notatus on the Korean Peninsula has originated from the paleo-Yellow River system, ii) the populations located in the rivers flowing into the west coast have genetic and phylogenetic signatures of estuary coalescence, iii) the populations of rivers flowing into the south coast where the continental shelf is not well-developed have signatures of history isolated from the western populations, and iv) the populations of the rivers flowing to the south coast, which probably formed through small-scale migration, have signatures of historical genetic drift.

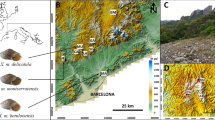
The collection sites of eight Rhodeus notatus populations from the Korean Peninsula. The locations of the reference species (pink: Chinese R. notatus; yellow: R. fangi; green; R. atremius; and blue: R. suigensis) used for comparison were also indicated on the East Asia map. The drainages on the peninsula were denoted by different colors. Rhodeus notatus populations comprise, from the west coastal region (western population group): HG, GE, MG; and from the south coastal region (southern population group): YS, TJ, SJ, NA1 and NA2. Geographic information of sampling localities is detailed in Table 1. The map was generated by Adobe Illustrator CC 2015 using a GIS shape file retrieved from inland water database in DIVA GIS (http://www.diva-gis.org/) and modified in QGIS v 2.16.3 in accordance with the guidelines suggested in the websites.
Results
Mitochondrial analysis
Our mitochondrial analysis revealed low to moderate levels of genetic variation within and among populations of R. notatus. A total of 9, 15, 12, 8, 10 and 15 unique haplotypes were identified from the sequence alignments of COI (KY628232 – KY628240), NADH1 (KY628271 – KY628285), NADH2 (KY628259 – KY628270), 16 S (KY628241 – KY628248), 12 S (KY628249 – KY628258) and cyt b (KY628286 – KY628300), respectively (Supplementary Table S1). All populations except Seomjin (SJ), Nakdong (NA) 1 and NA2 contained their own unique haplotypes in all six mitochondrial loci (Supplementary Table S1). A total of 28 haplotypes were detected when the sequences of six mitochondrial loci were concatenated (Supplementary Table S2). Genetic diversity, estimated from concatenated sequences, varied considerably across populations, with haplotype number (hn) ranging from two in Youngsan (YS) and NA1 to six in Han (HG) and Tamjin (TJ); further, haplotype diversity (h) varied between 0.200 (±0.154; YS) and 0.844 (±0.103; HG and TJ; Table 1).
We produced a haplotype network that illustrates genetic structure among the R. notatus populations we studied (Fig. 2a; Supplementary Fig. S1). All six mitochondrial loci data showed that haplotypes of populations SJ, NA1 and NA2 formed a cluster, around which population YS was closely located (Fig. 2a; Supplementary Fig. S1). Populations HG, Geum (GE) and Mangyoung (MG; western population group) were also closely related together despite the presence of strong genetic differentiation between them (Fig. 2a). In all loci except 12 S, population NA2 contained haplotypes that were distinct from the cluster of SJ, NA1 and NA2 and placed close to the haplotypes of western population group (Supplementary Fig. S1). Although population TJ geographically belongs to the southern population group, its haplotypes were quite distinct from those in other populations within this group (Supplementary Fig. S1).

(a) The unrooted haplotype network generated based on the concatenated sequences of six mitochondrial loci used. (b) Time-calibrated Bayesian tree reconstructed by BEAST 2.3.2 using cyt b sequences of R. notatus and various acheilognathid species. This tree is an excerpt from the original tree made with all reference species (Supplementary Fig. S2 and Table S5). The 95% highest posterior confidence intervals of divergence times were estimated on all major nodes with blue bar. The biogeographic history of R. notatus and the closely related species was inferred under the dispersal-extinction-cladogenesis (DEC) model of geographic range evolution implemented in RASP 3.2. The colors of the nodes represent the most likely ancestral areas, which are the same color as the nine regions shown on the map.
In our phylogenetic trees reconstructed using six mitochondrial loci, all Korean R. notatus haplotypes were monophyletic (Supplementary Fig. S2). Among the Korean populations of R. notatus, western population group, populations HG, GE and MG, and populations SJ, NA1 and NA2 independently formed monophyletic clades (Supplementary Fig. S2). Populations TJ and YS clustered with populations SJ, NA1 and NA2 (Supplementary Fig. S2). When using only cyt b, the western population group and clades of SJ-NA1-NA2 were clearly revealed, and populations TJ and YS clustered with SJ-NA1-NA2 (Supplementary Fig. S3), suggesting that the populations TJ and YS were highly differentiated from these two clades as predicted from our haplotype network analysis (Fig. 2a; Supplementary Fig. S1).
In the BEAST26 tree reconstructed using cyt b data, all Korean R. notatus haplotypes were divided into western (HG, GE and MG) and southern population groups (TJ, YS, SJ, NA1 and NA2; Fig. 2b; Supplementary Fig. S3). Like the results of haplotype network analysis, some haplotypes of population NA2 were clustered together with the haplotypes of western population group (Fig. 2b). Korean R. notatus was resolved as the most likely sister taxon of Chinese R. notatus (Fig. 2b; Supplementary Fig. S3). The haplotypes of R. fangi were recovered as a sister group of Chinese and Korean R. notatus, with Japanese R. atremius and R. suigensis being placed at the ancestral position in the species complex (Fig. 2b; Supplementary Fig. S3). The root node for R. notatus species complex was estimated to be about 4 MYA (Fig. 2b; Supplementary Fig S3). The results of our BEAST tree and dispersal-extinction-cladogenesis (DEC) model analyses showed that R. notatus populations on the Korean Peninsula originated from China at about 1.25 MYA (Fig. 2b; Supplementary Fig S3). These analyses could also be used to reconstruct the historical processes of gradual dispersion and population formation. Right after the colonization on the west coast (population HG), R. notatus individuals likely migrated to the place where population TJ was located, at about 1.1 MYA (Fig. 2b; Supplementary Fig. S3). The individuals of population HG and population TJ appear to have migrated to form the populations of western and southern population groups, respectively (Fig. 2b; Supplementary Fig. S3).
Mismatch distribution analysis (Supplementary Fig. S4) and extended Bayesian skyline plot (EBSP; Fig. 3) were performed for the two genetic clusters, western population group and the cluster of YS, SJ, NA1 and NA2 (southern genetic cluster; NA2 haplotypes clustered with western population group were not included) that were revealed in our haplotype network and BEAST tree analyses. The existence of strong genetic structure within western population group and southern genetic cluster likely produced multiple separate peaks in the mismatch distribution analysis (Supplementary Fig. S4). As expected from multiple peaks in the mismatch distribution graphs, both western population group and southern genetic clusters showed strong positive values of Tajima’s D and Fu’s Fs (Table 1). In our EBSP, the effective population size greatly decreased around 100,000 years ago in southern genetic cluster, whereas no visible change was observed in western population group (Fig. 3). Except population NA2, no population showed significant values of Tajima’s D and Fu’s Fs (Table 1). Significant positive values shown in population NA2 appear to be due to the existence of two different groups of haplotypes (Table 1; Supplementary Table S1).

The results of extended Bayesian skyline plot analysis performed based on the concatenated sequences of six mitochondrial loci for western population group (HG, GE and MG; a) and southern genetic cluster (YS, SJ, NA1 and NA2; b) of Rhodeus notatus.
Microsatellite analysis
Microsatellite loci exhibited extensively different levels of diversity. For example, Ak424 and Ak462 showed considerably high genetic diversity with 22 and 25 alleles, respectively, but only two alleles were observed in RC625 (Supplementary Table S3). As shown by the low heterozygosity of Ak424, the level of heterozygosity was not completely correlated with the allelic diversity (Supplementary Table S3). No significant signature of genotypic linkage disequilibrium was revealed from Fisher’s exact tests, indicating no possibility of physical linkage between loci or substructuring within populations. With the exception of Ak424, FIS values did not deviate significantly from zero (Supplementary Table S3). However, Ak424 was included in our further analyses, because such a result was thought to be due to the influence of the significant heterozygote deficiency in only a few populations (e.g., NA1) caused by the presence of null alleles, according to the results of Micro-Checker27. The overall level of interpopulation genetic differentiation was substantial with global FST = 0.361 and RST = 0.894 (see also Supplementary Table S3). The RST values were significantly higher than the mean permuted RST (and FST) values with a few exceptions (Supplementary Table S3), indicating a significant contribution of stepwise mutations to the interpopulation genetic differentiation.
The genetic diversity of R. notatus varied across populations. A considerably higher level of genetic diversity was observed in populations TJ and YS, whereas population HG showed extremely lower diversity estimates than others (Table 2). At the population level, no significant heterozygote deficiency was observed (Table 2). In our maximum likelihood estimation of relatedness among individuals within populations using ML-Relate28, a lower level of family relationship was found in populations MG and TJ than others (Table 2). No signature of genetic bottleneck was found, with the exception of population NA1 that exhibited the signature of genetic bottleneck upon Wilcoxon’s heterozygosity excess test (Table 2). M ratio values varied between 0.446 and 0.650 (Fig. 4). Three populations in southern population group, SJ, NA1 and NA2, showed clearly lower values than the Mc values estimated for each population (Fig. 4).

M ratio calculated from the microsatellite genotyping. Each circle indicates the average M ratio of each population of Rhodeus notatus. The solid and dot lines indicate the Mc threshold calculated from parameter θ = 1 and θ = 10, respectively.
The overall pairwise FST and RST values among populations were substantially high with some exceptions (Table 3), as predicted from the great global FST and RST values (Supplementary Table S3). Populations HG and GE in western population group showed extremely high FST and RST values in all pairwise comparisons (Table 3). Populations SJ, NA1 and NA2 exhibited a quite low genetic differentiation from each other, and populations TJ and YS seemed to have relatively higher genetic affinity to those populations (Table 3). Overall, multilocus RST values were significantly higher than FST and mean permuted RST values (RST = 0.894, pRST = 0.598; P = 0.006), suggesting the significant contribution of stepwise mutations to interpopulation genetic differentiation at the microsatellite loci. However, the average pairwise RST values were much lower than those of pairwise FST in the comparisons among populations of southern population group (Table 3). It was difficult to determine the optimal number of genetically distinguishable groups for our Bayesian Structure analysis (Fig. 5a). Most populations seem to have distinctly different genetic characteristics, although populations SJ, NA1 and NA2 tended to be closely tied together (Fig. 5b). Populations MG and TJ also showed relatively high genetic affinity with each other in this analysis (Fig. 5b). Upon PCA analysis, populations were likely subdivided into three groups (HG, GE-MG and the remainder), though a slight overlap occurred due to the genetic affinity between populations MG and TJ (Supplementary Fig. S5).

Population structure of Rhodeus notatus estimated from microsatellite genotyping. (a) The delta K method indicated that the most reliable number of clusters was two or six. (b) The population structure estimated in Structure indicated the existence of variable numbers of genetic clusters (K = 2 to 7).
An Approximate Bayesian Computation (ABC) approach was applied to identify historical pathway from western population group to population TJ using DIYABC29. Five possible scenarios were designed and tested with logistic regressions of simulated against observed data (Fig. 6). These five scenarios represent three different hypotheses about the origin of population TJ; the origin in scenarios 2 and 4, scenarios 3 and 5, and scenario 6 were MG, HG and GE, respectively (Fig. 6a). Our DIYABC analysis revealed that the most likely was scenario 2, followed scenario 4, while possibilities for other scenarios were negligible (Fig. 6), indicating that population TJ or the southern population group split from an ancestor of MG. These results were correlated with the overall pattern of pairwise FST and RST values (Table 3; Supplementary Fig. S5).

The results of Approximate Bayesian Computation (ABC) analysis implemented in DIYABC to identify the origin of population TJ from western population group. (a) Six (including null) biogeographic scenarios were designed and tested in DIYABC analysis. (b) The most likely scenario with the highest posterior probability was identified by performing a weighted logistic regression of each scenario probability to compare the deviations between simulated and observed summary statistics. The most likely was scenario 2, followed scenario 4, while possibilities for other scenarios were negligible.
Discussion
The average intrapopulation genetic diversity of the eight Rhodeus notatus populations examined in this study is quite low, given that fewer than two haplotypes per mitochondrial locus were observed in a population. Although no direct comparison was attempted using the same loci, the average number of microsatellite alleles per locus in a population was much below the known freshwater fish average21,30,31,32. Tanakia somjinensis, also a member of Acheilognathidae, has a much higher microsatellite diversity than R. notatus29. Given that T. somjinensis is a highly restricted Korean endemic species and is protected by Korean authority33, careful monitoring and management strategies for R. notatus will likely be required.
The results of our phylogenetic analysis revealed that two Japanese species, R. suigensis and R. atremius were the earliest branches from the clade of R. notatus species complex, suggesting that this complex originally evolved near Japan, or that peripheral speciation occurred around Japan from a widely distributed common ancestor. This prediction aligns with the previous claims that there have been widespread migrations of freshwater species between China and Japan via the Korean Peninsula34,35,36. In addition, the clear phylogenetic division of the Japanese species from others may be related to the geological process in which Japanese Archipelago has become completely separated from the continent by the sea. It can be assumed from our data that R. notatus on the Korean Peninsula originated through the paleo-Yellow River, based on the results of historical distribution estimation using our DEC model analysis and its phylogenetic affinity to the Chinese populations of R. notatus and R. fangi.
Given that Chinese R. notatus haplotypes are distinct from all Korean R. notatus haplotypes, it is difficult to determine the origin of R. notatus on the Korean Peninsula using only the degree of genetic affinity to Chinese haplotypes. The presence of strong genetic structure in the western population group demonstrates that there has been little active gene flow among the populations formed by paleo-yellow confluence. From the results of our haplotype network, DEC model and DIYABC analyses, we attempted to reconstruct the historical pathway to form southern population group from western population group. Solely based on our DEC model results, some individuals from population HG further migrated south to form a peri-Tamjin River population (TJ). It might be possible that populations GE and MG might have formed from the dispersal from population HG; both our haplotype network and microsatellite analyses indicate that the southern population group and population TJ had the closest genetic affinity with population MG. These results were also supported by DIYABC simulation and showed, at least indirectly, the historical trace of a dispersal over time from north to south on the west coastal continental shelf and to southern coastal rivers through population TJ.
The genetic diversity of population TJ was high compared to other populations in both mitochondrial and microsatellite loci. Although the mitochondrial diversity of population HG was also high, its microsatellite diversity was substantially lower than others, with half of the loci being completely fixed to a single allele. Models of neutral evolution suggest that genetic diversity is roughly proportional to the size of a population37,38; however, our data do not suggest that population TJ has even been particularly large throughout its history. This suggests that the region encompassing population TJ might have received migrants from several western coastal populations on the Korean Peninsula. Two slightly differentiated haplogroups were present in the monophyletic population TJ can be considered as evidence of gene flow from multiple isolated regions. Although the Korean Peninsula has never been covered with glaciers during Pleistocene glacial advances, cold and arid weather had persisted long even in the central region39 where the populations HG and GE are located. Under these historical circumstances, there may have been massive migration of freshwater species to more favorable regions, and large populations may have been formed in the southwestern corner.
Although population YS is also located on the southwestern corner of the Korean Peninsula, no clear signature of historical gene flow was identified from the western population group. Our genetic analyses revealed that population YS showed some degree of genetic affinity with population TJ and with the southern populations, SJ, NA1 and NA2. This result suggested the presence of a biogeographic pathway from population TJ to YS and then to the southern populations. The biogeographic route of freshwater systems along the south coast has never been reported to date, and in this respect, our results are of great importance.
As noted above, unlike the west coast of the Korean Peninsula, the continental shelf is not well developed around the south coast, and opportunities for past rivers to share estuaries with sea level fluctuation may not have existed. Indeed, studies have reported on the signature of gene flow by watershed capture between southern-flowing rivers25,40. Massive gene flow might not have been possible through watershed capture, as predicted in the introduction. The results of our various analyses help prove this assumption. First, our M-ratio data showed the signature of historical bottlenecks in populations SJ, NA1 and NA2. Second, at a regional level, the relative contribution of stepwise mutations to genetic differentiation among populations of southern population groups was much less pronounced; it is more likely that genetic drift was the cause of the differentiation among these populations. Finally, as evidenced by our EBSP, the populations of southern population group had likely undergone historical size decline.
Population NA2 contained rather unusual haplotypes that were genetically close to those of the western population group, especially population HG. It is not reasonable to suspect that such a genetic composition was caused by anthropogenic introduction of this species living in the Han River into the Nakdong River, because these two populations did not share the identical haplotypes. In addition, such a genetic pattern was not observed in our microsatellite analysis. Considering that the watershed locations of the Han and Nakdong Rivers are spatially proximal, it is possible that these haplotypes of population NA2 were naturally distributed; however, no direct evidence has yet been reported in support of this hypothesis.
We were able to address our major questions regarding the geographic origins of extant diversity among populations of R. notatus. First, our study revealed that R. notatus on the Korean Peninsula originated through the paleo-Yellow River system from China. Second, our genetic data demonstrated that the rivers flowing into the west and southwest coasts had formed confluence with the paleo-Yellow River system. However, the presence of a strong and complicated genetic structure within the western population group implies that historical gene flow among populations was not highly active, probably because long-term perfect confluence has not been formed, and the topographical structure on the continental shelf has strongly influenced on the pattern of gene flows. Third, the rivers flowing to the south coast seem to have been completely isolated from the confluence on the west and southwest, considering the great genetic difference between the western and southern population groups. Our data demonstrate that populations on the southwest corner had served as a biogeodispersal passage from west to south on the peninsula. Finally, migration among the populations of southern population group might have been possible only by small-scaled watershed captures, which was supported by the signature of historical genetic drift found in populations SJ, NA1 and NA2 from our results. This study represents the first use of molecular genetics to characterize the process of shaping contemporary distribution of freshwater species in the southwestern part of the Korean Peninsula. The inferences about the various fine-scale biogeographic pathways obtained here can be verified by a comparative study in the future with taxa having similar geographical distribution, for example, R. sinensis of the same genus.
Materials and Methods
Sampling and extraction
Rhodeus notatus individuals used in our genetic studies were 248 specimens stored at the Department of Life Sciences, Yeungnam University. Those specimens were collected from eight populations on seven different river basins flowing into west coast (western population group: HG, GE and MG) and south coast (southern population group: TJ, YS, SJ, NA1 and NA2; Fig. 1) from 2009 to 2011 in accordance with the Inland Water Fisheries Act and Wildlife Protection and Management Act of the Republic of Korea. The entire procedure of this study was approved by the Yeungnam University Institutional Animal Care and Use Committee (Protocol # 2015013). Small fin-clips cut from caudal fin of all individuals were used as tissue samples for our genetic analysis. Genomic DNA was extracted using a Wizard Genomic DNA purification kit (Promega, Madison, WI, USA).
Mitochondrial sequencing
Six mitochondrial loci, cytochrome oxidase subunit 1 (COI), cytochrome b (cyt b), NADH dehydrogenase subunit 1 (NADH1), NADH dehydrogenase subunit 2 (NADH2), 12 S rRNA (12 S) and 16 S rRNA (16 S), were sequenced for 10 selected R. notatus individuals per population. Information on the mitochondrial primer sets used in this study41,42,43,44,45 was provided in Supplementary Table S4. PCR was conducted using a 25 µl mixture consisting of 10–50 ng DNA, 1× Taq buffer (containing 2.5 mM MgCl2), 0.25 mM dNTPs, 1 μM of each primer and 0.25 units of Taq DNA polymerase (Solgent Inc, Daejeon, South Korea). Thermal cycling was composed of an initial denaturation at 95 °C for 5 min, 35 cycles of a denaturation at 95 °C for 30 sec, an annealing at 54 or 56 °C (see Supplementary Table S4) for 30 sec, at an extension at 72 °C for 45 sec, and an extra extension at 72 °C for 10 min. The PCR products were electrophoresed on 1.5% agarose gels to check the quality, purified using Wizard Genomic DNA purification kit (Promega, Madison, WI, USA) and sequenced by Genotech Inc (Daejeon, South Korea) on an ABI3730XL (Applied Biosystmes, Foster City, CA, USA) with BigDye Terminator 3.1 Cycle Sequencing Kit (Applied Biosystems).
Mitochondrial analyses
All mitochondrial sequences were rechecked through BLAST searches and aligned using Geneious 9.1.846. The coding loci, including COI, cyt b, NADH1 and NADH2, were examined against the inferred reading frame for the corresponding proteins using MEGA 6.0647. The intrapopulation genetic diversity was quantified by the number of haplotypes (hn), haplotype diversity (h)48 and nucleotide diversity (π)48 estimated using DnaSP 5.1049. The unrooted haplotype network was generated for the six mitochondrial loci and the concatenated sequences of all loci based on the connection limit above 0.95 in probability using TCS 1.26350 to analyze the distribution of haplotype diversity.
Phylogenetic trees were generated under two different algorithms, Bayesian inference (BI) and maximum likelihood (ML). All six mitochondrial loci were used in the reconstruction of phylogenetic trees. BI tree analysis was performed using MrBayes 3.251, and GTR + I + G was chosen as the best-fit substitution model by jModelTest 252 as the best-fitting nucleotide substitution model. BI analysis consisted of two parallel runs of 80 million Markov Chain Monte Carlo (MCMC) generations with sampling every 1,000 steps. The consensus tree for each data set was generated after removing the first 25% of sampled trees as burn-in. The Bayesian posterior probabilities (in percent) were presented as the node confidence in the tree. The ML tree analysis was conducted using IQ-Tree 1.3.10 with 10,000 ultrafast bootstrap replicates53. The best-fit substitution model was chosen to be GTR + I + G based on Bayesian Information Criterion (BIC) using ModelFinder54 implemented in IQ-Tree. Cyt b sequences were used in divergence time estimation using BEAST 2.3.026. This mitochondrial locus was used for this analysis because the cyt b sequence was most commonly reported in bitterling and cyprinid species that could be used as comparative materials from China and Japan and outgroups; see Supplementary Table S5. For the BEAST analysis, two independent runs of MCMC were performed based on 100 million generations, with sampling every 1,000th generations, using GTR + I + G selected by jModelTest 2 as the best-fitting nucleotide substitution model. A Yule tree prior and an uncorrelated lognormal relaxed-clock were implemented on this analysis. Calibration of the molecular clock was conducted by using the earliest fossil record data of Acheilognathidae55,56 for the basal node of this family (23.0 Mya; normal distribution). Tracer 1.65757 was used to examine the convergence of run parameters by determining whether ESSs (effective population sizes) exceeded 200. The maximum clade credibility tree with median heights was generated using TreeAnnotator 2.3.058, after discarding the first 25% of trees as burn-in. FigTree 1.4.259 was used to visualize the topology of the consensus tree generated with divergence times. The biogeographic history of R. notatus and the closely related species was explored under the dispersal-extinction-cladogenesis (DEC) model of geographic range evolution60 implemented in RASP 3.261. The DEC model was used to assess the likelihood of all possible ancestral distributions at a given node57. A biogeographic model was constructed by coding each species or population used in the BEAST analysis as occurring in one of the nine regions including the seven drainage basins in which Korean R. notatus individuals were collected, China and Japan. The tests of DEC were performed based on the parameters obtained from our BEAST analysis, and the results were visualized on the tree using Affinity Designer 1.6.1 (Serif Ltd; https://affinity.serif.com/en-us/).
All six mitochondrial loci were used to find the best approximation regarding the historical demographic pattern for each R. notatus population and genetic cluster based on three different methods. First, Tajima’s D62 and Fu’s Fs63 were calculated using DnaSP. Second, mismatch distribution analysis was carried out to show the observed distribution of pairwise differences between mitochondrial sequences under the model of population expansion64 using Arlequin 3.565 for each genetic cluster. Finally, extended Bayesian skyline plot (EBSP) was created using BEAST for each genetic cluster66 to evaluate historical demographic changes of effective population size over historical time under the HKY model and strict-clock model with mutation rates (1% per million years)67. The EBSP analysis was performed for 10 million generations with sampling every 1,000 steps and discarding the first 25% as burn-in. Tracer was used to assess the convergence between runs by examining whether ESSs exceeded 200 as an indicator.
Microsatellite genotyping
All samples were genotyped using eight previously reported microsatellite primer sets (Supplementary Table S3)26,68,69. The 5′ end of each forward primer was fluorescently labelled with either FAM, HEX or NED (Applied Biosystems Life Technologies, Carlsbad, CA, USA). PCR was conducted using a 10 µl mixture containing 1 µl of extracted DNA, 1× Taq buffer (containing 2.5 mM MgCl2), 0.25 mM dNTPs, 1 µM of each primer and 0.15 units of Taq DNA polymerase (Solgent). Thermal cycling consisted of an initial denaturation at 95 °C for 5 min, 35 cycles of a denaturation at 95 °C for 30 sec, an annealing at 54–58 °C for 30 sec, an extension at 72 °C for 45 sec, and an final extension at 72 °C for 10 min. The fluorescently labeled PCR products were genotyped on an ABI 3730xl Genetic Analyzer by Biomedic Inc (Bucheon, South Korea) and scored using GeneMapper 3.7 (Applied Biosystems). Allele size was determined using the Peak Scanner 1.0 (Applied Biosystems).
Microsatellite analyses
Genetic diversity for each locus and population was quantified with the mean or total number of alleles per locus, allelic richness, observed and expected heterozygosities and fixation index (FIS) estimated in Arlequin, Fstat 2.9.3.270 and Genepop 4.271. Genepop was used to detect whether the genotype frequencies were deviated from Hardy-Weinberg equilibrium for each locus–population combination using the Fisher’s exact test based on Markov chain parameters with 1000 batches and 10,000 iterations per batch72. Fisher’s exact test of linkage disequilibrium between pairs of loci was performed with the Markov chain algorithm under the null hypothesis of independence using Genepop. Microsatellite genotypes were checked using Micro-Checker 2.2.327.
The expected percentage of family (full-sib, half-sib and parent-offspring) relatedness for each population was determined using ML-Relate. A significant excess of expected heterozygosity under the mutation-drift equilibrium relative to expected heterozygosity under Hardy-Weinberg equilibrium was checked based on the Wilcoxon sign-rank test under the TPM (two phase model) with a setting of 70% stepwise mutations model (SMM) and 30% infinite alleles model (IAM) using Bottleneck v1.273. In addition, Bottleneck was used to examine mode-shift deviation from the typical L-shaped distribution of allelic frequencies74. Both Bottleneck analyses were used to test whether populations may have recently undergone significant size reduction. The M ratio, which is the mean ratio of the number of alleles to the range in allele size75, and a critical value of M (Mc) were quantified for each population using Arlequin to test whether populations have historically experienced a significant reduction in size75. Mc was calculated with default values and θ (=4Neμ) = 1 and 10 under a TPM constraining the model by defining 80% of mutations as conforming to a stepwise mutation model and 20% as an infinite allele model. Critical significance values in all statistics were corrected for multiple comparisons based on the Bonferroni procedure76.
Genetic distance among populations was quantified using global FST77 and RST78, as well as pairwise FST77 and RST79 estimated by Arlequin and Genepop. A randomization procedure of allele sizes was then conducted using SPAGeDi v1.180, whereby the different allele sizes found at a locus were subjected to 2,000 random permutations to examine whether the allele sizes from stepwise mutation contributed to population differentiation81. Bayesian clustering analysis with the software Structure v2.3.482 was used to estimate the degree of structuring among populations. Before analysis, the putative numbers of genetically distinguishable groups (K) were predicted by the delta K method83 implemented in Structure Harvester v0.6.9484. Ten independent MCMC runs were performed for each K with 4 × 105 iterations after a burn-in of 105 iterations. To visualize the relationship between individuals and populations based on the best linear combination of allele frequencies, principal component analysis (PCA) was implemented using GENETIX v4.0585. To test competing hypotheses about biogeographic pathways leading to population TJ from western population groups showing strong genetic structure, an Approximate Bayesian Computation (ABC) approach was taken, implemented in the software DIYABC v.2.1.029. A total of five scenarios with a null hypothesis were tested (Fig. 6), and the factor to distinguish between scenarios was tree topology. A total of 1,000,000 simulated datasets were grouped according to each scenario. The probability of each scenario was measured by performing a weighted logistic regression of each scenario probability to compare the deviations between simulated and observed summary statistics to find the most likely scenario.
References
Castric, V. & Bernatchez, L. Individual assignment test reveals differential restriction to dispersal between two salmonids despite no increase of genetic differences with distance. Mol. Ecol. 13, 1299–1312 (2004).
Crispo, E., Bentzen, P., Reznick, D. N., Kinnison, M. T. & Hendry, A. P. The relative influence of natural selection and geography on gene flow in guppies. Mol. Ecol. 15, 49–62 (2006).
Caldera, E. J. & Bolnick, D. I. Effects of colonization history and landscape structure on genetic variation within and among threespine stickleback (Gasterosteus aculeatus) populations in a single watershed. Evol. Ecol. Res. 10, 575–598 (2008).
Bermingham, E. & Martin, A. P. Comparative mtDNA phylogeography of neotropical freshwater fishes: testing shared history to infer the evolutionary landscape of lower Central America. Mol. Ecol. 7, 499–517 (1998).
Sivasundar, A., Bermingham, E. & Ortí, G. Population structure and biogeography of migratory freshwater fishes (Prochilodus: Characiformes) in major South American rivers. Mol. Ecol. 10, 407–417 (2001).
Suk, H. & Neff, B. Microsatellite genetic differentiation among populations of the Trinidadian guppy. Heredity 102, 425–434 (2009).
Whiteley, A. R. et al. Genetic variation and effective population size in isolated populations of coastal cutthroat trout. Conserv. Genet. 11, 1929–1943 (2010).
Griffiths, D. Pattern and process in the ecological biogeography of European freshwater fish. J. Anim. Ecol. 75, 734–751 (2006).
Jeon, H. & Suk, H. Y. Pseudo but actually genuine: Rhodeus pseudosericeus provides insight into the phylogeographic history of the Amur bitterling. Anim. Cells Syst. 18, 275–281 (2014).
Thomaz, A. T., Malabarba, L. R. & Knowles, L. L. Genomic signatures of paleodrainages in a freshwater fish along the southeastern coast of Brazil: genetic structure reflects past riverine properties. Heredity 119, 287–294 (2017).
Jung, J., Song, K. H., Lee, E. & Kim, W. Mitochondrial genetic variations in fishes of the genus Carassius from South Korea: proximity to northern China rather than Japanese Islands? Hydrobiologia 635, 95–105 (2009).
Jang, J. E. et al. Genetic diversity and genetic structure of the endangered Manchurian trout, Brachymystax lenok tsinlingensis, at its southern range margin: conservation implications for future restoration. Conserv. Genet. 18, 1023–1036 (2017).
Jeon, H., Kim, D., Lee, Y. J., Bae, H. & Suk, H. Y. The genetic structure of Squalidus multimaculatus revealing the historical pattern of serial colonization on the tip of East Asian continent. Sci. Rep. 8, 10629, https://doi.org/10.1038/s41598-018-28340-x (2018).
Chough, S. K., Kwon, S., Ree, J. & Choi, D. K. Tectonic and sedimentary evolution of the Korean peninsula: a review and new view. Earth-Sci. Rev. 52, 175–235 (2000).
Kim, I. Illustrated encyclopedia of fauna & flora of Korea, vol. 37, Freshwater Fishes (Ministry of Education, 1997).
Takehana, Y., Uchiyama, S., Matsuda, M., Jeon, S. & Sakaizumi, M. Geographic variation and diversity of the cytochrome b gene in wild populations of medaka (Oryzias latipes) from Korea and China. Zool. Sci. 21, 483–491 (2004).
Yoo, D. et al. Seismic stratigraphy and depositional history of late Quaternary deposits in a tide-dominated setting: An example from the eastern Yellow Sea. Mar. Petrol. Geol. 73, 212–227 (2016).
Zhang, D. et al. Pleistocene sea level fluctuation and host plant habitat requirement influenced the historical phylogeography of the invasive species Amphiareus obscuriceps (Hemiptera: Anthocoridae) in its native range. BMC Evol. Biol. 16, 174, https://doi.org/10.1186/s12862-016-0748-3 (2016).
Kim, D., Hirt, M. V., Won, Y. & Simons, A. M. Small fishes crossed a large mountain range: Quaternary stream capture events and freshwater fishes on both sides of the Taebaek Mountains. Integra. Zool. 12, 292–302 (2017).
Chang, C. et al. Phylogenetic relationships of Acheilognathidae (Cypriniformes: Cyprinoidea) as revealed from evidence of both nuclear and mitochondrial gene sequence variation: evidence for necessary taxonomic revision in the family and the identification of cryptic species. Mol. Phylogenet. Evol. 81, 182–194 (2014).
Miyake, T. et al. The genetic status of two subspecies of Rhodeus atremius, an endangered bitterling in Japan. Conserv. Genet. 12, 383–400 (2011).
Kimura, S. & Nagata, Y. Scientific name of Nippon-baratanago, a Japanese bitterling of the genus Rhodeus. Jap. J. Ichthyol. 38, 425–429 (1992).
Arai, R., Jeon, S. & Ueda, T. Rhodeus pseudosericeus sp. nov., a new bitterling from South Korea (Cyprinidae, Acheilognathinae). Ichthyol. Res. 48, 275–282 (2001).
Yu, Y., Yi, W., Ma, Z., Yang, R. & Shen, J. The complete mitochondrial genome of Rhodeus fangi (Cypriniformes, Cyprinidae, Acheilognathinae). Mitochondrial DNA Part A. 27, 284–285 (2016).
Kim, I. & Park, J. Freshwater fishes of Korea (Kyohaksa, 2002).
Bouckaert, R. et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10, e1003537, https://doi.org/10.1371/journal.pcbi.1003537 (2014).
van Oosterhout, C., Hutchinson, W. F., Wills, D. P. M. & Shipley, P. Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4, 535–538 (2004).
Kalinowski, S. T., Wagner, A. P. & Taper, M. L. ML‐Relate: a computer program for maximum likelihood estimation of relatedness and relationship. Mol. Ecol. Resour. 6, 576–579 (2006).
Cornuet, J. M. et al. DIYABC v2.0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30, 1187–1189 (2014).
Shirai, Y., Ikeda, S. & Tajima, S. Isolation and characterization of new microsatellite markers for rose bitterlings, Rhodeus ocellatus. Mol. Ecol. Resour. 9, 1031–1033 (2009).
Ward, R. D., Woodwark, M. & Skibinski, D. O. F. A comparison of genetic diversity levels in marine, freshwater, and anadromous fishes. J. Fish Biol. 44, 213–232 (1994).
DeWoody, J. A. & Avise, J. C. Microsatellite variation in marine, freshwater and anadromous fishes compared with other animals. J. Fish Biol. 56, 461–473 (2000).
Jeon, H. et al. Development of novel microsatellite loci and analyses of genetic diversity in the endangered Tanakia somjinensis. Biochem. Syst. Ecol. 66, 344–350 (2016).
Sakai, H., Yamamoto, C. & Iwata, A. Genetic divergence, variation and zoogeography of a freshwater goby, Odontobutis obscura. Ichthyol. Res. 45, 363–376 (1998).
Yu, D., Chen, M., Tang, Q., Li, X. & Liu, H. Geological events and Pliocene climate fluctuations explain the phylogeographical pattern of the cold water fish Rhynchocypris oxycephalus (Cypriniformes: Cyprinidae) in China. BMC Evol. Biol. 14, 225, https://doi.org/10.1186/s12862-014-0225-9 (2014).
Park, S., Yoo, D., Lee, C. & Lee, E. Last glacial sea-level changes and paleogeography of the Korea (Tsushima) Strait. Geo-Mar. Lett. 20, 64–71 (2000).
Gilpin, M. & Soulé, M. Conservation biology: the science of scarcity and diversity. (Sinauer, 1986).
Frankham, R. Genetics and extinction. Biol. Conserv. 126, 131–140 (2005).
d’Alpoim Guedes, J., Austermann, J. & Mitrovica, J. X. Lost foraging opportunities for East Asian hunter‐gatherers due to rising sea level since the Last Glacial Maximum. Geoarchaeology 31, 255–266 (2016).
Yang, E. C., Lee, S. Y., Lee, W. J. & Boo, S. M. Molecular evidence for recolonization of Ceramium japonicum (Ceramiaceae, Rhodophyta) on the west coast of Korea after the last glacial maximum. Bot. Mar. 52, 307–315 (2009).
Palumbi, S. et al. The simple fool’s guide to PCR (Version 2.0) (Department of Zoology and Kewalo Marine Laboratory, University of Hawaii, 1991).
Palumbi, S. Nucleic acids II: the polymerase chain reaction (ed. Hillis, D. M., Moritz, C., Mable, B. K.) 205–247 (Sinauer, 1996).
Brito, R. M., Briolay, J., Galtier, N., Bouvet, Y. & Coelho, M. M. Phylogenetic relationships within genus Leuciscus (Pisces, Cyprinidae) in Portuguese fresh waters, based on mitochondrial DNA cytochrome b sequences. Mol. Phylogenet. Evol. 8, 435–442 (1997).
Thacker, C. E. Molecular phylogeny of the gobioid fishes (Teleostei: Perciformes: Gobioidei). Mol. Phylogenet. Evol. 26, 354–368 (2003).
Perdices, A., Cunha, C. & Coelho, M. Phylogenetic structure of Zacco platypus (Teleostei, Cyprinidae) populations on the upper and middle Chang Jiang (=Yangtze) drainage inferred from cytochrome b sequences. Mol. Phylogenet. Evol. 31, 192–203 (2004).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Nei, M. Molecular evolutionary genetics (Columbia university press, 1987).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Clement, M., Posada, D. & Crandall, K. A. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659 (2000).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772 (2012).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–74 (2015).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Tomoda, Y., Kodera, H., Nakajima, T. & Yasuno, T. Fossil freshwater fishes from Japan. Mem. Geol. Soc. Jpn. 14, 221–243 (1977).
Yasuno, K. Fossil pharyngeal teeth of the Rhodeinae fish from the Miocene Katabira Formation of the Kani Group, Gifu Prefecture, Japan. Bull. Mizunami Fossil Mus. 11, 101–105 (1984).
Rambaut, A., Suchard, M. A., Xie, D. & Drummond, A. J. Tracer 1.6, available at, http://beast.bio.ed.ac.uk/tracer (2014).
Rambaut, A. & Drummond, A. J. TreeAnnotator v.2.3.0., available at, http://beast2.org (2014).
Rambaut, A. FigTree v.1.4., available at, http://tree.bio.ed.ac.uk/sofware/fgtree/ (2012).
Ree, R. H. & Smith, S. A. Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst. Biol. 57, 4–14 (2008).
Yu, Y., Harris, A. J., Blair, C. & He, X. RASP (Reconstruct Ancestral State in Phylogenies): a tool for historical biogeography. Mol. Phylogenet. Evol. 87, 46–49 (2015).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Fu, Y. X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147, 915–925 (1997).
Rogers, A. R. & Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 9, 552–569 (1992).
Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Zardoya, R. & Doadrio, I. Molecular evidence on the evolutionary and biogeographical patterns of European cyprinids. J. Mol. Evol. 49, 227–237 (1999).
Dawson, D. A., Burland, T. M., Douglas, A., Le Comber, S. C. & Bradshaw, M. Isolation of microsatellite loci in the freshwater fish, the bitterling Rhodeus sericeus (Teleostei: Cyprinidae). Mol. Ecol. Notes 3, 199–202 (2003).
Kim, W. et al. Isolation and inheritance of microsatellite loci for the oily bittering (Acheilognathus koreensis): applications for analysis of genetic diversity of wild populations. Anim. Cells Syst. 16, 321–328 (2012).
Goudet, J. FSTAT, a program to estimate and test gene diversity and fixation indices (version 2.9.3), available at, http://www2.unil.ch/popgen/softwares/fstat.htm (2001).
Raymond, M. & Rousset, F. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J. Hered. 86, 248–249 (1995).
Guo, S. W. & Thompson, E. A. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48, 361–372 (1992).
Piry, S., Luikart, G. & Cornuet, J. BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 90, 502–503 (1999).
Luikart, G., Sherwin, W., Steele, B. & Allendorf, F. Usefulness of molecular markers for detecting population bottlenecks via monitoring genetic change. Mol. Ecol. 7, 963–974 (1998).
Garza, J. & Williamson, E. Detection of reduction in population size using data from microsatellite loci. Mol. Ecol. 10, 305–318 (2001).
Rice, W. R. Analyzing tables of statistical tests. Evolution 43, 223–225 (1989).
Weir, B. S. & Cockerham, C. C. Estimating F‐statistics for the analysis of population structure. Evolution 38, 1358–1370 (1984).
Rousset, F. Equilibrium values of measures of population subdivision for stepwise mutation processes. Genetics 142, 1357–1362 (1996).
Michalakis, Y. & Excoffier, L. A generic estimation of population subdivision using distances between alleles with special reference for microsatellite loci. Genetics 142, 1061–1064 (1996).
Hardy, O. J. & Vekemans, X. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Resour. 2, 618–620 (2002).
Hardy, O. J., Charbonnel, N., Freville, H. & Heuertz, M. Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163, 1467–1482 (2003).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Earl, D. A. & vonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–36−0.695 (2012).
Belkhir, K., Borsa, P., Chikhi, L., Raufaste, N. & Bonhomme, F. GENETIX 4.05, Windows TM software for population genetics (Laboratoire génome, populations, interactions, CNRS UMR 5000, 2004).
Acknowledgements
We thank Seong-Jang Cho, Akimitsu Hanado and Inoue Yutaka for providing field assistance. We also thank anonymous reviewers and the editor for constructive comments on the manuscript. This research was performed under the support of the grants provided from the National Research Foundation of Korea (Grant No. 2015R1D1A2A01058987 and 2018R1D1A1A02086230).
Author information
Authors and Affiliations
Contributions
H.W. and H.Y.S. wrote the main manuscript text. H.J. provided key ideas for experimental design and analysis. H.W. and H.J. collected the data. H.W. and H.Y.S. prepared tables. H.W. and H.J. prepared figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Won, H., Jeon, HB. & Suk, H.Y. Evidence of an ancient connectivity and biogeodispersal of a bitterling species, Rhodeus notatus, across the Korean Peninsula. Sci Rep 10, 1011 (2020). https://doi.org/10.1038/s41598-020-57625-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-57625-3
This article is cited by
-
Characterization of the complete mitogenome of the endangered freshwater fish Gobiobotia naktongensis from the Geum River in South Korea: evidence of stream connection with the Paleo-Huanghe
Genes & Genomics (2022)
-
Phylogeography of the Korean endemic Coreoleuciscus (Cypriniformes: Gobionidae): the genetic evidence of colonization through Eurasian continent to the Korean Peninsula during Late Plio-Pleistocene
Genes & Genomics (2022)
-
Differential patterns of diversity at neutral and adaptive loci in endangered Rhodeus pseudosericeus populations
Scientific Reports (2021)
-
Conservation issues partly solved on “Suigenzenitanago” (Rhodeus smithii complex) impaired by the taxonomic confusion
Ichthyological Research (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
