
Abstract
Pinctada fucata martensii, is an economically important marine bivalve species cultured for seawater pearls. At present, we know little about the molecular mechanisms of the insulin signalling pathway in this oyster. Herein, we cloned and analysed an insulin-like peptide (PfILP) and its signalling pathway-related genes. We detected their expression levels in different tissues and developmental stages. Recombinant PfILP protein was produced and found to significantly increase primary mantle cell activity and induce the expression of the proliferating cell nuclear antigen (PCNA) gene. PfILP could also regulate the 293T cell cycle by stimulating the S phase and inhibiting the G1 and G2 phases. Recombinant PfILP protein induced the expression of its signalling pathway-related genes in mantle cells. In vitro co-immunoprecipitation analysis showed that PfILP interacts with PfIRR. PfILP activated expression of the pfIRR protein, and also activated the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways by stimulating phosphorylation of MAPK and AKT. Further analysis showed that PfILP up-regulated glycogen synthesis-related genes glycogen synthase kinase-3 beta (GSK-3β), protein phosphatase 1 (PP1) and glucokinase (GK) at the mRNA level, as well as the expression of the PP1 protein, and phosphorylation of GSK-3β. These results confirmed the presence of a conserved insulin-like signalling pathway in pearl oyster that is involved in cell activity, glycogen metabolism, and other physiological processes.
Similar content being viewed by others


Introduction
The insulin-relaxin superfamily is composed of two subfamilies: the insulin and insulin-like growth factor (IGF) subfamily, and the relaxin and insulin-like (INSL) subfamily1. These proteins exhibit a highly structural conservation. They share conserved cysteine residues required for the formation of disulphide bridges which are the hallmark of this superfamily2. The insulin-relaxin superfamily has a variety of functions; they can activate both PI3K/AKT and MAPK pathways that are composed of multiple effectors3. These pathways control cell growth, proliferation, differentiation and apoptosis, cell adhesion, embryo development, organ formation, bone regeneration, and other physiological processes4,5,6. Insulin is a key peptide hormone that can activate the PI3K and MAPK pathways to regulate metabolism and growth in metazoans1,7,8,9. In vertebrates, the function of insulin has been extensively studied since it is central to regulate the glycogen, fat, and protein metabolism2,10,11.
Insulin-like peptides (ILPs) are an ancient protein family, present in vertebrates and invertebrates, that regulate diverse physiological processes. In invertebrates, multiple ILPs have been identified, and bombyxin was the first reported ILP from the silk moth Bombyx mori12,13. An abundance of ILPs have since been identified in various invertebrates species, including the branchiopod crustacean Daphnia pulex14, parasitic platyhelminths15, the nematode Caenorhabditis elegans16 and various molluscs17,18, but primarily in arthropods19.
In insects, the structure and functiona of ILPs is similar to vertebrate insulins and relaxin20. In addition to sharing similar functions with vertebrate peptides, insect ILPs also regulate neurotransmitters and growth factors19,21,22. Signalling of ILPs is broadly studied in the fruit fly Drosophila melanogaster. Eight genes encoding ILPs (ILP1-8) in its genome were found20,23,24. These ILPs are coupled to insulin-like receptor, and then activate downstream signal network25,26. Besides, Dilp8 can bind GPCR-namely LGR3 and LGR4 to mediate the regeneration checkpoint developmental delay and growth coordination in D.melanogaster27,28. Insulin/ILP signalling pathways then regulates cell proliferation, differentiation and growth of organisms29,30,31.
In molluscs, ILPs have been identified in the Lymnea stagnalis32,33, Aplysia californica17, Anodonta cygnea34, and the Sinonovacula constricta13. However, most previous studies have only deal with the primary characterization of gene products and that more functional data are needed. Regarding ILP receptors, a unique pfIR receptor (PfIRR) was identified in P. fucata martensii and found to be mainly expressed in the testis and adductor muscle35. Moreover, an IGF binding protein gene (Pfigfbp) from P. fucata martensii was characterised, and found to be transcribed mainly in the foot36. However, no insulin-like analogues or peptides were found in P. fucata martensii, and effectors of the insulin-like signalling pathway remain poorly documented. In addition, it is unknown whether PfILPs has similar functions to vertebrates insulins or IGFs. Furthermore, whether PfILP can act like vertebrate insulin/IGFs and activate similar downstream signalling pathways in P. fucata martensii remains unknown.
To investigate these questions, in the present work we identified the first ILP and six effectors in the pearl oyster P. fucata martensii. We analysed their phylogenetic relationships with other homologous proteins and determined their spatio-temporal expression by quantitative real-time PCR (qPCR). We probed the interactions between recombinant PfILP and pfIRR, and the effects of PfILP-mediated pfIRR stimulation in the activation of the MAPK and PI3K signalling cascades in primary P. fucata martensii mantle cells. We further examined the biological activity of PfILP using in vitro phosphorylation assays and investigated its modulatory roles in glycogen metabolism and the cell cycle.
Results
Identification of seven ILP signalling pathway genes
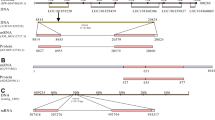
Seven potential effectors related to ILP signalling (based on sequence homology)- Pfirs1, Pfpik3r1, Pfakt, Pfsos2, Pfrap-1, Pfraf and PfILP- were identified from P. fucata martensii(Supplementary Table S1). BLASTp analyses showed high sequence homology between these six genes (except the PfILP) and genes known to be involved in the insulin signaling pathway (Supplementary Table S1). Sequence alignment demonstrated the levels of high identity; From an evolutionary perspective, six ILP signalling pathway genes are thought to have evolved from an ancestral gene, respectively, which explains the common features (Supplementary Figs. 1–6). For PfILP genes, it has four exons and three introns in genomic (Fig. 1A). PfILP protein comprises a signal peptide followed by a B-chain, a connecting peptide (C-peptide), and an A-chain (Fig. 1B). PfILP includes six cysteines in the A and B chain, allowing the formation of disulphide bridges, and two putative disulphide bonds were assumed to form two inter-chain bridges across the two chains, and one intra-chain bond on the A-chain (Fig. 1B) that is essential for tertiary folding. The 3D structure of PfILP is consistent with this phenomenon (Fig. 1C). Multiple sequence alignment of PfILP showed that their sequences are poorly conserved, especially among invertebrates, except for the six cysteine sites, which are strictly conserved (Supplementary Fig. 7). The GenBank number is in Supplementary Table S2. The phylogenetic tree shows that PfILP sequence identified in P. fucata martensii is orthologous to the previously identified mollusks ILPs in Crassostrea gigas, Crassostrea virginica, Mizuhopecten yessoensis, Sinonovacula consricta (Fig. 1D). The GenBank number is in Supplementary Table S3. The identified ILP signalling-related components provide valuable information for future studies of this pathway in P. fucata martensii.

Basic information of PfILP. (A) Schematic diagram of the PfILP gene. The gene contains four exons and three introns. (B) Schematic representation of the predicted primary structure of the PfILP protein. (C) Predicted three-dimensional model of PfILP based on human insulin-like growth factor 1 (IGF1; PDB id 2gf1). Conserved cysteine residues and the resulting disulphide bridges are marked. The model was constructed using Swiss-Model and edited using PyMOL Viewer. (D) Maximum likelihood phylogenetic tree of PfILP from P. fucata martensii with homologs from other species. PfILP from P. fucata martensii is marked with a green circle. Numbers at tree nodes indicate the bootstrap percentage after 1000 replicates.
The expression patterns of seven ILP signalling pathway genes in different tissues and developmental stages
Seven tissues from pearl oysters or different developmental stages were selected to determine the basal transcriptional levels by real-time PCR. Most of them are highly expressed in the foot or in D-shaped larvae whereas, in contrast in adductor muscle or 32-cells embryos there is apparently contrasting expression patterns (Fig. 2). The results showed that the expression patterns of the seven genes in tissues or developmental stages were totally same, which suggests that they might be involved in same physiological activities of in P. fucata martensii.

Expression analyses of seven ILP signalling pathway genes. Expression levels of PfILP, Pfirs1, Pfpik3r1, Pfakt, Pfsos2, Pfrap-1 and Pfraf in healthy tissues (A) and developmental stage (B) of P. fucata maretnsii measured by real-time quantitative real-time PCR. The 18S rRNA gene was used as an internal control and calculated using the 2−ΔΔCT method. Each vertical bar represents the mean ± SEM (n = 3).
Production of recombinant PfILP
The expression vector containing a cDNA encoding the mature PfILP polypeptide (no signal peptide) fused to a His-tag was constructed and transformed into E. coli Transetta (DE3) cells, and expression of recombinant PfILP protein was induced by IPTG. The results showed that most of the protein was present in insoluble inclusion bodies (Fig. 3A). Negative control cells containing vector alone did not yield overexpression bands. The His-tagged PfILP protein was retrieved from inclusion bodies via solubilisation before purification. Refolding PfILP protein sample was filtered to remove impurities from the resin by repetitive washing. And then a single protein band of 26 kDa was greatly enriched in the final column eluate (Fig. 3B). Subsequent immunoblotting performed with anti-His antibody revealed that the purified protein was specifically labelled (Fig. 3C).

Purification of recombinant PfILP. (A) SDS-PAGE (12%) analysis of recombinant PfILP after 1 mM IPTG induction. Lane 1, soluble fraction after ultrasonication; Lane 2, insoluble fraction after ultrasonication precipitation; Lane 3, total protein from induced E. coli cells harbouring pET28a. (B) Inclusion body proteins were washed, dissolved, refolded and purified. Lanes 1–3, recombinant PfILP eluted with 250 mM imidazole. (C) Purified PfILP blotted with a rabbit anti-His IgG antibody (lane 1–4). Each set of bands for each protein is from the same gel.
Cell viability following treatment with recombinant PfILP
A large quantity of cells were migrated out from explant at day 7. Cells almost covered plates at 2 weeks (Fig. 4A). Trypan blue staining demonstrated that more than 90% of cells survived up 2 weeks (Fig. 4B). The mitogenic effect of PfILP was examined by measuring the cell viability of primary cells using a Cell Counting Kit-8. The results clearly showed that PfILP improved mantle cell viability at a concentration of 0.25 μg/ml till top at 1 μg/ml (Fig. 4C). Additionally, the PCNA gene was up-regulated maximally by PfILP at a dose of 1 μg/ml (Fig. 4D).

Effects of PfILP on cell activity and PCNA gene expression. (A) Living conditions of primary P. fucata martensii mantle cells over 2 weeks. At day 15, more cells migrated; (B) Cell suspension stained with trypan blue. (C) Cells were treated with different doses of PfILP protein. *significantly different from controls at p < 0.05; **extremely significantly different from controls at p < 0.01 (n = 3). (D) Analysis of PCNA transcript levels following treatment with different concentrations of PfILP. *p < 0.05, **p < 0.01.
PfILP regulates the expression of pfIRR
To verify the interaction between PfILP and pfIRR, in vitro Co-IP analysis was performed using P. fucata martensii mantle cells. The results showed that PfILP was co-immunoprecipitated by pfIRR (Fig. 5A), and pfIRR was co-immunoprecipitated by PfILP (Fig. 5B). Additionally, we investigated the expression of pfIRR in mantle cells following treatment with PfILP. The pfirr transcript was significantly up-regulated following treatment with 1.0 μg/ml or 2.0 μg/ml PfILP (Fig. 5C). The pfIRR protein expression was examined by determining the ratio of pfIRR/GAPDH. As shown in Fig. 5D, PfILP induced pfIRR protein expression within 5 min, levels peaked at 10 min, and induction lasted for 1 h. When treated for 30 min, PfILP induced pfIRR expression at a dose of 0.25 μg/ml, expression was maximal with a dose of 0.5 μg/ml (Fig. 5E) and induction was observed up to a dose of 2.0 μg/ml.

Effects of PfILP on PfIRR. (A,B) Co-IP analysis of the interaction between PfILP and PfIRR in vitro; (C) Quantitative relative expression of Pfirr in mantle cells after a 24 h incubation with various concentrations of PfILP. Each bar represents the mean ± S.E.M. (n = 3). *p < 0.05, **p < 0.01. (D,E) Western blotting analysis of pfIRR protein expression following treatment with PfILP for different timepoints and at different concentrations. GAPDH was used as an internal reference. Each set of bands for each protein is from the same gel.
PfILP activates MAPK and PI3K/Akt signalling pathways
To study the effects of PfILP on activation of the MAPK and PI3K signal transduction pathways, the seven genes (Pfirs1, Pfpik3r1, Pfakt, Pfsos2, Pfrap-1, Pfraf and Pferk) associated with the insulin-like signalling pathway of P. fucata martensii were examined. Additionally, phospho-p44/42 MAPK, p44/42/MAPK, phospho-Akt and Akt also were examined. As the treatment concentration of PfILP increased, Pfirs1, Pfpik3r1 and Pfakt transcripts were upregulated, and levels peaked at a dose of 2.0 μg/ml. By contrast, Pfsos2 and Pferk transcripts were not significantly changed. The Pfrap-1 transcript was upregulated at a dose of 0.25 μg/ml, and the Pfraf transcript was upregulated at a dose of 2.0 μg/ml (Fig. 6A).

Effects of PfILP on ILP signalling pathway genes and protein expression in cultured primary mantle cells. (A) Quantitative relative expression of Pfirs1, Pfpik3r1, Pfakt, Pfsos2, Pfrap-1, Pfraf and Pferk in mantle cells after a 24 h incubation various concentrations of PfILP. Each bar represents the mean ± S.E.M. (n = 3). *p < 0.05, **p < 0.01. (B–E) Mantle cells were analysed by western blotting to detect the phosphorylation of Akt/PKB at residue T308, and p44/42 MAPK, as well as the amounts of Akt/PKB and p44/42 MAPK. Each set of bands for each protein is from the same gel.
Phosphorylation of Akt was examined by determining the ratio of phosphorylated to total Akt. As shown in Fig. 6B, PfILP induced the phosphorylation of Akt (on residue T308) from 5 min to 240 min, and phospho-Akt levels peaked at 60 min. PfILP increased Akt (T308) phosphorylation with increasing dose from 0.25 μg/ml to 1.0 μg/ml, but this declined at a dose of 2.0 μg/ml (Fig. 6C). PfILP also induced MAPK phosphorylation within 5 min, and the induction lasted for 240 min (Fig. 6D). Again, activation time-dependently increased from 0 to 240 min. When treated for 30 min, PfILP dose-dependently induced phosphorylation of p44/42 MAPK at a concentration ranging from 0 to 1.0 μg/ml (Fig. 6E). These results clearly show that recombinant PfILP activates both MAPK and PI3K/Akt signalling pathways.
PfILP influences the expression of glycogen-related genes and proteins
To investigate the participation and conservation of carbohydrate/glycogen metabolism in response to PfILP induction, we analysed three glycogen-related genes involved in PfILP signalling. The results showed that the relative expression levels of GSK-3β, GK and PP1 were significantly increased in mantle cells after PfILP stimulation (Fig. 7A).

Effects of PfILP on glycogen metabolism-related genes and protein expression in cultured primary mantle cells. (A) Quantitative relative expression of GK, GSK-3β and PP1 in mantle cells after a 24 h incubation with PfILP at various concentrations. Each bar represents the mean ± S.E.M. (n = 3). *p < 0.05, **p < 0.01. (B–E) Phospho-GSK-3β and PP1 protein levels were analysed by western blotting following treatment with PfILP at different concentrations or for different durations. GAPDH was used as an internal reference. Each set of bands for each protein is from the same gel.
Levels of phospho-GSK-3β and PP1 proteins were measured by western blotting, and phospho-GSK-3β levels increased gradually and reached a maximum at a dose of 0.5 μg/ml PfILP (Fig. 7B). With increasing PfILP concentration, the phosphorylation level of GSK-3β began to dose-dependently decline from 0.5 μg/ml to 2.0 μg/ml PfILP. PfILP induced GSK-3β phosphorylation within 5 min, and the induction lasted for 1 h (Fig. 7C). When treated for 30 min, PP1 expression dose-dependently increased from 0.25 μg/ml to 0.75 μg/ml. However, PP1 expression was decreased at a dose of 1.0 μg/ml and 2.0 μg/ml (Fig. 7D). At the same concentrations, PfILP time-dependently decreased PP1 expression from 0 to 240 min, except for the 30 min timepoint (Fig. 7E).
Recombinant PfILP affects cell cycle progression
Due to the weak proliferation of mantle cells, 293 T cells were used to investigate the effect of PfILP on cell cycle phase distribution. Cells were treated with recombinant PfLIP protein, stained with FxCycle PI/Rnase Staining Solution, and analysed by FACS. As shown in Fig. 8, a 5 μg/ml dose of recombinant PfLIP protein caused a significant decrease in the number of cells in G1 and G2 phases, with a corresponding significant accumulation of cells in S phases. These results indicate that PfILP promotes the transition of 293 T cells from G1 to S, and decreases the number of cells in G2. Thus, PfLIP promotes cell cycle progression.

Flow cytometry analysis of the effect of PfILP on the cell cycle of 293 T cells. Cells were treated with different doses of PfILP for 36 h, and the cycle distribution was analysed by FACS analysis. Each bar corresponds to the mean ± SEM for three independent experiments. *p < 0.05, **p < 0.01.
Discussion
The insulin/insulin-like signalling pathway is evolutionarily conserved in invertebrate and vertebrate animals37,38. Several key insulin pathway components including PfILP, Pfirs1, Pfpik3r1, Pfakt, Pfsos2, PfRap-1 and PfRaf have been cloned from P. fucata martensii. PfILP shares the typical characteristics of ILP proteins, including six conserved cysteine residues that form disulphide bridges, and a similar 3D structure (Fig. 1). Previous studies showed that the insulin-relaxin superfamily have a similar modular organisation as their precursor, including an N-terminal signal peptide, A, B and C domains2,13. Post-translational modification leads to cleavage of the signal peptide and the C-peptide, resulting in a mature hormone consisting of the A and B chains. The six conserved cysteine residues in ILPs form one intra-chain (within the A-chain) and two inter-chain disulphide bridges that play a key role in protein function by cross-linking peptides19,32. Herein, six potential effectors of signalling, sharing varying degrees of conservation, were investigated. In vertebrates, these effectors are related to the MAPK pathway (Pfsos2, PfRap-1 and PfRaf) or the PI3K pathway (Pfirs1, Pfpik3r1 and Pfakt), suggesting both pathways may be conserved in molluscan species.
Analysis of the expression of Pfirs1, Pfpik3r1, Pfakt, Pfsos2, Pfrap-1, Pfraf and PfILP revealed that they were expressed during different developmental stages and in various tissues (Fig. 2). These results indicate that these proteins encoded by these genes may have broad functions. ILP signalling pathway genes display similar expression patterns in oyster tissues. It should be noted that PfILPs (including Pfirs1, Pfpik3r1, Pfakt, Pfsos2, PfRap-1 and PfRaf) were highly expressed in the foot. In oysters, the neuroendocrine system is relatively developed in the foot. In mollusc species, growth, reproduction, and their associated metabolic processes are known to be subjected to neuroendocrine control mechanisms, and nervous ganglia are crucial centres for the production of regulatory molecules3. However, they were expressed primarily in D-shape larvae in the present study. The D-shape larval stage is a period in which embryonic development and organ formation is relatively fast. Furthermore, the D-shape larval stage is crucial for soft tissue and shell growth. High expression of ILP signalling pathway genes during this developmental stage implies that they may play a crucial role.
In this study, we explored a mantle primary cell culture as an in vitro model to investigate the effect of the insulin-like peptides PfILP on P. fucata martensii mantle cell development and metabolism. The recombinant mature PfILP peptide (without the signal peptide) increased mantle cell viability (Fig. 4A), and upregulated the PCNA gene. This phenomenon show that PfILP may promote cell proliferation. In different gastropod species, neuroendocrine factors participate in soft growth and shell growth by stimulating cell proliferation and protein synthesis39,40,41,42,43.
In brain, ILPs are specifically synthesised and released into the hemolymph, and then transported to target cells, where they interact with insulin receptors, triggering downstream signalling pathways44,45,46. Using Co-IP assays, PfILP and pfIRR were shown to interact with each other (Fig. 5A,B). Moreover, we investigated if the binding of PfILP to ILP receptors (pfRR) could activate the receptors and elicit downstream signalling cascades in P. fucata martensii. Early research shows that the activation of IGF receptors can cause the MAPK and PI3K/Akt pathways cascades47,48. In our previous study, human IGF-1 was shown to interact with P. fucata martensii pfIRR and to activate MAPK and PI3K/Akt pathways in P. fucata martensii oocytes49. Furthermore, in Sparus aurata, insulin signalling pathway-related transcripts were affected by IGF-I or IGF-II50. In our study, most ILP signalling pathway-related genes in P. fucata martensii was up-regulated in response to PfILP. Moreover, we demonstrate that PfILP also induced the expression of pfRR (Fig. 5C–E) and activated PI3K/Akt and MAPK pathways by stimulating phosphorylation of Akt and MAPK, although their threshold concentrations and durations differed (Fig. 6). Previous studies reported that Akt activation stimulates myogenic differentiation, whereas MAPK activation stimulates mitogenesis48,51,52. These results showed that PfILP can activate downstream intracellular signalling pathways in P. fucata martensii.
Early research found that exogenous insulin regulate glycogen metabolism in invertebrates such as the white shrimp Penaeus vannamei53 and the lobster Panulirus argus54. HrIGF-I through coupled PfIRR to activate the MAPK and PI3K signaling pathways, and then regulate glycogen metabolism in P. fucata martensii51. GSK3 can regulate the activity of GS to synthesis Glycogen55,56. Insulin can inhibit GSK3 to activate GS by, and then activate PP157. PP1 is an important regulator in blood glucose levels as well as glycogen metabolism in the liver57. And GK act as a glucose sensor by catalysing the phosphorylation of glucose to glucose-6-phosphat in carbohydrate metabolism58. In our work, PP1, GSK-3β and GK mRNA levels were upregulated significantly by PfILP (Fig. 7). These results suggest that PfILP regulates glycogen metabolism pathways like vertebrate ILP. Furthermore, cell cycle analysis showed that PfILP influenced 293 T cell growth by promoting cell cycle progression (Fig. 8).
In summary, our discovery of PfILPs and six effectors in a mollusc species strengthens the hypothesis that the insulin signaling pathway has a common ancestor between vertebrates and invertebrates. Our findings can help us better understand the regulatory mechanisms and biological roles of insulin-like signalling pathways in P. fucata martensii and other invertebrates.
Materials and Methods
Pearl oysters (2-year-old) were obtained from the Daya Bay Marine Biology Research Stations of the Chinese Academy of Sciences (Shenzhen, Guangdong, P.R. China) and maintained in an aerated indoor cement ponds for 1 week under controlled temperature (20 ± 2 °C) and fed Chlorella vulgaris every day. Animal experimentation in this study was approved by Experimental Animal Ethics of Chinese Academy of Sciences.
RNA extraction
Total RNA were extracted using a TRIzol (Magen, Guangzhou, China). The integrity of RNA was determined by a 1% agarose gel electrophoresis. The quality and quantity of RNA were measured with a Quawell Q5000 (Thermo, California, USA). First-strand cDNA synthesis was performed using a SMART RACE cDNA Amplification Kit (Clontech, Palo Alto, CA, USA).
For analysis of gene expression in different tissues, mantle, digestive gland, gonad, adductor muscle, foot, heart, and gill were extracted as described above. The embryo development stages samples-the polar body, 2-cell, 32-cell, blastocyst, trochophore, D-shaped larvae, and umbo larvae stage were also extracted as above. Nine pearl oysters were randomly divided into three replicates, and equal quantities (500 ng) of total RNA from different tissues were reverse-transcribed into cDNA templates for ReverTra Ace qPCR using RT Master Mix with gDNA Remover (Toyobo, Osaka, Japan) according to the manufacturer’s instructions.
Cloning of ILP signalling elements
To clone ILP signalling elements from P. fucata martensii, multiple pairs of primers (Supplementary Table S4) were designed according to partial fragments from the transcriptome and used to amplify full-length cDNAs. PCR products were cloned into the pGM-T Fast Vector (TIANGEN, Beijing, China) and the resulting constructs were transformed into Escherichia coli DH5α cells for DNA sequencing.
Sequence characterisation and phylogenetic analysis
The cDNA sequences were evaluated by TBLASTX (NCBI; http://www.ncbi.nlm.nih.gov/blast/). The PfILP genomic sequence was obtained by searching genomic database (https://www.ncbi.nlm.nih.gov/genome/?term=Pinctada+fucata+martensii) using the full-length cDNA sequence. The genomic and cDNA sequences were then compared to derive the genomic structure of this gene (https://www.ncbi.nlm.nih.gov/sutils/splign/splign.cgi?textpage=online&level=form).DNAStar software was used to predicte the open reading frame (ORF). The simple modular architecture research tool (SMART; http://marinegenomics.oist.jp/pearl/viewer?-project_id=36) was used to analyse the amino acid structure. ExPASy tools (http://cn.expasy.org/tools/pi_tool.html) was used to analyse the molecular weight and isoelectric point (pI). Signal sequence prediction was performed using the SignaIP4.0 Server (http://www.cbs.dtu.dk/services/SignalP/). The tertiary structure of ILPs was predicted using SWISS-MODEL (https://www.swissmodel.expasy.org/interactive) and edited using PyMOL Viewer (www.pymol.org). Multiple sequence alignment was performed using CLC Main Workbench 7.7.3 software (CLC Bio, Aarhus, Denmark) to highlight regions of conservation. MEGA 6.0 software with the maximum likelihood method (1000 bootstrap replicates) was used to construct the phylogenetic trees59.
Real-time quantitative PCR (qPCR) analysis
Gene expression levels was determined by qPCR using a Roche LightCycler480 instrument (Roche, Basel, Switzerland). The 18S rRNA gene was used as an internal control, and primers were used for qPCR are listed in Supplementary Table S4. QPCR was performed using SYBR Green (Toyobo) and the expression levels were calculated based on the 2−ΔΔCT method. Each sample was tested with three replicates. The primers used for qPCR are listed in Supplementary Table S4.
Plasmid construction, and expression and purification of recombinant PfILP
The cDNA encoding PfILP mature polypeptide was amplified using sequence specific primers (Supplementary Table S4) containing EcoR I and Hind III restriction sites. After double digestion with EcoR I and Hind III, the products was cloned in-frame into the EcoR I/Hind III sites of the pET-28α expression vector to generate an N-terminal polyhistidine-tagged version of PfILP lacking its signal peptide (amino acids 1–32). The recombinant plasmid were transformed into E. coli Transetta (DE3) cells which grown overnight in liquid Luria-Bertani (LB) medium supplemented with 100 mg/ml kanamycin and 100 mg/ml penicillin under constant shaking (220 rpm) at 37 °C. The cultures was then diluted 1:50 into fresh LB with penicillin and kanamycin, and grown until an OD600 of 0.5–0.7 under the same conditions. The bacterial was then induced with 1 mM isopropyl-β-D- thiogalactopyranoside (IPTG) and incubation continued at 37 °C or a further 12 h. The bacterial cells were pelleted by centrifugation at 8,000 × g for 5 min. Inclusion bodies were washed twice with solution I (100 mM NaCl, 50 mM Tris-HCl, 1 mM Ethylenediaminetetraacetic acid (EDTA), 1 mM Dithiothreitol (DTT), 0.5% Triton X-100, pH 8.0) and solutions II (1 mM EDTA, 50 mM Tris-HCl, 1 mM DTT, pH 8.0) for 30 min each time, respectively. Inclusion bodies were then solubilised in 50 ml of denaturing buffer comprising 100 mM Tris-HCl, 8 M urea, 1 mM glycine, 1 mM EDTA, 100 mM 2-Hydroxy-1-ethanethiol (p-ME), 10 mM DTT, 1 mM reduced glutathione (GSH), 0.1 mM oxidised glutathione (GSSG) and 10 mM imidazole (pH 9.0), and incubated at 4 °C for 24 h under constant rotation. Dissolving solution were dialysed at 4 °C and five concentrations of urea (6 M, 4 M, 2 M, 1 M and 0.5 M) in Tris-HCl buffer (20 mM Tris-HCl, 60 mM glycine, 0.5 mM DTT, 0.08 mM GSSG, 0.8 mM GSH, 5% glycerine, 6 M urea, pH 9.0) were tested. Refolded samples were centrifuged at 12,000 × g for 10 min at 4 °C and the supernatant was used for further purification. Recombinant protein was purified using a HisPurTM Ni-NTA Resin (Thermo Fisher Scientific, USA) according to the manufacturer’s protocol. The purity of eluted samples was analysed by 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and stained with Coomassie Brilliant Blue R-250. Protein concentrations were determined by Modified BCA Protein Assay Kit (Sangon Biotech, Shanghai, China) following the manufacturer’s instructions.
Primary culturing of mantle cells
Oysters were maintained in a sterile seawater tank containing 0.024 g/l ampicillin and 0.02 g/l gentamicin for two days prior to experimentation. The outside of the shell was rapidly wiped with 72% ethanol. Shells were opened and the mantle was dissociated. The primary mantle cells were cultured according to a previous study60.
Cell Counting Kit-8 assays and PCNA expression analysis
Cell Counting Kit-8 assays were performed to test the effects of recombinant PfILP on mantle cell growth. Mantle cells (2000 cells/well) were isolated and seeded in 96-well plastic plates and cultured with 2.5% fetal calf serum. After culturing for 24 h, various concentrations (0, 0.25, 0.5, 1, 1.5, and 2 μg/ml) of recombinant PfILP was added and culturing continued for a further 24 h. Cell proliferation was then assayed using the Cell Counting Kit-8 following the manufacturer’s instructions. PCNA (Proliferating cell nuclear antigen, from pearl oyster) gene expression levels was measured as an indicator of cell proliferation status.
Effects of PfILP on PfIRR and Western blot assays
Co-immunoprecipitation (Co-IP) analysis of the interaction between PfILP and PfIRR was carried out using the Pierce Classic IP Kit (#26146; Pierce, Rockford, IL, USA) according to the manufacturer’s instructions. Mantle cells were treated with 1 mM PfILP overnight at 4 °C, and Mantle cells were lysed and immunoprecipitated using a Pierce Classic IP Kit (#26146; Pierce, Rockford, IL, USA). Lysates were co-immunoprecipitated using anti-His-PfILP antibodies, subjected to SDS-PAGE (10%) and detected by western blotting with an anti-pfIRR antibody (10 μg) from our lab. Co-IP lysates obtained using anti-pfIRR antibodies (10 μg) were detected by western blotting with anti-His-PfILP (10 μg). Relative expression levels of Pfirr were analysed after a 24 h incubation with PfILP at various concentrations. pfIRR protein expression levels were analysed after a 1 h incubation with PfILP at various concentrations, or with 1 μg/ml PfILP for various times, by western blotting according described previously60. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a reference.
PfILP stimulation in primary mantle cells
First, mantle cells (5 × 105 cells/well) were seeded in 24-well plates (Corning, NY, USA). One group were incubated for 24 h with recombinant PfILP protein and RNA was subsequently extracted and subjected to qPCR analysis. Primers used for genes involved in the PfILP signalling pathway (pfirr, Pfirs1, Pfpik3r1, Pfakt, Pfsos2, Pfrap-1, Pfraf, Pferk, GK, GSK-3β and PP1), as well as the 18S rRNA gene, are listed in Supplementary Table S4.
The other group of mantle cells were incubated with PfILP protein (1 μg/ml) for various time periods (0, 5, 10, 30, 60 or 240 min) then analysed by western blotting to detect the phosphorylation of Akt/PKB at residue T308, as well as the amount of Akt/PKB, phosphorylated p44/42 MAPK, p44/42 MAPK, GSK-3β, PP1 and GAPDH. Mantle cells were incubated with PfILP at different concentrations (0, 0.25, 0.5, 0.75, 1.0 or 2.0 μg/ml) for 30 min. Lysates were subjected to western blotting with appropriate antibodies, and the densitometry ratios of phospho-p44/42/total p44/42, phospho-Akt/total Akt, GSK-3β/GAPDH, and PP1/GAPDH were measured using Lane 1D software from Minichemi 610.
Cell cycle analysis
Because mantle primary cells display weak cell proliferation, we used human 293 T cells instead, and these cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum at 37 °C with 5% CO2. After 3 days, 293 T cells (5 × 104 cells/well) were seeded in 24-well plates (Falcon, Corning, NY, USA). When cells had grown to 80%, the medium was discarded, and different concentrations of recombinant PfILP protein were added with 2% serum medium and cultured at 37 °C with 5% CO2 for 36 h. Phosphate-buffered saline (PBS) was used instead of recombinant PfILP protein in the negative control group. Samples were collected and washed twice with PBS, resuspended in 1.5 ml of pre-cooled 80% ethanol, mixed, and incubated overnight at 4 °C in the dark. Samples were collected and washed twice with PBS, then mixed with 0.5 ml FxCycle PI/RNase Staining Solution (Thermo Fisher Scientific) and incubated at room temperature for 30 min in the dark. Samples were collected and filtered through a 40 μm cell strainer and analysed by FACScan flow cytometry (Philadelphia, Pennsylvania, USA) at 488 nm.
Statistical analysis
All data graph were analysed by GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA). Values were displayed the means ± SEM and analyzed by Student’s t-test. SPSS software (version 18.0) (SPSS, Chicago, IL, USA) was used for the statistical analysis. A p-value < 0.05 or <0.01 was considered statistically significant.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Chan, S. J. & Steiner, D. F. Insulin through the ages: phylogeny of a growth promoting and metabolic regulatory hormone1. Am. Zool. 40, 213–222, https://doi.org/10.1093/icb/40.2.213 (2000).
Lecroisey, C., Pétillon, Y. L., Escriva, H., Lammert, E. & Laudet, V. Identification, evolution and expression of an insulin-like peptide in the cephalochordate Branchiostoma lanceolatum. PLoS One. 10, e0119461 (2015).
Jouaux, A. et al. Identification of ras, pten and p70s6k homologs in the pacific oyster Crassostrea gigas and diet control of insulin pathway. Gen. Comp. Endocrinol. 176, 28–38 (2012).
Kenyon, C. J. The genetics of ageing. Nature. 464, 504–512 (2010).
Greer, K. A., Hughes, L. M. & Masternak, M. M. Connecting serum IGF-1, body size, and age in the domestic dog. Age. 33, 475–83 (2011).
Bartke, A., Sun, L. Y. & Longo, V. Somatotropic signaling: trade-offs between growth, reproductive development, and longevity. Physiol. Rev. 93, 571–598 (2013).
Badisco, L., Wielendaele, P. V. & Broeck, J. V. Eat to reproduce: a key role for the insulin signaling pathway in adult insects. Front. Physiol. 4, 202 (2013).
Min, C. L. et al. Identification of insulin-like peptide 1 (ILP1) gene and its expression in response to different food sources in the intertidal copepod Tigriopus japonicus. Fish. Sci. 81, 495–504 (2015).
Lochhead, P. A., Coghlan, M., Rice, S. Q. & Sutherland, C. Inhibition of GSK-3 Selectively Reduces Glucose-6-Phosphatase and Phosphatase and Phosphoenolypyruvate Carboxykinase Gene Expression. Diabetes. 50, 937–946 (2001).
Ferguson, R. D., Gallagher, E. J., Scheinman, E. J., Damouni, R. & Leroith, D. Chapter two–the epidemiology and molecular mechanisms linking obesity, diabetes, and cancer. Vitam. Horm. 93, 51–98 (2013).
Landsberg, L. Insulin resistance and the metabolic syndrome. Diabetologia. 48, 1244–1246 (2005).
Nagasawa, H. et al. Amino-terminal amino acid sequence of the silkworm prothoracicotropic hormone: homology with insulin. Science. 226, 1344–1345 (1984).
Niu, D. H. et al. Identification, expression, and innate immune responses of two insulin-like peptide genes in the razor clam Sinonovacula constricta. Fish. Shellfish. Immun. 51, 401–404 (2016).
Dircksen, H. et al. Genomics, transcriptomics, and peptidomics of daphnia pulex neuropeptides and protein hormones. J. Proteome Res. 10, 4478–4504 (2011).
Wang, S. et al. Identification of putative insulin-like peptides and components of insulin signaling pathways in parasitic platyhelminthes using genome-wide screening. FEBS J. 281, 877–893 (2013).
Li, C. & Kim, K. Neuropeptides. Wormbook. 198, 1–36 (2008).
Floyd, P. D. et al. Insulin prohormone processing, distribution, and relation to metabolism in Aplysia californica. J. Neurosci. 19, 7732–7741 (1999).
Smit, A. B. et al. Towards understanding the role of insulin in the brain: lessons from insulin-related signaling systems in the invertebrate brain. Prog. Neurobiol. 54, 35–54 (1998).
Wu, Q. & Brown, M. R. Signaling and function of insulin-like peptides in insects. Annu. Rev. Entomo. 51, 1–24 (2006).
Brogiolo, W. et al. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr. Biol. 11, 213–221 (2001).
Veenstra, J. A. Do insects really have a homeostatic hypotrehalosaemic hormone? Biol. Rev. 64, 305–316 (2010).
Vafopoulou, X. The coming of age of insulin-signaling in insects. Front. Physiol. 5, 216 (2014).
Colombani, J., Andersen, D. S. & Léopold, P. Secreted peptide dilp8 coordinates Drosophila tissue growth with developmental timing. Science. 336, 582–585 (2012).
Gronke, S., Clarke, D. F., Broughton, S., Andrews, T. D. & Partridge, L. Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet. 6, e1000857 (2010).
Zhang, L. & Wang, G. Insulin-like peptides in invertebrates and their signaling pathways-take insects. Biotechnol Bull. 33–42 (2014).
Horton, A. A. et al. Identification of three single nucleotide polymorphisms in Anopheles gambiae, immune signaling genes that are associated with natural plasmodium falciparum, infection. Malar. J. 9, 160 (2010).
Vallejo, D. M., Juarez-Carreno, S., Bolivar, J., Morante, J. & Dominguez, M. A brain circuit that synchronizes growth and maturation revealed through Dilp8 binding to Lgr3. Science 350, aac6767 (2015).
Jaszczak, J. S., Wolpe, J. B., Bhandari, R., Jaszczak, R. G. & Halme, A. Growth coordination during drosophila melanogaster imaginal disc regeneration is mediated by signaling through the relaxin receptor Lgr3 in the prothoracic gland. Genetics. 204, 703–709 (2016).
Mizoguchi, A. & Okamoto, N. Insulin-like and IGF-like peptides in the silkmoth Bombyx mori: discovery, structure, secretion, and function. Front. Physiol. 4, 217–217 (2013).
Nässel, D. R., Liu, Y. & Luo, J. Insulin/IGF signaling and its regulation in Drosophila. Gen. Comp. Endocrinol. 221, 255–266 (2015).
Murphy, C. T. & Hu, P. J. Insulin/insulin-like growth factor signaling in C. elegans. Wormbook. 1–43 (2013).
Claeys, I. et al. Insulin-related peptides and their conserved signal transduction pathway. Peptides. 23, 807–816 (2002).
Smit, A. B. et al. Growth-controlling molluscan neurons produce the precursor of an insulin-related peptide. Nature. 331, 535–538 (1988).
Shipilov, V. N., Shpakov, A. O. & Rusakov, Y. I. Pleiotropic action of insulin_like peptides of mollusk, Anodonta cygnea. Ann. Ny. Acad. Sci. 1040, 464–465 (2005).
Shi, Y., Guan, Y. Y. & He, M. X. Molecular identification of insulin-related peptide receptor and its potential role in regulating development in Pinctada fucata. Aquaculture. 408-409, 118–127 (2013).
Zhang, H., Shi, Y. & He, M. X. Molecular identification of an insulin growth factor binding protein (IGFBP) and its potential role in an insulin-like peptide system of the pearl oyster, Pinctada fucata. Comp. Biochem. Physiol. B-Biochem Mol. Biol. 214, 27–35 (2017).
Pertseva, M. N. & Shpakov, A. O. Conservatism of the insulin signaling system in evolution of invertebrate and vertebrate animals. J. Evol. Biochem. Physiol. 38, 547–561 (2002).
Pinero Gonzalez, J., Carrillo Farnes, O., Vasconcelos, A. T. & Gonzalez Perez, A. Conservation of key members in the course of the evolution of the insulin signaling pathway. Biosystems. 95, 7–16 (2009).
Geraerts, W. P. Control of growth by the neurosecretory hormone of the light green cells in the freshwater snail Lymnaea stagnalis. Gen. Comp. Endocrinol. 29, 61–71 (1976).
Geraerts, W. P. The role of the lateral lobes in the control of growth and reproduction in the hermaphrodite freshwater snail Lymnaea stagnalis. Gen. Comp. Endocrinol. 29, 97–108 (1976).
Gomot, A. & Gomot, L. Neurohormonal control of body and shell growth of the snail Helix. Bulletin de l’Institut Oceanographique (Monaco) 0 (SPEC ISSUE 14 PART 2). 14, 141–149, https://eurekamag.com/research/021/405/021405487.php (1995).
Gomot, A., Gomot, L., Marchand, C. R., Colard, C. & Bride, J. Immunocytochemical localization of insulin-related peptide(s) in the central nervous system of the snail helix aspersa muller: involvement in growth control. Cell Mol. Neurobiol. 12, 21–32 (1992).
Widjenes, J. & Runham, N. W. Studies on the control of growth in Agriolimax reticulatus (Mollusca, Pulmonata). Gen. Comp. Endocrinol. 31, 154–156 (1977).
Géminard, C., Rulifson, E. J. & Léopold, P. Remote control of insulin secretion by fat cells in Drosophila. Cell Metab. 10, 199–207 (2009).
Iwami, M. Bombyxin: an insect brain peptide that belongs to the insulin family. Zool. Sci. 17, 1035–1044 (2000).
Nässel, D. R., Kubrak, O. I., Liu, Y., Luo, J. & Lushchak, O. V. Factors that regulate insulin producing cells and their output in Drosophila. Front. Physiol. 4, 252 (2013).
White, M. F. Insulin signaling in health and disease. Science. 302, 1710–1711 (2003).
Liu, M. & Zhang, S. Amphioxus IGF-like peptide induces mouse muscle cell development via binding to IGF receptors and activating MAPK and PI3K/AKT signaling pathways. Mol. Cell Endocrinol. 343, 45–54 (2011).
Shi, Y. & He, M. X. PfIRR Interacts with HrIGF-I and Activates the MAP-kinase and PI3-kinase Signaling Pathways to Regulate Glycogen Metabolism in Pinctada fucata. Sci. Rep. 6, 22063 (2016).
Azizi, S. et al. IGF-I and IGF-II effects on local IGF system and signaling pathways in gilthead sea bream (Sparus aurata) cultured myocytes. Gen. Comp. Endocrinol. 232, 7–16 (2016).
Castillo, J., Ammendrup-Johnsen, I., Codina, M., Navarro, I. & Gutiérrez, J. IGF-I and insulin receptor signal transduction in trout muscle cells. Am. J. Physiol-Regul Integr. Comp. Physiol. 290, R1683–R1690 (2006).
Coolican, S. A., Samuel, D. S., Ewton, D. Z., McWade, F. J. & Florini, J. R. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J. Biol. Chem. 272, 6653–6662 (1997).
Gutiérrez, A., Nieto, J., Pozo, F., Stern, S. & Schoofs, L. Effect of insulin/IGF-1 like peptides on glucose metabolism in the white shrimp Penaeus vannamei. Gen. Comp. Endocrinol. 153, 170–175 (2007).
Gallardo, N. et al. Isolation and biological characterization of a 6-kda protein from hepatopancreas of lobster Panulirus argus with insulin-like effects. Gen. Comp. Endocrinol. 131, 284–290 (2003).
Bacca, H. et al. Molecular cloning and seasonal expression of oyster glycogen phosphorylase and glycogen synthase genes. Comp. Biochem. Physiol. B Biochem Mol. Biol. 140, 635–646 (2005).
Frame, S. & Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 359, 1–16 (2001).
Cohen, P. T. Protein phosphatase 1–targeted in many directions. J. Cell Sci. 115, 241–256 (2002).
Kawai, S., Mukai, T., Mori, S., Mikami, B. & Murata, K. Hypothesis: structures, evolution, and ancestor of glucose kinases in the hexokinase family. J. Biosci. Bioeng. 99, 320–330 (2005).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. and Evolution. 30, 2725–2729 (2013).
Zhang, H. et al. Molecular cloning and characterization of a putative mitogen-activated protein kinase (erk1/2) gene: involvement in mantle immunity of Pinctada fucata. Fish. Shellfish. Immunology. 8, 63–70 (2018).
Acknowledgements
This study was supported by the earmarked fund for Modern Agro-industry Technology Research System (grant no. CARS-49), Science and Technology Planning Project of Guangdong Province, China (No. 2017B030314052), the PhD Start-up Fund of Natural Science Foundation of Guangdong Province, China (2015A030310362), and the Science and Technology Planning Project of Guangdong Province, China (Grant No. 2014B030301064).
Author information
Authors and Affiliations
Contributions
H.Z. and M.X.H. planned and designed the research. H.Z. performed experiments, analyzed data and wrote the paper. M.X.H. revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, H., He, M. The role of a new insulin-like peptide in the pearl oyster Pinctada fucata martensii. Sci Rep 10, 433 (2020). https://doi.org/10.1038/s41598-019-57329-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-57329-3
This article is cited by
-
Polymorphism in the Ras and β-Glucosidase Genes and Their Association with Growth Traits in the Pacific Oyster Crassostrea gigas
Journal of Ocean University of China (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
