
Abstract
Analyses of circulating tumor cells have been shown to be effective for the detection of cancer relapse and prognosis prediction. However, research regarding its utility in sarcoma remains scarce. In this study, the microfluidic chip-type cell sorter On-chip Sort was used to construct a system for detecting circulating sarcoma cells (CSCs). A pilot study using normal fibroblast or sarcoma cell lines was designed to establish a reliable protocol to separate CSCs by On-chip Sort. A single CSC was separated and recovered from 10 ml of whole blood from a patient with locally advanced myxofibrosarcoma. The nonsynonymous mutation for KMT2B p.Ile2602Val identified in the formalin-fixed paraffin-embedded tumor sample was also confirmed in the CSC. Use of the developed protocol may allow CSCs to become an early predictor for metastasis and recurrence of sarcoma. Further, it may aid in optimizing post-operative therapies for patients without metastasis.
Similar content being viewed by others

Introduction
Soft tissue sarcoma (STS) is a rare malignant tumor of various histological types that arises from bones and soft tissues1. High-grade STS is managed using surgical resection, radiation, and chemotherapy2. Despite these multidisciplinary therapies, metastasis remains a critical issue3. Indeed, the 5-year overall survival (OS) rate of advanced STS is only 30%, while that of localized STS is 70%4.
Although newer molecular-targeting drugs, such as imatinib, pazopanib, and olaratumab, have been approved for the treatment of STS5,6, the response rates are not very high7. Therefore, doxorubicin, which was developed about 30 years ago, is still used as the first-line treatment. However, doxorubicin treatment is often associated with severe adverse effects, such as neutropenia and a neutropenic fever8. It is therefore controversial to administer chemotherapy to patients without metastasis.
Detecting early recurrence and metastasis is essential to providing chemotherapy in a timely manner. Radiographic imaging, including computed tomography (CT) and magnetic resonance imaging (MRI), are routinely used for follow-up after STS surgery9. Metastasic loci often spread throughout the body before such imaging systems detect metastatic regions. Therefore, high-sensitivity methods for detecting recurrence and metastasis are needed.
Recently, several biomarkers have been established for predicting metastasis and assessing tumor activity. These include circulating tumor cells (CTCs), cell-free DNA (cfDNA), and circulating microRNAs10,11. The number of CTCs is a particularly strong prognostic factor. CTCs can be detected in the peripheral blood of patients with metastatic and localized tumors12. The detection of CTCs has been performed across various tumor types, especially in epithelial carcinomas, such as lung cancer, breast cancer, and prostate cancer13,14. However, research regarding its utility in sarcoma remains scarce15.
The CELLSEARCH System (Veridex, NJ, USA) is the only assay for CTC enumeration approved by the U.S. Food and Drug Administration16. In this system, cell-surface expression of epithelial cell adhesion molecule‐1 (EpCAM) is used to detect CTCs. EpCAM is a standard marker of epithelial tumors. However, EpCAM is not generally expressed in STS, making an alternative method necessary to collect circulating sarcoma cells (CSCs). Although the confirmation of fusion genes in CSCs have been examined17, detection of somatic mutations, such as single nucleotide variants or small insertion/deletion, has not yet been performed.
On-chip Sort (On-chip Biotechnologies, Tokyo, Japan) is a cell sorter that uses a disposable microfluidics chip and includes a collection reservoir to store the target cells18,19. The principle of On-chip Sort comprises flow cytometry using the FISHMAN-R system20. Unlike commonly used cell sorters, this device can sort repeatedly without damaging cells. When a highly concentrated sample is analyzed, the target is reliably recovered without loss of target cells, but with a significant number of nonspecific cells. However, the target recovery rate improved when the cell concentration was sufficiently reduced after repeated sorting to accurately differentiate the target cells. Use of this device for the collection of CTCs has been validated for lung cancer21,22.
The current study aimed to establish a protocol for CSC collection and molecular profiling using On-chip Sort. The Todai OncoPanel (TOP), a multi-gene panel test that examines 464 genes23, was used to confirm that CSCs harbored somatic mutations that were identical to the original tumor.
Results
Protocol for CSC enrichment, enumeration, sorting, and sequencing
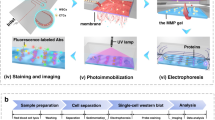
An overview of the CSC analyses used in this study is shown Fig. 1. Blood cell depletion was carried out by autoMACS Pro Separator using CD45 MicroBeads and CD235a MicroBeads. The CD45 antigen is expressed in all cells of hematopoietic origin except erythrocytes, platelets, and their precursor cells. The CD235a antigen (glycophorin A), a single-pass transmembrane glycoprotein, is expressed in mature erythrocytes and erythroid precursor cells.

Protocol for CSC enrichment, enumeration, sorting, and sequencing. An overview of CSC analysis in this study. The whole blood obtained from a sarcoma patient was enriched using autoMACS Pro Separator (Step 1). CSCs were fixed and stained with antibodies specific for sarcoma or WBCs (Step 2). CSC enumeration (Step 3) and sorting (Step 4) were carried out using the On‐chip Sort system. Genome sequencing was performed after WGA of CSCs. The permission for the usage of image of On-chip Sort was obtained from On-chip Biotechnologies.
The remaining cells were fixed and stained with antibodies specific for sarcoma, vimentin, or white blood cells (WBCs), such as CD45 and CD14. The CD14 antigen, a component of the LPS receptor complex, is strongly expressed in most monocytes and macrophages and weakly expressed in neutrophils and some myeloid dendritic cells. Conversely, vimentin is expressed in a wide range of cell types. These include stromal cells, fibroblasts, endothelial cells, and neuronal precursor cells. It was hypothesized that CD45/14 (–)/vimentin (+) fractions in peripheral blood may specifically contain CSCs.
CSC enumeration and sorting were carried out using the On‐chip Sort system (On-chip Biotechnologies). Genome sequencing was performed after whole-genome amplification (WGA) of CSCs (Fig. 1).
A pilot study using the human fibroblast cell line BJ was designed as follows. Five hundred BJ cells were spiked in 3.0 × 105 of WBCs. The sample was then fixed and stained with anti-CD45 mouse monoclonal antibody (mAb) conjugated to allophycocyanin (APC), anti-CD14 mouse mAb conjugated to APC, 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI), and anti-vimentin Alexa Flour 488-conjugated rabbit mAb. We intended to enrich spiked BJ cells (skin fibroblasts) by reducing WBCs (CD45-positive), macrophages, and monocytes (CD14-positive). To completely remove macrophages and monocytes, we utilized an APC-conjugated anti-CD45 Ab combined with an anti-CD14 Ab. A CD45/14 (−)/vimentin (+) fraction from the sample was purified with the On‐chip Sort system. Sorting was repeated three times to enrich CSCs. After purification, a total of 326 cells were gated as CD45 (−)/vimentin (+), and aliquoted into 10 tubes (containing ~30 cells per tube) (Fig. 2A). One of those tubes was subjected to WGA.

Protocol establishment and validation using a normal human fibroblast cell line. (A) A total of 500 BJ cells were spiked into WBCs from healthy volunteers (3.0 × 105 cells). The samples were then stained with DAPI, CD45, CD14, and vimentin and sorted three times using On-chip Sort. The first gate was DAPI (+)/vimentin (+). The second and third gates were CD45 (−)/vimentin (+), resulting in 326 sorted cells. (B) The STRs of the WGA sample were determined to investigate the composition of the sorted cells. BJ bulk: DNA extracted from bulk BJ cells, WBC bulk: DNA extracted from bulk WBCs, BJ 30 cells: WGA DNA of sorted BJ cells using On-chip Sort.
The short tandem repeats (STRs) determined in the WGA sample were used to investigate the composition of the sorted cells. The STR locus D5S818 of the BJ genome contains 12 repetitions of “AGAT” in both alleles, whereas WBCs have 10 repetitions in one allele and 13 repetitions in the other. As shown in Fig. 2B, Sanger sequencing of the polymerase chain reaction (PCR) products for the D5S818 locus revealed that 85.7% of the sorted fraction were identified as BJ cells.
WBC removal using autoMACS Pro Separator
The necessity of WBC depletion from the whole blood sample before running On-chip Sort was next explored. Approximately 50 cells of the rhabdomyosarcoma cell line RH30 were spiked into 2 ml of whole blood collected from a healthy volunteer. One sample underwent red blood cell (RBC) lysis without a negative selection for WBCs. The sample was then fixed, stained, and run through On-chip Sort. However, RH30 cells were not well-separated from the WBC group (Fig. 3A). In contrast, RH30 cells were clearly identified after removing WBCs from the sample using autoMACS Pro Separator (Fig. 3B).

WBC removal using autoMACS Pro Separator. (A) The sample underwent RBC lysis without WBC depletion before running On-chip Sort. RH30 cells were not well-separated from WBCs. (B) After using autoMACS Pro Separator to remove WBCs from 2 ml of whole blood, RH30 cells and WBC were clearly separated by On-chip Sort. (C) Sanger sequence was performed to confirm the TP53 c.817C > T mutation (p.Arg 273 Cys), a known mutation in the RH30 cell line. The electropherograms of bulk WBCs (left panel), bulk RH30 cells (middle panel), and sorted 15 cells of RH30 (right panel) are shown.
Forty-seven cells gated as CD45 (−)/vimentin (+) were collected and divided into three tubes. One of those tubes was subjected to WGA. Sanger sequencing was performed to confirm the TP53 c.817C > T DNA mutation (protein mutation: p.Arg273 Cys) in the amplified DNA, which is known to be present in the RH30 genome. The electrophoretogram of the sorted cells was the same as that of bulk RH30 cells (Fig. 3C). Thus, depleting WBCs before loading the sample onto On-chip Sort improved the separation accuracy for sorting spiked sarcoma cells from whole blood samples.
Method for gating CSCs using WBCs from the same patient on On-chip Sort
To construct an appropriate gate for vimentin, RH30 was always analyzed as a positive control cell line in parallel to the specimens of interest. The CSC gates for the flow cytometer were set to contain ≥90% of RH30 cells (G1) and no WBCs of the patient (G2). WBCs were separated in advance from 10 ml of whole blood using autoMACS Pro Separator. The first round of sorting was performed using the G1 gate, and the second and the third rounds were performed using the G2 gate (Fig. 4).

CSC sorting in a 60-year-old man with myxofibrosarcoma. A CSC-specific gate including ≥90% of RH30 cells (G1) was created using simultaneously prepared RH30 cells as a vimentin (+) control. A gate that included no WBCs from the patient (G2) was created using the patient’s WBCs as a vimentin (−) control. WBCs were separated in advance from 10 ml of whole blood by autoMACS Pro Separator. The first round of sorting using the G1 gate resulted in 51 cells. The second and the third rounds using the G2 gate resulted in four cells and one cell, respectively.
Detection of CSCs in a myxofibrosarcoma patient
A 60-year-old man with myxofibrosarcoma (histological grade: Fédération Nationale des Centers de Lutte Contre le Cancer grade 3, disease stage: American Joint Committee on Cancer/Union for International Cancer Control tumor-node-metastasis stage III) underwent preoperative chemotherapy, followed by surgical excision and post-operative chemotherapy. A blood sample was collected from the patient before the preoperative chemotherapy, 3 months post-, and 9 months post-surgery. No clear metastases were observed at any point during blood sampling (Fig. S1).
Erythrocytes and WBCs were removed from the collected blood specimens. The samples were then fixed and stained with antibodies for CD45, CD14, DAPI, and vimentin. CSC enumeration and sorting was performed using On-chip Sort. A single CSC was sorted at 3 months after surgery (Fig. 4), while no CSCs were detected before treatment or at 9 months after surgery (Fig. S2).
Formalin-fixed paraffin-embedded (FFPE) tumor tissue, normal tissue, CSC, and cfDNA sequencing
To determine whether alterations in the genomes of primary tumor cells can be detected in CSCs and cfDNA, we analyzed genomic alterations in single CSCs and cfDNAs three months after surgery and compared them with those found in the primary tumor FFPE sample. Genomic DNA of tumor or normal tissues isolated from the FFPE and peripheral blood specimen of the patient were subjected to extensive sequencing for cancer-related genes using the TOP panel23. A subsequent bioinformatics analysis revealed cancer-associated somatic mutations, including KMT2B c.7804A > G DNA (protein: p.Ile2602Val) and MGA c.3628C > G DNA (protein: p.Arg1210Gly) with allele frequencies of 13.06% and 6.12%, respectively (Table 1).
Targeted deep sequencing of KMT2B was performed in the CSC DNA and cfDNA obtained 3 months after surgery. Importantly, the KMT2B c.7804A > G mutation was confirmed to be present in the CSC [variant allele frequency (VAF) of 34.9%], whereas KMT2B c.7804A > G, c.7804A > C, and c.7804A > T were also observed in the cfDNA at 0.18%, 0.011%, and 0.015%, respectively (Table 1). To verify that the KMT2B mutations observed in cfDNA at low VAFs were true positive mutations, targeted deep sequencing was performed in two giant cell tumor of bone (GCTB) specimens that did not have KMT2B c.7804A > G in the original tumors. The KMT2B c.7804A > G mutation was also found in the GCTB tumors at VAFs of 0.19% and 0.20% (Table 1). Therefore, the KMT2B mutations found at low VAFs were likely to be sequence errors.
Discussion
The current study established a new protocol for CSC detection using a microfluidic chip-type cell sorter, the On-chip Sort. Several reports have described detection of CSCs using different devices, such as a size-exclusion microfiltration system24 and flow cytometry25,26 with specific markers. However, the utility of size-exclusion is limited by the lack of a histology-specific marker. Flow cytometry is limited due to the heterogeneous expression of target markers, which may affect the detection sensitivity. Even specific markers, such as CD99 for Ewing’s sarcoma, show background expression on normal hematopoietic cells27.
As there were no standard markers suitable for CSC isolation, vimentin was used to target mesenchymal cells28. However, WBCs are also known to express vimentin to some extent. Thus, the number of WBCs was first reduced using a negative selection with the autoMACS Pro Separator system. Then, the CD45 (−)/vimentin (+) fractions were separated using On-chip Sort. Simultaneous analyses of RH30 as a vimentin (+) control and WBCs as a negative control from the same patient allowed for precise gating of the target fraction. Theoretically, the current protocol may be applicable for the isolation of any type of CSCs.
A previous report used real-time PCR to confirm the fusion genes of CSCs29. Although fusion genes are found in about one-third of sarcomas, no specific genetic abnormalities can be identified in the remaining two-thirds of sarcomas, including myxofibrosarcoma. Somatic mutations identified by the TOP panel can be utilized to confirm any type of CSCs. Reports on sarcoma cfDNAs have been published30,31,32, and efforts have been made to identify sarcoma mutations in cfDNA by improving sequencing techniques33. However, cfDNA remains difficult to detect in pre-metastatic cases. The current study showed that, in contrast to the CSC analysis, cfDNA was unable to detect the somatic KMT2B mutation found in the sarcoma. MicroRNAs have also attracted interest in the study of STS34,35, although their utility in clinical practice has not been demonstrated. By improving the sorting method, CSCs can be identified in patients before metastasis. These findings suggested that CSCs might be an early predictor of metastasis and recurrence in sarcoma and could help inform decisions concerning post-operative radiation therapy and chemotherapy for patients without metastasis. Further studies are needed to compare the limit of detection between CSCs and cfDNA analyses.
Presence of CTCs (three cells or more) was shown to predict shorter progression-free survival/OS for colon cancer patients36. While one CSC was detected at 3 months after surgery, the patient in the current study did not demonstrate any signs of metastases after 9 months of surgery. This may suggest that a few remaining CSCs are insufficient to colonize any metastatic sites. Quantitative evaluation of CSCs with the current protocol might help to stratify the prognosis of patients with few CSCs. The accuracy of separating CSCs can be further improved by adding other selection windows, such as CD99 for Ewing sarcoma or MDM2 for liposarcoma. The clinical utility of the current improved protocols must be tested in larger cohorts.
One of the limitations of this study is the sample preparation for the pilot study. The 65% recovery in this pilot experiment using the BJ cell line was relatively lower than that of the other platforms because the number of manually prepared BJ cells used for the spike was not 500. The other pilot experiment using the RH30 cell line shows that the isolation efficiency was sufficiently high (~90%). To calculate the accuracy of enrichment, we should use a flow cytometer to acquire an exact cell number for the spike experiment.
Overall, the accuracy in CSC collection and enumeration was markedly improved using the On-chip Sort system. The protocol suggested in this study may prove to be a fundamental tool for CSC collection. Future work may apply this protocol to laboratory testing for monitoring the recurrence and molecular profile of tumors, which will improve patient prognosis and maximize opportunities for treatment.
Methods
Cell preparation
BJ cells and RH30 cells (ATCC, Manassas, VA, USA) were cultured in EMEM and RPMI-1640 mediums, respectively. Media contained 10% fetal bovine serum and 1% penicillin-streptomycin solution. Cells were collected when they reached 70–80% confluence.
Patients and tumor samples
This project was approved by the ethics review board of Juntendo University Hospital (No. 2018169). Written informed consent was obtained from the patients whose samples were analyzed in the present study. Samples were collected from a myxofibrosarcoma patient who received chemotherapy and two GCTB patients at the Department of Orthopedic Surgery in Juntendo University Hospital. A 10 ml sample of whole blood and FFPE were collected at the time of diagnosis. In the myxofibrosarcoma patient, blood samples were collected at three points: before treatment was started, 3 months post-, and 9 months post-surgery. In addition, whole blood was collected from healthy participants. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Immunomagnetic enrichment
Blood samples (10 ml) were collected with blood collection tubes (EDTA-2K) and used within 3 h. Whole blood was separated into two 15 mL Conical Centrifuge Tubes (Thermo Fisher Scientific, Waltham, MA, USA) and 5 ml of autoMACS Pro Separator Running Buffer was added to each sample. The samples were then centrifuged at 400 g for 10 min to separate the plasma without damaging the buffy coat. CD45 Micro Beads were added to each 5 ml sample at a concentration ratio of 1:20. The sample was then stirred by MACSmix Tube Rotator (Miltenyi Biotec, Bergisch Gladbach, Germany) for 15 min. After stirring, Running Buffer was added to bring the sample up to 13 ml to flush the CD45 MicroBeads attached to the tube wall. The samples were then centrifuged at 400 g for 5 min to separate the plasma without damaging the buffy coat. The WBCs were then separated out using autoMACS Pro Separator. The negative fraction was mixed with 4 ml RBC Lysis Buffer (TONBO Bioscience, San Diego, CA, USA) and 28 ml UltraPure DNase/Rnase-Free Distilled Water (Thermo Fisher Scientific). The samples were then stirred using MACSmix Tube Rotator for 10 min. The samples were then centrifuged at 400 g for 5 min to separate the supernatant. Running buffer (10 ml) was added to the samples, and the samples were centrifuged again at 400 g for 5 min and the supernatant was removed. Both samples were then mixed together and running buffer was added to bring the sample up to 500 µl. PE anti-human CD235a (Glycophorin A) antibody was also added at a concentration of 1:50. The sample was incubated for 10 min. Anti-PE MicroBeads (Miltenyi Biotec) were added to the sample at a concentration of 1:5 and incubated for 10 min. Running buffer was added up to 5 ml and the sample was then placed in autoMACS Pro Separator. The negative fraction was centrifuged at 300 g for 10 min and the supernatant was removed. Running buffer was added to bring the sample up to 500 µl, and the sample was centrifuged at 300 g for 10 min and the supernatant was removed. Running buffer was added up to 100 µl to load the sample onto On-chip Sort.
CSCs enumeration and sorting using the On-chip Sort system
A 100-µl aliquot of the sample was fixed and stained. For fixing, 100 μl of Solution C (On-chip Biotechnologies) was added to the sample and incubated for 10 min. For cell permeabilization, 100 μl Solution D (On-chip Biotechnologies) was added to the sample and incubated for 20 min. Then 400 μl of On-chip T buffer was added to the sample before centrifuging at 400 g for 5 min. The supernatant was aspirated down to 100 μl. Sample staining was then performed using anti-CD45 mouse mAb conjugated to APC (1:40 dilution; Miltenyi Biotec), anti-CD14 mouse mAb (61D3) conjugated to APC (1:40 dilution; Cell Signaling Technology, Danvers, MA), Solution E-1 (DAPI) (1:100 dilution; On-chip Biotechnologies), anti-vimentin rabbit mAb (D21H3) conjugated to Alexa Flour 488 (1:50 dilution; Cell Signaling Technology), and Solution B for Fc receptor blocking (1:100 dilution; On-chip Biotechnologies). The sample was then incubated for 30 min. Enumeration and sorting of the cells were performed using On-chip Sort according to the manufacturer’s instructions19.
WGA and PCR for genotyping
Cells sorted by On-chip Sort were transferred from the collection reservoir of the chip to a 200 μL PCR tube and centrifuged at 400 g for 10 min. Excess liquid was removed after centrifugation, leaving 2 μL of the liquid. WGA was performed using the SMARTer PicoPLEX WGA Kit (Takara Bio Inc., Shiga, Japan) following the manufacturer’s protocol. The amplified products were subjected to Sanger sequencing or next-generation sequencing (NGS).
DNA extraction
Tumor tissue genomic DNA was extracted from FFPE samples using the Gene Read DNA FFPE Kit (Qiagen, Hilden, Germany). Genomic DNA of WBCs was extracted by QIAamp DNA Mini Kit (Qiagen). cfDNA was extracted from plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen). All kits were used according to the manufacturer’s protocols.
Sanger sequencing
For capillary sequencing with a 3130xl Genetic Analyzer (Thermo Fisher Scientific), 10 ng of template DNA is used to amplify the STR locus of the D5S818 and TP53 gene mutation (p.Arg 273 Cys) with GoTaq G2 Hot Start Green Master Mix (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The following primer sets were used: the STR locus of D5S818, 5′-GGGTGATTTTCCTCTTTGGT-3′ (sense) and 5′-TGATTCCAATCATAGCCACA-3′ (antisense), and TP53 gene mutation (p.Arg 273 Cys), 5′-GGGACAGGTAGGACCTGATTTCC-3′ (sense) and 5′-GTGGTGAGGCTCCCCTTTCTTG-3′ (antisense).
Mutational profiling of primary tumor by TOP
The detailed protocol and the content of gene panel were described in previous paper23. In brief, gDNA (500 ng) of tumor FFPE and normal blood samples were subjected to target fragment enrichment using a SureSelectXT Custom kit (Agilent Technologies). Custom-made probes were designed to hybridize and capture the gDNA of the target genes listed by TOP. Massive parallel sequencing of the isolated fragments was performed using a HiSeq. 2500 (Illumina, San Diego, CA, USA).
Targeted deep sequencing of CSCs and cfDNA
The library for deep sequencing was generated using the NEBNext Ultra DNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s instructions. WGA DNA of CSCs (100 ng) or cfDNA (100 ng) was subjected to PCR with the following primers for the KMT2B mutation (p.Ile2602Val): 5′-CCTGTTCTGTAAGCGCAACATCG -3′ (sense) and 5′-CTTGTCAGTCAACACCGAGCG-3′ (antisense). The PCR products were purified using AMPure beads (Beckman Coulter, Brea, CA, USA) for library preparation. The library quality was assessed using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific) and the Agilent 2200 TapeStation System (Agilent, Foster City, CA, USA). The library was sequenced on MiSeq (Illumina) using a Reagent Micro Kit V2 (300-cycles) with the 150-bp paired-end option.
NGS analysis
Target sequencing reads of gDNA from FFPE, blood, CSC, and cfDNA were independently aligned to the human reference genome (University of California Santa Cruz Genome Browser assembly ID: hg38) using Burrows-Wheeler Aligner (http://bio-bwa.sourceforge.net/) and Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml). For target sequencing using FFPE, PCR duplicate reads were removed using BamUtil (https://genome.sph.umich.edu/wiki/BamUtil). Somatic mutations of gDNA were identified using MuTect2 (GATK 4.0.11.0, https://software.broadinstitute.org/gatk/), VarScan2 (http://dkoboldt.github.io/varscan/) and an in-house pipeline. Mutations were discarded if: (i) the read depth was <100 or VAF was <0.01, (ii) they were supported by only one strand of the genome, or (iii) they were present in normal human genomes in either the 1000 Genomes Project dataset (http://www.internationalgenome.org/) or the in-house database. Gene mutations were annotated by SnpEff (http://snpeff.sourceforge.net/). Mutations of CSC and cfDNA were identified by an in-house method using SAMools (http://samtools.sourceforge.net/).
References
Jo, V. Y. & Fletcher, C. D. WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology 46, 95–104, https://doi.org/10.1097/pat.0000000000000050 (2014).
Clark, M. A., Fisher, C., Judson, I. & Thomas, J. M. Soft-tissue sarcomas in adults. The New England journal of medicine 353, 701–711, https://doi.org/10.1056/NEJMra041866 (2005).
In, G. K., Hu, J. S. & Tseng, W. W. Treatment of advanced, metastatic soft tissue sarcoma: latest evidence and clinical considerations. Therapeutic advances in medical oncology 9, 533–550, https://doi.org/10.1177/1758834017712963 (2017).
Miller, B. J., Lynch, C. F. & Buckwalter, J. A. Conditional survival is greater than overall survival at diagnosis in patients with osteosarcoma and Ewing’s sarcoma. Clinical orthopaedics and related research 471, 3398–3404, https://doi.org/10.1007/s11999-013-3147-8 (2013).
Ratan, R. & Patel, S. R. Chemotherapy for soft tissue sarcoma. Cancer 122, 2952–2960, https://doi.org/10.1002/cncr.30191 (2016).
Nakano, K. & Takahashi, S. Current Molecular Targeted Therapies for Bone and Soft Tissue Sarcomas. International journal of molecular sciences 19, https://doi.org/10.3390/ijms19030739 (2018).
Nakano, K. et al. Differences in the responses to pazopanib and the prognoses of soft tissue sarcomas by their histological eligibility for the PALETTE study. Japanese journal of clinical oncology 45, 449–455, https://doi.org/10.1093/jjco/hyv022 (2015).
Karch, A., Koch, A. & Grunwald, V. A phase II trial comparing pazopanib with doxorubicin as first-line treatment in elderly patients with metastatic or advanced soft tissue sarcoma (EPAZ): study protocol for a randomized controlled trial. Trials 17, 312, https://doi.org/10.1186/s13063-016-1434-x (2016).
Casali, P. G., Jost, L., Sleijfer, S., Verweij, J. & Blay, J. Y. Soft tissue sarcomas: ESMO clinical recommendations for diagnosis, treatment and follow-up. Annals of oncology: official journal of the European Society for Medical Oncology 19(Suppl 2), ii89–93, https://doi.org/10.1093/annonc/mdn101 (2008).
Falzone, L. et al. Integrated analysis of colorectal cancer microRNA datasets: identification of microRNAs associated with tumor development. Aging 10, 1000–1014, https://doi.org/10.18632/aging.101444 (2018).
Dawson, S. J. et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. The New England journal of medicine 368, 1199–1209, https://doi.org/10.1056/NEJMoa1213261 (2013).
Rahbari, N. N. et al. Compartmental differences of circulating tumor cells in colorectal cancer. Annals of surgical oncology 19, 2195–2202, https://doi.org/10.1245/s10434-011-2178-1 (2012).
Lianidou, E. S., Mavroudis, D., Sotiropoulou, G., Agelaki, S. & Pantel, K. What’s new on circulating tumor cells? A meeting report. Breast cancer research: BCR 12, 307, doi:10.1186/bcr2601 (2010).
Cristofanilli, M. et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. The New England journal of medicine 351, 781–791, https://doi.org/10.1056/NEJMoa040766 (2004).
Chang, L., Asatrian, G., Dry, S. M. & James, A. W. Circulating tumor cells in sarcomas: a brief review. Med Oncol 32, 430, https://doi.org/10.1007/s12032-014-0430-9 (2015).
Andreopoulou, E. et al. Comparison of assay methods for detection of circulating tumor cells in metastatic breast cancer: AdnaGen AdnaTest BreastCancer Select/Detect versus Veridex CellSearch system. International journal of cancer 130, 1590–1597, https://doi.org/10.1002/ijc.26111 (2012).
Benini, S. et al. Detection of circulating tumor cells in liquid biopsy from Ewing sarcoma patients. Cancer management and research 10, 49–60, https://doi.org/10.2147/cmar.S141623 (2018).
Sawada, T. et al. Sensitive cytometry based system for enumeration, capture and analysis of gene mutations of circulating tumor cells. Cancer science 107, 307–314, https://doi.org/10.1111/cas.12868 (2016).
Watanabe, M. et al. A novel flow cytometry-based cell capture platform for the detection, capture and molecular characterization of rare tumor cells in blood. Journal of translational medicine 12, 143, https://doi.org/10.1186/1479-5876-12-143 (2014).
Takao, M. & Takeda, K. Enumeration, characterization, and collection of intact circulating tumor cells by cross contamination-free flow cytometry. Cytometry. Part A: the journal of the International Society for Analytical Cytology 79, 107–117, https://doi.org/10.1002/cyto.a.21014 (2011).
Watanabe, M. et al. Multicolor detection of rare tumor cells in blood using a novel flow cytometry-based system. Cytometry. Part A: the journal of the International Society for Analytical Cytology 85, 206–213, https://doi.org/10.1002/cyto.a.22422 (2014).
Watanabe, M. et al. Isolation and molecular analysis of circulating tumor cells from lung cancer patients using a microfluidic chip type cell sorter. Cancer science 109, 2539–2548, https://doi.org/10.1111/cas.13692 (2018).
Kohsaka, S. et al. Comprehensive assay for the molecular profiling of cancer by target enrichment from formalin-fixed paraffin-embedded specimens. Cancer science, https://doi.org/10.1111/cas.13968 (2019).
Hayashi, M. et al. Size-based detection of sarcoma circulating tumor cells and cell clusters. Oncotarget 8, 78965–78977, https://doi.org/10.18632/oncotarget.20697 (2017).
Dubois, S. G., Epling, C. L., Teague, J., Matthay, K. K. & Sinclair, E. Flow cytometric detection of Ewing sarcoma cells in peripheral blood and bone marrow. Pediatric blood & cancer 54, 13–18, https://doi.org/10.1002/pbc.22245 (2010).
Vo, K. T. et al. Impact of Two Measures of Micrometastatic Disease on Clinical Outcomes in Patients with Newly Diagnosed Ewing Sarcoma: A Report from the Children’s Oncology Group. Clinical cancer research: an official journal of the American Association for Cancer Research 22, 3643–3650, https://doi.org/10.1158/1078-0432.Ccr-15-2516 (2016).
Dworzak, M. N. et al. Flow cytometric assessment of human MIC2 expression in bone marrow, thymus, and peripheral blood. Blood 83, 415–425 (1994).
Li, M. et al. A novel function for vimentin: the potential biomarker for predicting melanoma hematogenous metastasis. Journal of experimental & clinical cancer research: CR 29, 109, https://doi.org/10.1186/1756-9966-29-109 (2010).
Hoshino, M. et al. Molecular analyses of cell origin and detection of circulating tumor cells in the peripheral blood in alveolar soft part sarcoma. Cancer genetics and cytogenetics 190, 75–80, https://doi.org/10.1016/j.cancergencyto.2008.11.014 (2009).
Butler, T. M. et al. Exome Sequencing of Cell-Free DNA from Metastatic Cancer Patients Identifies Clinically Actionable Mutations Distinct from Primary Disease. PloS one 10, e0136407, https://doi.org/10.1371/journal.pone.0136407 (2015).
Hemming, M. L. et al. Detection of Circulating Tumor DNA in Patients With Leiomyosarcoma With Progressive Disease. JCO precision oncology 2019, https://doi.org/10.1200/po.18.00235 (2019).
Eastley, N. C. et al. Circulating tumour-derived DNA in metastatic soft tissue sarcoma. Oncotarget 9, 10549–10560, https://doi.org/10.18632/oncotarget.24278 (2018).
Abbosh, C., Birkbak, N. J. & Swanton, C. Early stage NSCLC - challenges to implementing ctDNA-based screening and MRD detection. Nature reviews. Clinical oncology 15, 577–586, https://doi.org/10.1038/s41571-018-0058-3 (2018).
Smolle, M. A. et al. MicroRNAs in Different Histologies of Soft Tissue Sarcoma: A Comprehensive Review. International journal of molecular sciences 18, https://doi.org/10.3390/ijms18091960 (2017).
Kubota, D. et al. miR-125b and miR-100 Are Predictive Biomarkers of Response to Induction Chemotherapy in Osteosarcoma. Sarcoma 2016, 1390571, https://doi.org/10.1155/2016/1390571 (2016).
Cohen, S. J. et al. Prognostic significance of circulating tumor cells in patients with metastatic colorectal cancer. Annals of oncology: official journal of the European Society for Medical Oncology 20, 1223–1229, https://doi.org/10.1093/annonc/mdn786 (2009).
Acknowledgements
This study was supported by The National Cancer Center Research and Development Fund 31-A-1, by grants from the Program for Integrated Database of Clinical and Genomic Information under grant number JP18kk0205003, Project for Cancer Research And Therapeutic Evolution (P-CREATE) under the grant number JP18cm0106502, and the Practical Research for Innovative Cancer Control under grant number JP18ck0106252 from the Japan Agency for Medical Research and Development, AMED.
Author information
Authors and Affiliations
Contributions
S. Kohsaka and H. Mano conceived and designed the study. N. hasegawa and S. Kohsaka developed the method and protocol. N. hasegawa acquired the data. N. hasegawa, S. Kohsaka, T. Ueno and S. Kojima analyzed the data. N. hasegawa, S. Kohsaka and H. Mano wrote the main manuscript text. I. Nakamura, Kawazu, K. Akaike, T. Okubo, T. Takagi, Y. Suehara, T. Hayashi and T. Saito provided administrative, technical, or material support. S. Kohsaka, Y. Suehara, K. Kaneko and H. Mano supervised the study. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hasegawa, N., Takeda Nakamura, I., Ueno, T. et al. Detection of circulating sarcoma tumor cells using a microfluidic chip-type cell sorter. Sci Rep 9, 20047 (2019). https://doi.org/10.1038/s41598-019-56377-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-56377-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
