
Abstract
Cyclotron-produced astatine-211 (211At) shows tremendous promise in targeted alpha therapy (TAT) applications due to its attractive half-life and its 100% α-emission from nearly simultaneous branched alpha decay. Astatine-211 is produced by alpha beam bombardment of naturally monoisotopic bismuth metal (209Bi) via the (α, 2n) reaction. In order to isolate the small mass of 211At (specific activity = 76 GBq·µg−1) from several grams of acid-dissolved Bi metal, a manual milliliter-scale solvent extraction process using diisopropyl ether (DIPE) is routinely performed at the University of Washington. As this process is complex and time consuming, we have developed a fluidic workstation that can perform the method autonomously. The workstation employs two pumps to concurrently deliver the aqueous and organic phases to a mixing tee and in-line phase mixer. The mixed phases are routed to a phase settling reservoir, where they gravity settle. Finally, each respective phase is withdrawn into its respective pump. However, development of a phase boundary sensor, placed in tandem with the phase settling reservoir, was necessary to communicate to the system when withdrawal of the denser aqueous phase was complete (i.e., the intersection of the two phases was located). The development and optimization of the autonomous solvent extraction system is described, and the 211At yields from several ~1.1 GBq-level 211At processing runs are reported.
Similar content being viewed by others


Introduction
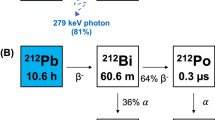
Several alpha-emitting radionuclides are being considered for use in targeted alpha therapy (TAT)1,2,3,4,5. Of these, cyclotron-produced astatine-211 (211At, t1/2 = 7.214 h) shows tremendous promise in TAT applications due to its attractive half-life and its 100% α-emission from nearly simultaneous branched alpha decay (211At (5.87 MeV, 41.8%) and 211Po (7.45 MeV, 58.2%))6. Several clinical trials have been approved using 211At-labeled monoclonal antibodies. In former trials, chimeric antitenascin monoclonal antibody 81C6 treatment was administered within surgical resection cavities of malignant brain tumor patients7 and 211At-MX35 F(ab′)2 was administered into the peritoneal cavity for treatment of ovarian cancer8. At present, clinical trials are underway at the Fred Hutch/University of Washington Cancer Consortium9 using radioimmunotherapy with 211At for treatment of blood-borne cancers (myeloid leukemia, acute lymphoblastic leukemia, and myelodysplastic syndrome)10.
Astatine-211 is produced using alpha beam bombardment of naturally monoisotopic bismuth (209Bi) via the (α, 2n) reaction11,12,13. The Scanditronix MC-50 cyclotron at the University of Washington (UW) is capable of producing >4 GBq 211At with good radionuclidic purity using a 29.0 MeV alpha particle beam11. With sufficient production capacity in place, separation of 211At from the target material is one of the major challenges for routine processing.
The earliest 211At separations were performed by distillation from molten Bi targets14,15,16,17,18 or by solvent extraction of acid-dissolved targets19,20, frequently with diisopropyl ether (DIPE)20,21,22,23,24,25. The UW has refined a solvent extraction method that employs DIPE to isolate 211At from dissolved Bi targets; it has been comprehensively described by Balkin et al.21. The method is performed manually in a glove box. It involves (1) dissolution of the Bi metal target in ≥10 M HNO3; (2) evaporation of the resulting solution to dryness, leaving a Bi oxynitrate saltcake; (3) dissolution of the salts in 8 M HCl; (4) solvent extraction of 211At into DIPE; (5) followed by repeated washing of the DIPE with 8 M HCl; and (6) back-extraction of the Bi-isolated 211At into 4 M NaOH. The manual solvent extraction process is performed by mixing the two phases with a magnetic stir bar in a 20 mL glass LSC vial. At the conclusion of each step, the aqueous (dense) phase is withdrawn using a disposable transfer pipette; the phase boundary between the aqueous and organic liquids is identified visually. While the method provides reliable 211At yields (78 ± 11%) with high purity and labeling performance, its execution requires considerable skill by a trained radiochemist.
We have investigated techniques to adapt the manual solvent extraction process to a fluidically automated regime. In general, solvent extraction processes can involve an array of phase mixing techniques, including shaking and stirring (most frequently performed in the laboratory), and centrifugal contacting and mixer/settler operations (typically performed at the industrial scale)26,27. In contrast, applications involving small scale, in-line solvent extraction processes are relatively scarce in the literature. Rodríguez et al. have recently reviewed strategies for in-line automation of solid-phase and liquid-liquid (L/L) extractions in radioanalytical applications28. These involve L/L microextraction and dispersive L/L microextraction. In one medical radionuclide application, in-line solvent extraction of 99mTc from dissolved, proton-bombarded 100Mo targets was performed using a stream of gas bubbles in a glass tube to mix the two liquid phases29.
In developing an automated solvent extraction system, the industrial-scale mixer/settler concept was adapted to an in-line, milliliter scale. Two pumps are used to deliver aqueous and organic phases concurrently into an in-line mixing device that induces a brief emulsification or a large/turbulent phase contact surface. We describe the performance of two in-line mixing devices: a glass bead-packed column and a serpentine (knotted tubing) mixer. Collection of the two mixed phases in a static phase settling reservoir on the other side of the mixing device allows the phases to gravity separate. The return of each phase into its respective pump is accomplished via the aspiration of the dense phase, followed by the light phase, from the phase settling reservoir. At this point, the organic phase (containing the extracted ion of interest) can be retained, while the post-contacted aqueous phase can be sent to waste. Then, the solvent extraction process can be resumed by performing the next step (e.g., organic phase washing with a fresh aliquot of acid).
A major challenge in automating a small-scale solvent extraction process is in accurately identifying the phase boundary, as will be discussed. Various optical methods have been previously employed to identify liquid phase boundaries, including the measurement of differences in refractive index, UV absorbance, and light reflection30,31. Given the dramatic differences in electrical conductivities between diisopropyl ether32 and the strongly acidic solutions required for Bi target dissolution and 211At solvent extraction33,34, we opted to develop a sensor that identifies the phase boundary by monitoring the phases’ ability to carry an electrical current.
Members of this research team have recently worked to develop fluidic systems to isolate 89Zr, a cyclotron-produced immunoPET radionuclide, from natY metal foil targets35,36,37. More recently, efforts have been underway to improve the efficiency of the complex 211At isolation method performed at UW by integrating laboratory automation into the process. Previously, an in-line Bi target dissolution system was demonstrated to produce reproducible 211At release profiles using optimized nitric acid concentrations, flow rates, and volumes38. Herein, we describe the development of an autonomous in-line system for 211At isolation from dissolved and prepared Bi cyclotron targets via the acid/DIPE solvent extraction process. An in-line mixer/settler system was employed to briefly emulsify the two concurrently-injected phases and then allow the phases to gravity separate in a phase settling reservoir (PSR). A phase boundary sensor (PBS) was configured at the outlet of the PSR to provide real-time identification of the location of the unpredictable aqueous/organic interface. With these components in place, the extraction of 211At into DIPE, and subsequent repetitive acid washing of the DIPE, was possible. The development and characterization of the system, and its performance on ~1.1 GBq levels of 211At, are described.
Results
In-line mixer/settler system characterization
While manual solvent extraction in the laboratory is typically accomplished by shaking/settling or stirring/settling operations, these methods are generally not conducive to in-line approaches. We evaluated the performance of two in-line phase mixers (a ~1 cm3 glass bead-packed column mixer and a 1.2 m long tubular “serpentine mixer”) to affect the isolation of 211At from dissolved Bi-containing solutions. The results were compared to those achieved by UW’s manual (stirring/settling) solvent extraction process.
Bi3+ ion removal from DIPE during load/wash steps
We first evaluated the decontamination of Bi3+ ions from DIPE during the solvent extraction process. Solutions containing ~3 g dissolved Bi metal in conc. HNO3 were evaporated to dryness, leaving a white Bi oxynitrate residue of undefined composition. The residue was dissolved in 8 M HCl.
The resulting Bi-bearing solution was initially processed with the forward-extraction steps using DIPE in a magnetically mixed 20 mL vial, as per the manual wet-chemistry 211At isolation method (see Balkin et al.21 and the Electronic Supplementary Materials (ESM) Section). The concentration of Bi3+ remaining in the organic phase after the manual load and three successive organic phase wash intervals was determined by inductively coupled plasma – optical emission spectroscopy (ICP-OES). The mass of Bi (µg) measured in the organic phase is presented in Fig. 1A, and the calculated Bi decontamination factor (DF) at each stage in the manual solvent extraction process is presented in Fig. 1B (where DF is defined as the mass ratio of Bi dissolved in the originally-contacted acidic solution and Bi measured in the organic phase). The results indicate post-load Bi3+ contamination levels at 428 ± 19 µg. Following three successive washes with 8 M HCl, the Bi3+ contamination levels had dropped to 5.7 ± 1.3 µg.

Comparison of Bi3+ contamination observed from manual solvent extraction method vs. in-line phase mixing methods. (A) Mass of Bi3+ determined in the DIPE phase following the initial sample load and after up to three wash cycles. (B) Calculated Bi3+ decontamination factors from an initial mass of ~3 g dissolved Bi metal in the load solution.
Next, in-line mixing systems employing column and serpentine mixers were assembled and tested with a fluidic system to determine their effectiveness in Bi3+ ion removal from the organic phase. Results are provided in Fig. 1. It was observed that both in-line mixing methods resulted in Bi3+ contamination levels in the post-load organic phase that were considerably lower compared to the manual method (60 ± 2 µg and 125 ± 5 µg for the column and serpentine mixers, respectively). By the end of the triple-wash cycle, contamination levels had dropped to 8.1 ± 0.6 µg and 12.8 ± 0.5 µg, respectively. The three methods had approximately the same effectiveness at Bi3+ ion removal across the load/wash sequence (each ending with <13 µg Bi); the manual solvent extraction method resulted in the highest Bi DF (~4.9 × 105), followed by the column and serpentine mixers (~3.5 × 105 and ~2.2 × 105, respectively).
211At solvent extraction during load/wash steps
Each of the two in-line mixing methods were subsequently evaluated for their effectiveness at 211At extraction into DIPE. A surrogate dissolved target solution was prepared by spiking freshly isolated 211At (~1.9 MBq) into freshly-dissolved Bi metal pieces (using 10 M HNO3). The solution was evaporated to dryness, and the salts were dissolved in 8 M HCl (vide supra). A pair of syringe pumps was used to deliver the 211At-/Bi-bearing solution and DIPE concurrently through a mixing tee and in-line mixer. At the conclusion of the delivery, the phases were allowed to gravity separate, and then a small volume of the DIPE solution (10 µL) was sampled and analyzed for 211At activity levels. Next, each phase-separated liquid was withdrawn into its respective syringe pump, and the phase mixing→phase settling→DIPE sampling process was repeated three more times. We observed that 211At extraction yields were nearly quantitative after the first pass, as shown in Table 1; the column and serpentine mixers provided extraction yields that were not statistically distinguishable from each other.
After confirming that both of the in-line mixers could effectively transfer 211At into the organic phase during a load sequence, the complete forward extraction load/wash process was evaluated for the manual method and the two in-line mixing assemblies. Surrogate dissolved targets were prepared with spikes of 211At (~1.9 MBq) added to Bi metal (~3 g), then dissolved and prepared as described above. Following each DIPE solution load and sequential wash step, a small volume of the aqueous phase was sampled and analyzed for 211At activity concentration. This activity was corrected for aqueous phase volume and compared to the known activity initially present in the prepared surrogate target solution. The performance of each method was evaluated in triplicate.
The results in Table 2 present the calculated 211At activity fraction remaining in the aqueous phase following each load and wash step (see Experimental Section and the ESM for details on load and wash step volumes for the in-line and manual methods, respectively). The manual method provided somewhat variable 211At extraction during load, with 10.5 ± 5.8% of 211At remaining in the aqueous phase. The subsequent 8 M HCl washes of the 211At-loaded DIPE resulted in minor and slightly increasing 211At losses at each stage. Overall, the manual method resulted in an extraction of all but 15.0 ± 5.9% of the 211At activity originally present.
The in-line mixing methods were evaluated next. Surrogate 211At-bearing dissolved target solution was delivered concurrently with DIPE two times through the in-line mixers prior to sampling for 211At activity determination. Passage of the aqueous and organic phases through the column and serpentine mixers during the dual load steps succeeded at extraction of all but 4.3 ± 0.5% and 5.8 ± 1.7% of the 211At, respectively. As with the manual method, each successive organic phase wash with 8 M HCl resulted in a small, incrementally increasing activity loss into the aqueous phase. Cumulative 211At losses for both in-line methods were below ~11%; they were within experimental uncertainty at ±2 s. However, given the column mixer’s slightly better performance relative to the serpentine mixer, it was selected to be integrated into the engineered fluidic system.
Phase boundary sensor (PBS) characterization
The development and integration of an aqueous/organic PBS into the fluidic system would make it possible for the apparatus to trigger an event when the aqueous/organic phase boundary is identified. In this manner, the unpredictable volumes of the aqueous and organic phases in the PSR could be accommodated. The system aspirates the (dense) aqueous phase into the aqueous solution handling pump until the phase boundary is sensed (i.e., all aqueous solution has been withdrawn from the PSR). At this point, the aqueous solution handling pump ceases aspirating, and the organic phase handling pump aspirates the remaining organic phase from the PSR.
The PBS was designed to trigger an event based on a change in the electrical resistivity measured when a highly conductive aqueous solution (acid) passed through the monitored flow channel, followed by a poorly conductive organic liquid (DIPE). The resistivity measured across two electrical leads spikes to a “high” state when the organic solution enters the zone between the two leads. Below, the behavior of the PBS in acid/organic solutions is reported.
Given its well-established electrical conductivity and chemical resilience to strong acids, Pt wire was chosen as an electrode material to be tested in the PBS for the acid/organic phases. Using a data-logging potentiostat set up in open circuit potential (OCP) mode, we evaluated the Pt wire sensor performance as a function of time while it was repeatedly exposed to strong HCl and acid-contacted DIPE. In OCP mode, the potentiostat applies no current or voltage; it simply monitors the voltage across a circuit (in our case, the Pt wires positioned within the PBS flow channel). Using the potentiostat in this manner, the integrated switching logic of the digital input signal on our solvent extraction platform was monitored (i.e., input voltage level).
A testing program was set up wherein a syringe pump was programmed to aspirate liquid from a phase-separated reservoir of 8 M HCl and DIPE while logging the voltage levels across the Pt leads via the potentiostat. Each trial consisted of three withdrawals of HCl/DIPE phases from the PSR; data traces across three trials were recorded. The potentiostat traces are shown in Fig. 2.

Voltage potentials measured across the Pt coil electrode terminals connected to the input logic board during alternating deliveries of 8 M HCl and DIPE solutions. Horizontal dashed line indicates approximate potential above which the sensor input logic is switched from “low” to “high” state. Arrows indicate a region of brief sensor instability upon exposure to acid.
When the Pt coil leads were exposed to acid, a low-resistivity condition was identified; the OCP recorded a low potential across the input logic circuit. When the input signal was below ~2.3 V potential (dashed line), the input circuit remained in the “low” state.
When the sensor signal rose above the ~2.3 V threshold potential, it switched to the “high” state. The high voltage is associated with a high resistivity between the Pt coil leads. Since DIPE is a vastly poorer electrical conductor than strong acid (vide supra), the resistivity across the sensor would be expected to increase significantly once the organic solution passed between the Pt coil electrodes within the monitored flow channel.
The data traces in Fig. 2 show that there is a brief period of sensor instability that occurs within a few seconds after 8 M HCl is introduced to the sensor (rapid fluctuation of signal above and below the sensor switching voltage). In order to assure that a premature switching event would not occur during aspiration of acid through the PBS, the software program was modified so that the PBS would exclude input data during this brief period of instability immediately following acid addition (3 s). Conversely, the voltage shift observed at the acid/DIPE interface did not exhibit the same instability; once the DIPE was in contact with the sensor, the voltage immediately went “high”, assuring that the switching mechanism would be promptly activated.
Fluidic system performance testing on cyclotron bombarded Bi targets
After evaluations of in-line solvent mixing and phase boundary sensing were completed, a fully autonomous solvent extraction system capable of emulating the manual wet chemical 211At isolation process was designed, constructed, and programmed. The programming of the system was initially refined while running non-radiological solutions, and then further refined on 211At-spiked solutions in the UW glovebox. Ultimately, a series of nine performance tests were made on freshly-bombarded Bi cyclotron targets that contained 1.07 ± 0.02 GBq of 211At (at end-of-bombardment (EOB)). The targets used in this series contained 4.8 ± 0.5 g of Bi metal.
For each GBq-level run, the Bi targets were dissolved using the automated target dissolution station that was previously described38. The dissolved Bi effluents were routed from the dissolution block to a heated distillation chamber that brought the solutions to dry Bi oxynitrate salts. After the salts were cooled, the fluidic system’s pump 1 injected 8 M HCl onto the salt residue; a combination of magnetic mixing and aspiration/dispensation of the HCl solution (pump 1) resulted in complete dissolution of the salts after several cycles had been performed. The prepared target solutions were then ready for in-line solvent extraction.
The loss of 211At to the aqueous phase was tracked at each stage of the processing runs (load and three successive washes). Each load and wash fraction was collected separately and analyzed after completion of the runs. The data presented in Fig. 3 shows the incremental 211At loss at each stage of the forward extraction method for each target.

Observed 211At losses in the aqueous phase following the prepared target/DIPE load step (4.7 ± 0.9% loss) and three successive washes with 8 M HCl (1.6 ± 0.6%, 2.4 ± 0.6%, and 2.7 ± 0.5% loss, respectively).
The data shows that the majority of the 211At loss occurred during the prepared target/DIPE mixing (“Load”) step. The average percentage of 211At remaining in the aqueous phase following the Load step was 4.7 ± 0.9%. These results compare favorably to the manual processing runs, wherein the 211At found in the post-contacted load solution was 10.5 ± 5.8% (Table 2).
After 211At loading, some of the DIPE-extracted 211At was incrementally lost to the three successive DIPE washes with 8 M HCl. The degree of 211At loss increased slightly with each wash, resulting in 1.6 ± 0.6%, 2.4 ± 0.6%, and 2.7 ± 0.5% lost in washes 1 through 3, respectively. These incremental 211At losses were likewise observed in our earlier trials when comparing the manual and in-line mixing methods (Table 2).
Overall, the total 211At loss to the aqueous phase was 11.4 ± 1.3% across the study. This was the fraction of 211At activity that was not retained in the organic phase (routed to waste) during the pump 1/2 forward extraction operations that involved the packed column-based mixer/settler system. The cumulative 211At recovery (obtained after target dissolution → target solution evaporation → recovered activity from the dissolved Bi salts → the above-described solvent extraction load/wash steps) was calculated to be 79.4 ± 4.5%.
Discussion
Necessity of the PBS
Development of a procedure that allows “automatic” solvent extraction in the isolation of 211At is difficult, if not impossible, given the unpredictable nature of the aqueous/organic phase volume changes encountered during the target preparation and solvent extraction processes. Although the DIPE and 8 M HCl reagents used in the method were pre-contacted with each other prior to use (to minimize volume change during execution of the method), the precise volume of dissolved target/HCl/DIPE in each phase was not known. In the manual method, these volume inconsistencies are inconsequential since phase separation at each step is accomplished by pro-active (visual) means.
Due to tolerances in production of the Al backing for the cyclotron targets, and machining the Bi metal melted onto the Al backing, the Bi metal targets used for 211At production had varying mass. In our initial studies, the Bi metal in the target assemblies ranged between 3.5 and 6.5 g. Later changes in machining tolerances, and adapting a weight-based removal of excess Bi from targets, reduced the Bi mass uncertainty substantially to 4.8 ± 0.5 g for targets employed in the GBq-level study. Because Bi metal mass in the target assemblies was determined by weight difference pre- and post-dissolution, the quantity of Bi metal could not be known in advance. Thus, dissolution of the Bi metal in nitric acid and subsequent heating/distillation of the acid resulted in unknown quantities of Bi-bearing salts. Further, differences in salt quantities affected the level of dryness obtained for a given thermal treatment interval, which in turn affected the chemical composition (e.g., bismuth:nitrate ratio and level of hydration) of the Bi oxynitrate salt39,40,41,42.
These unpredictable salt quantities and compositions resulted in unpredictable solution volumes when the salts were dissolved with 8 M HCl in preparation for 211At solvent extraction. The HCl-dissolved target solution volumes varied as a function of starting Bi metal mass and the HCl volume added (see data presented in the ESM).
Additionally, during the initial contact of the dissolved target and DIPE solutions, some volume change between the phases was observed, the degree of which was generally driven by the ionic strength of the dissolved target solution. Phase volume changes during the three DIPE solution wash steps were nominal, since the 8 M HCl solution used in the washes was equilibrated with DIPE prior to introducing the reagent into the fluidic system.
The general issues with “automatic” solvent extraction from unknown input volumes are illustrated in Fig. 4. The top row shows three hypothetical conditions encountered upon delivery of three dissolved Bi targets and a fixed organic solution volume to a PSR. “Heavy” (A), “normal” (B), and “light” (C) Bi targets result in decreasing aqueous phase volumes, respectively.

(Top) Illustration of the effects of “high” (A), “normal” (B), and “low” (C) mass Bi targets during automatic solvent extraction. Arrows indicate the liquid phase boundary based on a predictable, “normal” condition. (Bottom) Effects of the aspiration of a fixed aqueous phase volume based on a “normal” target.
The bottom row of Fig. 4 illustrates the effect of a fixed aqueous phase aspiration based on a pre-programmed “normal” target mass. In condition (B), the aspiration volume is sufficient to remove all of the dissolved target solution, thus enabling effective phase separation prior to initiation of the organic phase wash cycles. In condition (A), the same aspiration volume is insufficient to remove all of the dissolved target solution. This results in excess acid/Bi salt residing in the reservoir at the conclusion of the volume withdrawal. As a consequence, the subsequent wash steps with clean 8 M HCl are ineffective at removing Bi from the organic phase, since the remaining Bi-bearing target solution will simply be diluted at each wash step. In condition (C), the aspiration volume is excessive; all of the aqueous phase is withdrawn, along with a portion of the organic phase. Under this condition, some of the 211At-bearing organic phase is sent to waste along with the post-contacted dissolved target solution. The 211At yields will consequently suffer.
Given the unpredictable aqueous solution volumes going into the front end of the solvent extraction system, the process could not be achieved by performing a fixed sequence of pre-programmed (“automatic”) commands. Our response to this issue was to develop a sensor that could identify the phase boundary between the two immiscible liquids. With such a sensor, a fully “autonomous”, multi-step solvent extraction process became possible.
It is worth noting that the presented phase boundary sensing approach can conceivably result in the separation of arbitrary amounts of different aqueous/organic liquid phases from one another, so long as the liquid interface is well defined and the electrical conductivity of each liquid is sufficiently different.
211At yields obtained from the autonomous fluidic system
The solvent extraction steps encompassing the loading of 211At into the organic phase from the prepared target solution and the triple wash steps to remove Bi3+ ions from the organic phase resulted in 88.6 ± 1.3% 211At recovery in the DIPE. This was based on careful accounting of the 211At present in the load and triple wash solutions (11.4 ± 1.3% 211At loss in aqueous phase, Fig. 3).
When considering incremental 211At losses in the steps leading up to the conclusion of the solvent extraction process, the cumulative 211At yield was 79.4 ± 4.5%. This additional ~9.2% loss was attributed to several possible sources: (1) the inability to recover all 211At from the aluminum target assembly that backs the Bi metal layer; (2) 211At volatilization during the evaporation of HNO3 during the distillation step; (3) spatter of Bi oxynitrate salts on the upper walls of the distillation chamber caused by the boiling HNO3 during distillation; and (4) 211At-bearing residues remaining in the distillation chamber following aspiration of the HCl-dissolved Bi-bearing salts. A direct comparison of the 211At yields up to this point in the manual process is not possible, as Balkin et al.21 only reported mean yields in the final 211At product fraction (which included back-extraction of 211At into 4 M NaOH). However, our yields at the conclusion of the acid → DIPE cycles (79.4 ± 4.5%, n = 9) compare favorably with those reported by Balkin et al. for the entire 211At isolation process (78 ± 11%, n = 55). Further, the uncertainties associated with the autonomous vs. manual methods indicate that the presented method may be capable of providing more reproducible 211At yields (although the number of trials was fewer in the current evaluation).
Fluidic system expansion
The autonomous acid/DIPE solvent extraction system described herein was further expanded to include the capability to perform an autonomous DIPE/4 M NaOH back-extraction step, during which the isolated 211At in DIPE is transferred back to an aqueous phase. This back-extraction module allowed for execution of the complete, end-to-end solvent extraction process described by Balkin et al.21. The expanded system was comprised of three syringe pumps (pump 3 was for handling the NaOH-based back extractant) and a separate in-line mixer, PSR, and PBS. The PBS for DIPE/base phase identification employed the same electrical conductivity monitoring approach, although Pt electrodes were not practical and a new electrode material needed to be identified. Ultimately, the system dispensed the isolated 211At product in a small volume of 4 M NaOH. A description of the back-extraction module development and its performance in the end-to-end process with clinical levels of 211At will be described in a future article.
Methods
Reagents
Hydrochloric acid (HCl) and nitric acid (HNO3) were ACS Certified grade or higher (Fisher Scientific, Waltham, MA). Dilutions of these reagents were prepared from deionized water (≥18 MΩ∙cm) using a Barnstead Nanopure Diamond water purification system (Dubuque, IA). Scintillation cocktail was Ultima Gold™ AB (PerkinElmer, Waltham, MA).
Diisopropyl ether (DIPE) was Certified ACS grade (Fisher Scientific). For method development, DIPE was used as-is; for high-level (e.g., GBq-level) 211At isolation runs, DIPE was distilled prior to use to assure removal of reagent stabilizer (e.g., butylated hydroxytoluene (BHT) or hydroquinone).
Bi pellets of 99.999% purity, used in the manufacture of Bi target assemblies11, were acquired from Alfa Aesar (Ward Hill, MA). Simulated dissolved Bi metal targets employed Bi metal pieces of the same purity (Sigma-Aldrich, St. Louis, MO). Platinum wire was 0.25 mm dia. and had a metal purity of 99.9% (Alfa Aesar). Coiled Pt electrodes were prepared by wrapping the wire around an 18 gauge hypodermic needle. The electrodes were secured and sealed into the PBS housing with ferruled 1/4-28 fittings.
211At production
The Scanditronix MC-50 cyclotron at the University of Washington Medical Cyclotron Facility (UWMCF) routinely produces 211At via the 209Bi(α, 2n)211At reaction. It is capable of producing external alpha (He2+) beams in the energy range of 27.0–47.3 MeV. Gagnon et al. have described the design, preparation, and performance of the Bi targets11. For the nine Bi metal targets bombarded for the final fluidic system performance evaluation, the cyclotron ran an integrated target current of 38 µA·h, which required 0.75–1.2 h of beam time to produce 1.07 ± 0.02 GBq (28.8 ± 0.5 mCi) of 211At (EOB).
Manual method
The routine manual method for 211At isolation from solution-prepared cyclotron bombarded Bi metal targets by solvent extraction has been described by Balkin et al.21. A brief summary of the manual method (including target preparation and solvent extraction steps) is presented in the ESM.
Fluidic system components
Fluids are delivered using two 48,000 step digital syringe pumps (model V6, Norgren, Las Vegas, NV) coupled to 8-position distribution valves at their heads (Norgren). Pumps 1 and 2 are configured with 25 and 10 mL volume displacement (“zero dead volume, ZDV) syringes, respectively (Flex Fluidics, Las Vegas, NV). The dual pumps are assembled into a metal box provided by J-Kem Scientific (Model 2200, St. Louis, MO). Accessible from the rear of the box is the pump’s digital input/output board ports. The pumps’ distribution valves are plumbed using 0.75 mm ID × 1/16″ OD Teflon® FEP tubing. The tubing is connected between the distribution valves and the reagent reservoirs, waste receptacles, and phase mixing/settling hardware using polyether ether ketone (PEEK) or ethylene tetrafluoroethylene (Tefzel®, ETFE) ¼−28 flangeless nuts with Tefzel ferrules (Upchurch Scientific, Oak Harbor, WA). The fluidic system is controlled by a laptop PC using a PNNL-modified version of KemPump software (JKem Scientific).
In-line mixer/settler systems
The two liquid phases are delivered simultaneously from their respective syringe pump: aqueous solution from pump 1 and organic liquid from pump 2. The liquids intersect at a mixing tee and then pass into a phase mixer – a tortuous path through which the fluids are intimately co-mingled. Beyond the phase mixer is a PSR – a container used to collect the mixed phases and provide a static condition in which the phases can gravity separate (Fig. 5).

Schematic of a column (A) and serpentine (B) mixer positioned between a mixing tee and a conical vial used as a phase settling reservoir (PSR). Pump 1 delivers acidic solution, and pump 2 delivers organic liquid (DIPE) simultaneously into the mixing tee and in-line mixer. Following phase separation, the liquids are sequentially aspirated back into their respective pump (C).
The first phase mixer evaluated was a 1 cm3 SPE column (Sigma-Aldrich) packed with 212 µm dia. silanized glass beads (Sigma-Aldrich) (Fig. 5A). It was fitted with a custom-machined cap at the inlet end that enabled fluids to be delivered to the column via with a male luer adapter. Additionally, we evaluated a Super Serpentine Reactor™ (GlobalFIA, Fox Island, WA), which was a 1.2 m length of 0.75 mm ID/1/16″ OD Teflon FEP tubing (0.53 mL internal volume) braided tightly through a perforated plate (Fig. 5B).
The flow rates of each pump were programmed so that the total volume of each respective phase was delivered towards the mixing tee over an equal time period. Hence, the syringe volume:dispensation flow rate ratio was the same between aqueous and organic phase syringes: typically 1.25 min per full syringe stroke (20 mL∙min−1 for a 25 mL syringe; 8 mL∙min−1 for a 10 mL syringe). The two solutions were merged at the mixing tee, and were then passed through the in-line mixer. A quick dispensation of air (at same flow rates as above) assured that the fluids were “chased” through the apparatus at the conclusion of the syringe stroke.
The tortuous path of the serpentine mixer causes in-line mixing to occur43,44,45. For the column mixer, the two liquids were delivered to the column of glass beads, thus creating intensively mixed phases as they were driven through the bed of small spheres. Upon exiting either the serpentine or column mixer, the biphasic mixture was delivered to a PSR (centrifuge tube or syringe barrel), where the two phases quickly separated. The phase settling interval was 30 s, which was ample time for the organic/aqueous phases to separate and for most of the fine solution-entrained bubbles from the air push to rise to the surface of the DIPE.
Next, tubing that connected each syringe pump’s distribution valve to the bottom of the reservoir was used to withdraw first the (dense) aqueous phase, and then the organic phase, back into each respective pump (Fig. 5C). In this manner, the processing cycle was set to be repeated. Alternatively, the aqueous phase could be dispensed to waste, the syringe pump rinsed with clean 8 M HCl, and re-loaded with 8 M HCl rinse solution prior to the next phase mixing interval.
Phase boundary sensor (PBS)
The digital syringe pumps employed in the described fluidic processes are equipped with an external input signal processor board that allows voltage (0–5 V) to be monitored in real time; we took advantage of this feature to implement a PBS. The PBS body, which is machined out of a Teflon cylinder, is mounted to the base of a PSR (20 mL syringe barrel). The outlet of the PSR is connected to the inlet of the PBS with a luer/¼-28 coupler. Near the bottom of the PSR is a fluid channel in a “tee” configuration, which allows fluids to be withdrawn from the reservoir by either pump 1 or pump 2. Two electrodes project into the fluid channel, each held in place by ferruled ¼–28 fittings; they are positioned 2 cm apart. The electrodes are connected to the +5 V and ground terminals of the pump’s input signal processor. Aqueous and organic liquids are simultaneously passed through a mixing tee and phase mixer from pump 1 and pump 2. Upon exiting the phase mixer, they are collected in a PSR perched atop the PBS (Fig. 6A).

Sequence of steps in the automated biphasic liquid separation system. (A) Introduction of biphasic solution into the PSR from an in-line phase mixer and allowing phases to separate; (B) Withdrawal of aqueous phase to aqueous pump, PBS is in the “low” state; (C) Triggering of the PBS to the “high” state, and ceasing the PBS monitoring cycle; (D) Withdrawal of the remaining aqueous phase in the common fluid channel; (E) Withdrawal of organic phase to organic pump. Label descriptions are provided in Table 3.
The fluid handling software was programmed to read the signals from a simple logic gate binary system offered by the pump’s signal input board. After withdrawing 25 µL of aqueous solution through the PBS (at 40 mL∙min−1 flow rate), the input board’s signal is momentarily read. If the resistivity of the solution is small, then perform task 1; if the resistivity is large, then perform task 2. Task 1 instructs pump 1 to continue withdrawing solution through the sensor (at 25 µL increments), since acid is present between the two electrodes within the PBS’s flow channel (sensor reads “low”, Fig. 6B). Task 2 instructs the pump to cease withdrawing solution through the sensor once the organic phase has entered the sensor (e.g., the phase boundary has been detected, sensor reads “high” (Fig. 6C)). Once the phase boundary is sensed, pump 1 aspirates a small volume of the aqueous solution to clear the dead volume between the Pt electrodes and the tee at the base of the PBS (Fig. 6D). Finally, pump 2 withdraws the isolated organic phase from the PSR (Fig. 6E).
Engineered solvent extraction system and sample processing
The fully engineered autonomous solvent extraction system is comprised of two digital syringe pumps, a forward extraction mixer/settler system (1 cm3 column mixer), and a PBS (Fig. 7). The first pump handles the prepared target solution and acidic wash solutions; the second pump handles the DIPE solution. The syringe pumps were inverted, with the distribution valves below the syringe (note that Fig. 7 is not illustrated in this configuration). In this manner, liquid phases could be efficiently “chased” with air after each liquid delivery stroke.

Schematic of the dual-pump fluidic system used to perform autonomous solvent extraction for the isolation of 211At from dissolved Bi target solutions.
The system was designed to have the syringe pumps and all peripheral components of the mixer/settler arrangement organized in a small space immediately in front of the pumps. In this compact configuration, the tubing connections are kept as short as possible, and they are less likely to be damaged by the cumbersome glovebox gloves that are necessary to be used when 211At processing is underway. An image of the fluidic system is presented in the ESM.
The sequence of high-level steps performed by the fluidic system is described in Table 4, and the delivered reagents and volumes at each step in the process is presented in Table 5.
Step 1 is performed in a fixed time interval, while the other steps have varying time duration. Step 2 is performed until the salts are visually observed to be dry. This is primarily determined by observing the elapsed time between condensate droplets within a jacketed condenser positioned between the distillation and distillate chambers. Depending on the mass of Bi in the target assembly, the elapsed time of the combined target dissolution and nitric acid distillation was ~75–90 min (distillation performed at 180–190 °C). This automated acid distillation process takes longer than the manual distillation method (which requires ~30–45 min), since a temperature of 300 °C is used there21. The automated process cannot be performed at this high temperature, since severe Bi salt spatter begins to occur above ~200 °C. Salt spatter from acid boiling/bumping places much of the 211At-bearing Bi oxynitrate salt out of reach of the 8 M HCl used to subsequently dissolve the saltcake (per step 3), and therefore needs to be avoided.
Upon completion of the distillation step (per a prompt of the software by the operator), the Bi salt conversion (step 3) is initiated. First, the heating block is rapidly cooled (by remote gating of chiller fluid through the block) to 75 °C. During the block cooling interval, the program pauses until a 75 °C heating block temperature is reached. Next, the still-warm Bi oxynitrate saltcake is dissolved by cycled dispensation/aspiration of 8 M HCl from pump 1 to/from the distillation chamber. A number of these cycles is required ensure the saltcake is fully dissolved (and the number of cycles performed is calibrated to successfully dissolve a saltcake resulting from the heaviest possible Bi target). Prior to initiation of the first solvent extraction step (step 4), the program ensures that the heating block temperature has been reduced to at least 35 °C via a second block cooling interval.
At the initiation of step 4, pump 1 aspirates the HCl-dissolved 211At/Bi solution out of the distillation chamber and proceeds with the DIPE load step. During the solvent extraction process, the time to perform steps 4–5 varies slightly (14–17 min), depending on the volume of the dissolved Bi-bearing solution; a heavy Bi target solution, due to its larger volume, will require more time to pass through the PBS during the phase boundary determination cycles vs. a lighter target solution. Once the 211At-depleted Bi solution is sent to waste and the wash cycles begin (steps 6–8), the elapsed times are very similar, since the aqueous and organic volume changes at this point are minimal. The autonomous solvent extraction process (steps 4–9) ranged between 29 and 32 min. Overall, a complete processing run required ~2.1 to ~2.4 elapsed hours between initiation of the Bi target dissolution and dispensation of the isolated 211At product in DIPE. The autonomous solvent extraction operations represent only ~22% of the total 211At processing time.
211At activity quantification
All 211At activity levels were decay corrected to EOB. Direct measurements of 211At activity levels were obtained using a CRC-15R dose calibrator (Capintec, Inc., Ramsey, NJ). The instrument was calibrated for 211At by UW; a setting of 040 was employed for 211At-bearing solutions in polyethene vials. The setting was based on a cross-calibration to a high purity Ge (HPGe) detector (Ametek, Oak Ridge, TN) that had been calibrated against NIST-traceable gamma standards. It has been demonstrated that high Bi concentrations attenuate the weak 211At and progeny X-ray and gamma emissions, thus resulting in negatively biased activity measurements21. Therefore, UW-determined correction factors were employed for samples containing well-known Bi concentrations. Otherwise, 211At samples high in Bi were not reported by dose calibrator. Rather, liquid scintillation analysis (LSA) was employed.
LSA was performed on direct sample aliquots or serial dilutions of 211At-bearing samples, depending on 211At activity levels present in the sample. Typically, 20 µL aliquots of each fraction (direct or serial dilution) were withdrawn and added to 4 mL scintillation cocktail that was pre-dispensed into a 2-dram polyethylene scintillation vial. The prepared samples were typically allowed to decay through one or more half-lives prior to alpha decay measurement by LSA (TriCarb 1900CA, PerkinElmer). Only LS measurements that exhibited ≤3500 cps were used, in order to assure linear LS analyzer response. If the sample measurement exceeded this count rate, it was allowed additional time to decay and then recounted. The counting region between 40 and 2000 channels, with tSIE quench correction activated, assured integration of the 211At and 211Po alpha emissions (5.87 and 7.45 MeV, respectively). Since 211At branch decays to an alpha emission either via 211At directly, or through 211Po6, the sum of the two alpha peaks, adjusted to a 95% detection efficiency, yielded the activity of 211At in the sample. The LS detection efficiency was determined as the ratio of the count rate of the LS alpha counting region and the 211At disintegration rate reported by the cross-calibrated dose calibrator.
All system components and effluents that came in contact with 211At were sampled for 211At activity during or at the completion of each run. Low Bi-bearing samples were analyzed by dose calibrator and LSA. Values and uncertainties shown on individual run results are the mean ± 1s obtained between the two analytical instruments. When the average result over multiple 211At processing runs are reported, the uncertainty is the sample standard deviation (±1s) across that set of runs.
Bismuth analysis
The total mass of Bi in the isolated DIPE phase was determined following analysis of each sample by ICP-OES after undergoing the sample preparation method shown in Table 6.
Resulting solutions were analyzed using an iCap 6500 Duo (Thermo Scientific, Waltham, MA) ICP-OES with analysis performed in axial mode. An eight-point calibration curve was prepared for the Bi analysis by gravimetric dilutions of a 1000 µg/mL certified Bi standard purchased from High Purity Standards (Charleston, SC). Calibration curves used for the Bi standard concentrations had regression coefficients ≥0.9997; wavelengths of 190.2341, 222.8203, and 223.0602 nm were used in the analysis46. Analytical results were typically reported as being within ±5% at 2σ.
References
Ferrier, M. G., Radchenko, V. & Wilbur, D. S. Radiochemical aspects of alpha emitting radionuclides for medical application. Radiochim Acta 107, 1065–1085, https://doi.org/10.1515/ract-2019-0005 (2019).
Ferrier, M. G. & Radchenko, V. An Appendix of Radionuclides Used in Targeted Alpha Therapy. Journal of Medical Imaging and Radiation Sciences 50, Suppl. 1, S58–S65, https://doi.org/10.1016/j.jmir.2019.06.051 (2019).
Elgqvist, J., Frost, S., Pouget, J.-P. & Albertsson, P. The potential and hurdles of Targeted Alpha Therapy - Clinical trials and beyond. Front Oncol 3, https://doi.org/10.3389/fonc.2013.00324 (2014).
Sandrine, H.-M., Cyrille, A., Nicolas, V., Cathy, S. C. & Jacques, B. Alpha-Emitters for Immuno-Therapy: A Review of Recent Developments from Chemistry to Clinics. Curr Top Med Chem 12, 2642–2654, https://doi.org/10.2174/1568026611212230002 (2012).
Wilbur, D. S. Chemical and radiochemical considerations in radiolabeling with α-emitting radionuclides. Curr Radiopharm 4, 214–247, https://doi.org/10.2174/1874471011104030214 (2011).
National Nuclear Data Center. Information extracted from the NuDat 2 database, www.nndc.bnl.gov (2019).
Zalutsky, M. R. et al. Clinical experience with α-particle–emitting 211At: Treatment of recurrent brain tumor patients with 211At-labeled chimeric antitenascin monoclonal antibody 81C6. J Nucl Med 49, 30–38, https://doi.org/10.2967/jnumed.107.046938 (2008).
Andersson, H. et al. Intraperitoneal α-particle radioimmunotherapy of ovarian cancer patients: Pharmacokinetics and dosimetry of 211At-MX35 F(ab′)2—A phase I study. J Nucl Med 50, 1153–1160, https://doi.org/10.2967/jnumed.109.062604 (2009).
Fred Hutch/University of Washington Cancer Consortium., www.cancerconsortium.org (2019).
U.S. National Library of Medicine. Clinical Trials Database, www.clinicaltrials.gov (2019).
Gagnon, K. et al. Design and evaluation of an external high-current target for production of 211At. J Labelled Compd Radiopharm 55, 436–440, https://doi.org/10.1002/jlcr.2968 (2012).
Zalutsky, M. R. & Pruszynski, M. Astatine-211: Production and availability. Curr Radiopharm 4, 177–185, https://doi.org/10.2174/1874471011104030177 (2011).
Groppi, F. et al. Optimisation study of α-cyclotron production of At-211/Po-211g for high-LET metabolic radiotherapy purposes. Appl Radiat Isot 63, 621–631, https://doi.org/10.1016/j.apradiso.2005.05.041 (2005).
Lambrecht, R. M. & Mirzadeh, S. Cyclotron isotopes and radiopharmaceuticals- XXXV. Astatine-211. Int J Appl Radiat Isot 36, 443–450, https://doi.org/10.1016/0020-708X(85)90207-8 (1985).
Lindegren, S., Bäck, T. & Jensen, H. J. Dry-distillation of astatine-211 from irradiated bismuth targets: a time-saving procedure with high recovery yields. Appl Radiat Isot 55, 157–160, https://doi.org/10.1016/s0969-8043(01)00044-6 (2001).
Barton, G. W., Ghiorso, A. & Perlman, I. Radioactivity of Astatine Isotopes. Phys Rev 82, 13, https://doi.org/10.1103/PhysRev.82.13 (1951).
Aneheim, E. et al. Automated astatination of biomolecules – a stepping stone towards multicenter clinical trials. Scientific Reports 5, 12025, https://doi.org/10.1038/srep12025 (2015).
Appelman, E. H. The Oxidation States of Astatine in Aqueous Solution. J Am Chem Soc 83, 805–807, https://doi.org/10.1021/ja01465a014 (1961).
Johnson, G. L., Leininger, R. F. & Segre, E. Chemical Properties of Astatine. I. J Chem Phys 17, 1–10, https://doi.org/10.1063/1.1747034 (1949).
Neumann, H. M. Solvent Distribution Studies of the Chemistry of Astatine. J Inorg Nucl Chem 4, 349–353, https://doi.org/10.1016/0022-1902(57)80018-9 (1957).
Balkin, E. R. et al. Evaluation of a wet chemistry method for isolation of cyclotron produced [211At]astatine. Applied Sci 3, 636–655, https://doi.org/10.3390/app3030636 (2013).
Appelman, E. H. The Radiochemistry of Astatine. Report No. NAS-NS 3012, 37 (National Academy of Sciences National Research Council, Washington D.C., 1960).
Meyer, G. J. Astatine. J Labelled Compd Radiopharm 61, 154–164, https://doi.org/10.1002/jlcr.3573 (2018).
Ruth, T. J., Dombsky, M., D’Auria, J. M. & Ward, T. E. Radiochemistry of Astatine. Report No. NAS-NS 3064, 92 (National Academy of Sciences Nuclear Science Series, 1988).
Zona, C. et al. Wet-chemistry method for the separation of no-carrier-added 211At/211gPo from 209Bi target irradiated by alpha-beam cyclotron. J Radioanal Nucl Chem 276, 819–824, https://doi.org/10.1007/s10967-008-0638-2 (2008).
Kslik, V. S. Solvent Extraction: Classical and Novel Approaches. 1st edn, (Elsevier, 2011).
Rydberg, J., Cox, M., Musikas, C. & Choppin, G. R. Solvent Extraction Principles and Practice. 2nd edn, (CRC Press, 2004).
Rodríguez, R., Avivar, J., Leal, L. O., Cerdà, V. & Ferrer, L. Strategies for automating solid-phase extraction and liquid-liquid extraction in radiochemical analysis. Trends Anal Chem 76, 145–152, https://doi.org/10.1016/j.trac.2015.09.009 (2016).
Martini, P. et al. A solvent-extraction module for cyclotron production of high-purity technetium-99m. Appl Radiat Isot 118, 302–307, https://doi.org/10.1016/j.apradiso.2016.10.002 (2016).
Jordan, S., Moshiri, B. & Durand, R. Automation of Liquid-Liquid Extraction Using Phase Boundary Detection. JALA-J Lab Autom 7, 74–77, https://doi.org/10.1016/s1535-5535-04-00178-9 (2002).
Maslana, E., Schmitt, R. & Pan, J. A fully automated liquid-liquid extraction system utilizing interface detection. J Autom Methods Manag Chem 22, 187–194, https://doi.org/10.1155/s146392460000033x (2000).
Rider, J. A. Electrochemical Polarization in Isopropyl Ether. J Chem Phys 23, 61–64, https://doi.org/10.1063/1.1740564 (1955).
Haynes, W. M. Molar Conductivity of Aqueous HF, HCl, HBr, and HI in Handbook of Chemistry and Physics (ed Haynes, W. M) 5–75 (CRC Press, 2014–2015).
Haynes, W. M. Electrical Conductivity of Aqueous Solutions in Handbook of Chemistry and Physics (ed Haynes, W. M.) 5-73 (CRC Press, 2014–2015).
O’Hara, M. J., Murray, N. J., Carter, J. C., Kellogg, C. M. & Link, J. M. Hydroxamate column-based purification of zirconium-89 (89Zr) using an automated fluidic platform. Appl Radiat Isot 132, 85–94, https://doi.org/10.1016/j.apradiso.2017.10.048 (2018).
O’Hara, M. J., Murray, N. J., Carter, J. C. & Morrison, S. S. Optimized anion exchange column isolation of zirconium-89 (89Zr) from yttrium cyclotron target: Method development and implementation on an automated fluidic platform. J Chromatogr A 1545, 48–58, https://doi.org/10.1016/j.chroma.2018.02.053 (2018).
O’Hara, M. J., Murray, N. J., Carter, J. C., Kellogg, C. M. & Link, J. M. Tandem column isolation of zirconium-89 from cyclotron bombarded yttrium targets using an automated fluidic platform: Anion exchange to hydroxamate resin columns. J Chromatogr A 1567, 37–46, https://doi.org/10.1016/j.chroma.2018.06.035 (2018).
O’Hara, M. J. et al. An automated flow system incorporating in-line acid dissolution of bismuth metal from a cyclotron irradiated target assembly for use in the isolation of astatine-211. Appl Radiat Isot 122, 202–210, https://doi.org/10.1016/j.apradiso.2017.02.001 (2017).
Christensen, A. N., Chevallier, M.-A., Skibsted, J. & Iversen, B. B. Synthesis and characterization of basic bismuth(III) nitrates. J Chem Soc, Dalton Trans, 265–270, https://doi.org/10.1039/A908055D (2000).
Gattow, G. & Kiel, G. Über Wismutnitrate. IV. Darstellung und Eigenschaften von Bi(NO3)3 · 5 H2O. Zeitsch anorg allgemeine Chem 335, 61–73, https://doi.org/10.1002/zaac.19653350106 (1965).
Miersch, L., Rüffer, T., Schlesinger, M., Lang, H. & Mehring, M. Hydrolysis Studies on Bismuth Nitrate: Synthesis and Crystallization of Four Novel Polynuclear Basic Bismuth Nitrates. Inorg Chem 51, 9376–9384, https://doi.org/10.1021/ic301148p (2012).
Christensen, A. N. et al. In-situ X-ray powder diffraction studies of hydrothermal and thermal decomposition reactions of basic bismuth(iii) nitrates in the temperature range 20–650 °C. Dalton Trans, 3278–3282, https://doi.org/10.1039/B303926A (2003).
Naidoo, E. B. & van Staden, J. F. Super serpentine reactors in SIA. A comparative response and precision study. Instrum Sci Technol 29, 77–89, https://doi.org/10.1081/CI-100103456 (2001).
Liawruangrath, S., Som-aum, W. & Townshend, A. A comparison of enrichment factor of knotted and serpentine reactors using flow injection sorption and preconcentration for the off-line determination of some trace elements by inductively coupled plasma mass spectrometry. Talanta 58, 1177–1184, https://doi.org/10.1016/S0039-9140(02)00415-0 (2002).
Waiz, S. et al. Dispersion in open tubular reactors of various geometries. Anal Chim Acta 428, 163–171, https://doi.org/10.1016/S0003-2670(00)01230-7 (2001).
NIST Physical Measurement Laboratory. Information extracted from the Handbook of Basic Atomic Spectroscopic Data; NIST Standard Reference Database 108, https://www.nist.gov/pml/handbook-basic-atomic-spectroscopic-data (2019).
Acknowledgements
This research is supported by the U.S. Department of Energy Isotope Program, managed by the Office of Science for Nuclear Physics. PNNL was funded under grant LAB-12-743 and UW was funded under grant DE-SC0018013. The Pacific Northwest National Laboratory is operated for the U.S. Department of Energy by Battelle under contract DE-AC06-67RLO 1830.
Author information
Authors and Affiliations
Contributions
M.J.O. conceived and proposed the project in collaboration with D.S.W., and he oversaw system design, development, programming and conduct of experiments. A.J.K. co-developed the system and programming, and assisted with conduct of experiments. D.K.H. facilitated system set-up in UW glovebox and conducted manual solvent extraction experiments. Y.L. and D.K.H. performed radionuclide product quality assessments. E.F.D. operated the cyclotron to produce the radionuclide. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
O’Hara, M.J., Krzysko, A.J., Hamlin, D.K. et al. Development of an autonomous solvent extraction system to isolate astatine-211 from dissolved cyclotron bombarded bismuth targets. Sci Rep 9, 20318 (2019). https://doi.org/10.1038/s41598-019-56272-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-56272-7
This article is cited by
-
Guest Edited Collection: Radioisotopes and radiochemistry in health science
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
