
Abstract
In epilepsy patients, drug-resistant seizures often originate in one of the temporal lobes. In selected cases, when certain requirements are met, this area is surgically resected for therapeutic reasons. We kept the resected tissue slices alive in vitro for 48 h to create a platform for testing a novel treatment strategy based on neuropeptide Y (NPY) against drug-resistant epilepsy. We demonstrate that NPY exerts a significant inhibitory effect on epileptiform activity, recorded with whole-cell patch-clamp, in human hippocampal dentate gyrus. Application of NPY reduced overall number of paroxysmal depolarising shifts and action potentials. This effect was mediated by Y2 receptors, since application of selective Y2-receptor antagonist blocked the effect of NPY. This proof-of-concept finding is an important translational milestone for validating NPY-based gene therapy for targeting focal drug-resistant epilepsies, and increasing the prospects for positive outcome in potential clinical trials.
Similar content being viewed by others

Introduction
Epilepsy is a multifactorial neurological disease characterised by increased excitability in the brain, leading to pathological hyper-synchronised activity of neurons manifested as spontaneous recurrent seizures (SRS). In a majority of cases, the symptomatic treatment by currently available anti-epilepsy drugs (AEDs) eliminates SRS under the medication period1. However, AEDs have unwanted side effects, and most importantly, are ineffective in about 30–40% of epilepsy patients2,3. The temporal lobe is one of the most common structures for focal seizure origin, and about a third of these patients are AED resistant1. These patients are considered for resection of the epileptic tissue, provided the seizure-generating focal area is reliably identified and its removal poses no substantial risk for post-operative functional deficit. Once resected, the temporal lobe is taken for histopathological examination. The therapeutic surgery also provides a unique opportunity to use parts of the resected epileptic tissue for functional studies in vitro, utilizing various electrophysiological methodologies. These studies help in understanding pathophysiological mechanisms of ictogenesis and allow for testing of novel treatment strategies in human epileptic tissue4,5,6.
Neuropeptide Y (NPY) is a 36 amino acid long peptide7 widely expressed in the brain and released from presynaptic terminals mostly during high-frequency activity8,9. Overexpression of NPY has been shown to be highly effective at suppressing epileptic activity in various animal models of both acute and chronic seizures10,11,12,13,14 (Fig. 1). This effect of NPY on seizures is thought to occur through the activation of presynaptic NPY Y2 receptors11,15,16 that inhibit voltage gated calcium channels17, thereby reducing glutamate release probability in excitatory synapses18,19. The reduction in glutamate release during NPY application have also been documented in dentate granule cells during electrical stimulation of the lateral perforant path in human brain slices20. Previously, we have also shown that Y2 receptors are present in human hippocampal tissue resected from temporal lobe of drug-resistant epilepsy patients and that exogenous application of NPY decrease basal synaptic transmission in these slices through activation of Y2 receptors6. This and other studies suggested that gene therapy based on overexpression of NPY alone or in combination with Y2 receptors might provide an alternative treatment strategy for drug-resistant temporal lobe epilepsy (TLE)6,11,21. Although it has been shown that NPY application reduces glutamate release in human drug-resistant epileptic tissue6,20, an important step toward a clinical gene therapy trial (Fig. 1, step 3), namely direct evidence that NPY can also suppress seizure-like activity in human hippocampal slices, is still lacking.

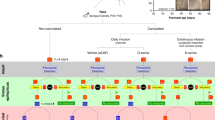
Translational roadmap for the use of human brain tissue as a step between animal research and clinical trials. Schematic illustration of how human-tissue resected from patients with epilepsy can be used to validate the effect of treatments found in animal models before proceeding to clinical trials for drug-resistant epilepsy, exemplified by NPY. (1) Basic research, with in vitro models, from several labs has shown that NPY has an inhibitory effect on epileptiform activity. (2) Chronic epilepsy models in vivo, such as the intrahippocampal kainate model, has provided key evidence demonstrating that NPY has an anti-epileptic effect in the chronic epileptic brain over a longer period of time. (3) The present study is a crucial validation step showing that NPY has an inhibitory effect on epileptiform activity in the target tissue, drug-resistant human hippocampal slices, minimising the risk for the next step of first-in-man clinical phase 1–2 study (step 4). The step 2 is interchangeable with step 3.
We addressed this question by inducing epileptiform seizure-like activity in human hippocampal slices while performing whole-cell patch-clamp recordings in granule cells of the dentate gyrus and tested the inhibitory effect of NPY on this activity.
We demonstrate, for the first time, that NPY effectively suppresses epileptiform discharges in human drug-resistant hippocampal slices, providing direct evidence that increased NPY signalling in human epileptic tissue is capable of inhibiting seizure-like activity.
Results
Resected human hippocampal tissue
Hippocampal tissue samples from ten patients undergoing surgery for drug-resistant TLE were collected for this study with their medical characteristics summarized in Table 1. Neuropathological examination of resected tissue with respect to hippocampal sclerosis and other pathologies were performed on hematoxylin-eosin- (H&E, Fig. 2a–c) and microtubule-associated protein 2- (MAP2, Fig. 2d–f) stained paraffin sections. The majority of resected tissue displayed hippocampal sclerosis (Table 1). Various degrees of degeneration were observed in the dentate gyrus (Fig. 2b,e) and in CA1 areas (Fig. 2c,f). Neuronal loss was particularly pronounced in the CA1 pyramidal cell layer. Within the dentate gyrus, neuronal cell loss was seen as a thinning of the granule cell layer towards both the hilus and molecular layer. No oedema was detected, indicating that these changes did not occur acutely, in the immediate time period prior to resection.

Neuropathological evaluation and granule cell layer with biocytin-filled cell: Example of (a–c) H&E and (d–f) MAP2 staining in hippocampal tissue resected from a patient with TLE. The sectioning and staining are made for neuropathological evaluation showing (a,d) the whole slice including dentate gyrus, CA3-CA1 and subiculum. The electrophysiological experiments were performed in the dentate gyrus magnified in B and E with the granule cell layer (gcl) identified, and further magnification inset showing individual granule cells. (c,f) In CA1, the neuronal cell loss is clear, the pyramidal cell layer has completely disappeared. After electrophysiological experiments the slices were fixated in PFA and later stained for (g) MAP2 (red) showing the granule cell layer with the recorded biocytin-filled granule cell (in green). (h) Higher magnification of the recorded neuron with typical granule cell morphology. Scale bar: 2 mm in a and d, 150 µm in b, c, e and f with 50 µm in inset, 200 µm in g and 50 µm in h.
NPY increases inter-spike interval, decreases number of paroxysmal depolarising shifts and the total number of action potentials
After 24 hours of incubation in the interface chamber, the slices were individually transferred to the dual-flow recording chamber. After acquiring whole-cell patch-clamp configuration of the dentate granule cell, resting membrane potential (RMP) was measured in continuous current-clamp mode to an average of −67.22 ± 1.16 mV (Table 2). RMP, intrinsic properties of the recorded cells (Table 2), their morphology and position in the dentate gyrus (Fig. 2a,b) confirmed that they were granule cells.
[0Mg2+]/4-aminopyridine (4-AP) – artificial cerebrospinal fluid (aCSF) solution was bath applied until stable epileptiform activity over several minutes was achieved, as judged by the appearance of regular paroxysmal depolarising shifts (PDS) in granule cells (Fig. 3a). Subsequently, NPY-[0Mg2+]/4-AP-aCSF was applied during 10 min, followed by a 10–30 min period of wash-out without NPY (Fig. 3a). To assess the possible effect of NPY on epileptiform activity, we measured (i) the time between action potentials (AP, inter-spike intervals), (ii) the number of PDS-events, (iii) the total number of APs and (iv) the average number of APs during individual PDS-events. These parameters were compared before, during and after NPY (Fig. 3) or NPY + BIIE0246 (Fig. 4) application (termed as baseline, NPY, NPY + BIIE0246 and washout periods, respectively). The results demonstrate that there was a significant increase in inter-spike intervals during NPY application, as measured by the Kolmogorov-Smirnov test of the cumulative probability (summarised cumulative probability plot of inter-spike interval: comparing baseline, n = 12, with NPY, n = 12, p < 0.0001, D = 0.1833, Kolmogorov-Smirnov test, Fig. 3e).

NPY-application suppresses epileptiform activity in human drug-resistant hippocampal slices. (a) Example recordings from dentate granule cells during epileptiform activity triggered by 0Mg2+/4-AP aCSF. Whole-cell recordings in current-clamp mode, arrows indicate the PDSs that are displayed in expanded time-scale to the right of each sweep. NPY application resulted in a marked reduction of APs and PDS frequencies compared to the baseline, and partial return to the baseline levels during the washout period. (b–d) The average values (±SEM) are displayed in orange, the individual values – in grey. (b) The number of PDS was reduced during the application of NPY (baseline: 47.42 ± 10.52 and NPY: 17.6 ± 6.06, n = 12, t-test, p = 0.0022). (c) The number of APs was reduced during the application of NPY (baseline: 1042 ± 205.1 and NPY: 371.8 ± 71.49, n = 12, t-test, p = 0.0013). (d) The number of APs during individual PDSs did not change during the application of NPY (baseline: 6.067 ± 1.66 and NPY: 7.94 ± 2.786, n = 12, t-test, p = 0.9436). (e) Cumulative plot of the time-interval between APs in baseline recordings (black) and recordings during NPY application (blue). A significant difference between baseline and NPY (p < 0.0001, D = 0.1833) was detected with the Kolmogorov-Smirnov test.

NPY application together with its Y2-receptor antagonist BIIE0246 has no effect on epileptiform activity. (a) Example recordings from dentate granule cells during epileptiform activity triggered by 0Mg2+/4-AP aCSF. Whole-cell recordings in current-clamp mode, arrows indicate the PDSs that are displayed in zoomed-in time-scale to the right of each sweep. NPY applied together with the Y2 receptor antagonist BIIE0246; under these conditions a reduction in AP or PDS frequency is not observed. (b–d) The average values (±SEM) are displayed in orange, the individual values – in grey. (b) The number of PDS is unchanged during the application of NPY + BIIE0246 (baseline: 35.75 ± 12.33 and NPY + BIIE0246: 26 ± 12.27, n = 8, t-test, p = 0.0834). (c) The number of APs is unchanged during the application of NPY + BIIE0246 (baseline: 633.6 ± 232.8 and NPY + BIIE0246: 650.4 ± 265.3, n = 9, t-test, p = 0.874). (d) The number of APs during the individual PDSs is unchanged during the application of NPY + BIIE0246 (baseline: 4.674 ± 1.028 and NPY + BIIE0246: 5.873 ± 1.361, n = 7, t-test, p = 0.094). (e) Cumulative plot of the time-interval between APs during the baseline recordings (black) and recordings during NPY + BIIE0246 application (blue). No difference between baseline and NPY + BIIE0246 (p = 0.2743, D = 0.0794) was detected with the Kolmogorov-Smirnov test.
We also observed a 60% reduction in the number of PDS-events during NPY application as compared to baseline (during baseline 47.4 ± 10.52 and NPY 17.5 ± 6.06, t(11) = 3.974, p = 0.0022. n = 12, t-test, Fig. 3b). Similarly, the total number of APs during the whole 300 s long analysis period was significantly decreased by 60% during NPY application as compared to baseline (during baseline 1042 ± 205.1 and NPY 371.8 ± 71.49, t(11) = 4.295, p = 0.0013. n = 12, t-test, Fig. 3c). In a subset of the cells, with successful recordings long enough to monitor the washout period, the number of PDS-events returned to baseline levels during washout (during baseline 44.3 ± 13.94 and washout 42.7 ± 21.44, t(6) = 1.115, p = 0.3075. n = 7, t-test, Fig. 3b). Similarly, the total number of APs was not different between baseline and washout (during baseline 853.5 ± 246.8 and washout 712.8 ± 235.1, t(7) = 0.2314, p = 0.8236. n = 8, t-test, Fig. 3c). In contrast, the average number of APs during each PDS-events was not significantly altered by NPY treatment (during baseline 6.1 ± 1.66 and NPY 7.9 ± 2.79, t(11) = 0.07241, p = 0.9436. n = 12, t-test, Fig. 3d).
Application of Y2 receptor antagonist blocks the effect of NPY
We hypothesised that the observed effect of NPY was due to decreased glutamate release from presynaptic terminals by activation of presynaptic Y2 receptors6,20. To address this question, NPY was applied together with the Y2 receptor antagonist BIIE0246 in a set of experiments (Fig. 4a). No significant difference could be detected in the inter-spike intervals during baseline and NPY + BIIE0246 application (summarised cumulative probability plot of inter-spike interval: comparing baseline, n = 9, with NPY + BIIE0246, n = 9, p < 0.2743, D = 0.0794, Kolmogorov-Smirnov test, Fig. 4e). Similarly, the number of PDS-events (during baseline 35.8 ± 12.33 and NPY + BIIE0246 26 ± 12.27, t(7) = 2.018, p = 0.0834. n = 8, t-test, Fig. 4b) or the number of AP (during baseline 633.6 ± 232.8 and NPY + BIIE0246 650.4 ± 265.3, t(8) = 0.1637, p = 0.874. n = 9, t-test, Fig. 4c) did not differ significantly between baseline and NPY + BIIE0246 application. The number of AP in each PDS was also not different from baseline (during baseline 4.7 ± 1.03 and NPY + BIIE0246 5.87 ± 1.36, t(6) = 1.988, p = 0.094. n = 7, t-test, Fig. 4d).
To verify that PDSs recorded in whole-cell mode in dentate granule cells reflected on-going epileptiform activity in the dentate gyrus, field postsynaptic potentials were recorded simultaneously with the whole-cell patch-clamp recordings in eight of the 21 slices, (three with NPY application and five with NPY + BIIE0246 application). Results showed that field epileptiform discharges in the granule cell layer corresponded with the PDS-events observed in whole-cell patch-clamp recordings (Fig. 5). There was a high degree of correlation between the number of PDSs in individual neurons and respective field potential discharges (r = 0.9643, p = 0.0028. n = 7, Spearman correction, Fig. 5a,d). Moreover, cross-correlation analysis revealed a time-locked temporal relationship between the PDSs in individual neurons and field discharges (Fig. 5b,c). In all three slices with NPY application, a decrease in the number of field epileptiform bursts was observed (139.7 ± 87.04 bursts during baseline and 50 ± 22.28 bursts during NPY, n = 3). In slices with NPY + BIIE0246 application, there was no overall average decrease in number of bursts during the application period (17 ± 12.03 bursts during baseline and 22 ± 14.54 bursts during NPY + BIIE0246, n = 5). These data support that PDSs recorded in individual neurons corresponded to epileptiform discharges in the dentate gyrus, thereby providing a reliable read-out for experimental outcomes, and suggesting suppression of epileptiform activity in dentate gyrus by NPY.

Simultaneous whole-cell current-clamp and field-recordings in the dentate gyrus during NPY application. (a) Representative recordings showing simultaneous whole-cell current-clamp recording and a field recording during baseline recording, NPY application, and washout period. Whole-cell recording from individual granule cell demonstrating 4-AP induced PDSs that are time-locked with field potential bursting. Both PDS and corresponding field potential bursting showed parallel reductions in frequency during NPY application. (b) Representative recordings showing an unfiltered PDS (top), a PDS low-pass filtered at 40 Hz with the best fit indicated by the red line (middle) and a low-pass filtered at 40 Hz field potential (bottom). (c) Cross-correlation analysis of whole-cell and field recordings showing a significant degree of time correlation, with the cross-correlation function estimate peaking close to 0 ms. The red trace is the average correlation estimate across recordings, the grey shadow represents SEM. (d) The number of LFPs plotted against the number of PDSs for each simultaneous whole-cell and field recording with the calculated Deming regression line plotted. The correlation between the number of LFPs and the PDSs is significant (p = 0.0028, n = 7, r = 0.9643) and the calculated regression line intersects the x-axis at 10.5.
Taken together, our results show that application of NPY significantly reduces on-going epileptiform activity in dentate granule cells of human hippocampal slices obtained from drug-resistant patients with TLE. The anti-epileptiform effect of NPY was no longer observed in the presence of the NPY Y2 receptor antagonist BIIE0246, suggesting that the Y2 receptor is mediating this NPY effect, most likely by decreasing glutamate release.
Discussion
We demonstrate that NPY application, in hippocampal slices surgically resected from patients with drug-resistant TLE, significantly reduces chemically induced epileptiform activity in the dentate gyrus. This novel finding suggests that in epileptic brain tissue, where conventional AEDs are ineffective, increasing the levels of NPY could be an alternative approach to achieve a therapeutic effect and suppress seizure activity.
Hippocampal slices from patients with drug-resistant TLE are emerging as an important in vitro platform to test new promising treatments established in animal models. It is highly warranted to test novel treatments for efficacy in human brain tissue resistant to conventional AEDs22,23. The positive outcome in epileptic resected tissue would increase the likelihood that the approach will be replicated in patients with drug-resistant epilepsy.
The 4-AP-in vitro model is widely used to induce epileptiform activity in the rodent as well as human temporal lobe slices24,25,26. 4-AP mainly acts by blocking voltage-gated potassium channels of the Kv-1 family, thereby broadening APs and leading to increased glutamate and GABA-release27,28,29. In previous studies, 4-AP application alone was unable to initiate interictal epileptiform activity in the human dentate gyrus22,30. However, when combined with low Mg2+ and/or high potassium, 4-AP induced stable interictal epileptiform bursting in the dentate gyrus24 observed with field recordings.
The effect of NPY on seizure-like activity was evaluated by whole-cell patch-clamp recordings of dentate granule cells, where effect on epileptiform activity was assessed by quantification of PDSs, cumulative probability of inter-spike intervals, plotted over time, as well as overall number of APs (the latter as a relative measure of cellular excitability). In all of the above-mentioned parameters we observed statistically significant changes during NPY application, indicating decreased excitability. Since we also demonstrate, as in several previous studies31,32,33, the correlation between PDSs in individual neurons with field discharges from the same area, one could conclude that NPY application has an inhibitory effect on epileptiform activity in the dentate gyrus of drug-resistant human epileptic tissue.
These in vitro findings in human hippocampal slices could be extrapolated, with certain degree of confidence, to the human brain in vivo. Indeed, there is a strong correlation between what has been observed in in vitro animal models of epileptiform activity with in vivo (both acute and chronic) epilepsy models in terms of NPY-mediated inhibition of seizure activity. We and others have shown that application of NPY onto rodent hippocampal slices has profound inhibitory effect on epileptiform activity induced by various means, including 4-AP10,11,12,13,14. These findings were validated and confirmed in vivo: It has been shown e.g. that overexpression of NPY bilaterally in the hippocampus significantly attenuates acute status epilepticus9, as well as decreases the SRSs after status epilepticus34. We have also shown that combined overexpression of NPY and its Y2 receptor bilaterally in the hippocampus has seizure-suppressant effect in electrical kindling and systemic Kainate models of acute seizures21. This seizure-suppressant effect in animals has been shown to be mediated mostly via activation of Y2 receptors15,21. Moreover, we have recently demonstrated that simultaneous unilateral overexpression of NPY and its Y2 receptor in the hippocampus has a disease-modifying effect, halting and even reversing the progressive increase of SRS frequency in the post-status epilepticus chronic epilepsy model (intrahippocampal kainate model)11. Thus, akin to rodent data our present in vitro study in human hippocampal slices predict that NPY might also exert its seizure-inhibitory effect in human chronic TLE. This hypothesis was supported by our previous study showing that NPY acting on Y2 receptors is responsible for attenuation of excitatory synaptic transmission in human drug-resistant epileptic hippocampal slices6. The important role of the Y2 receptors in suppressing seizure-like activity in human hippocampal tissue is now demonstrated for the first time in the present study.
What is the possible mechanism of seizure-suppressant effect of NPY in human epileptic hippocampal slices? One explanation would be analogous to what has been proposed in rodent studies: exogenous NPY acts via activation of Y2 receptors located on the presynaptic terminals of excitatory glutamatergic synapses to decrease glutamate release by negatively regulating voltage-gated calcium channels15,17. This ultimately leads to attenuation of epileptiform activity, which often is driven by excitatory glutamatergic transmission, particularly the fast interictal-like bursts in the 4-AP model27. Similarly, interictal discharges in human epileptic (subicular) slices has been shown to depend on glutamatergic (as well as GABAergic) signalling35. Our previous findings in human epileptic hippocampus demonstrate significant reduction of excitatory postsynaptic potentials in principal neurons by NPY application6. This effect was dependent on activation of presynaptic Y2 receptors on glutamatergic synapses, since addition of the specific Y2 receptor antagonist BIIE0246 blocked the effect6. In the same study, we also demonstrated that Y2 receptors are present and functional in human epileptic hippocampal tissue further supporting that in human epileptic brain tissue, NPY suppress epileptiform activity by decreasing glutamate release from excitatory presynaptic terminals.
The magnitude of the NPY effect in human slices was quite pronounced, showing on average 60% reduction in all but one of the parameters utilised to characterise epileptiform activity. Epileptiform activity, however, was not stopped completely. A possible explanation for this incomplete inhibition could be that 4-AP increases neurotransmitter release from both glutamatergic and GABAergic presynaptic terminals27, were GABA may provide depolarising synaptic inputs to principal neurons when altered distribution of Cl− ions across the membrane lead to reversal of chloride currents36,37 (reviewed by38). If one assumes that NPY predominantly affects glutamatergic inputs, the GABAergic inputs may still be responsible for driving remaining epileptiform activity.
Another interpretation could be that the partial effect of NPY is due its insufficient concentration within slices and/or in the perfusion medium. Indeed, we have observed only partial blockade of excitatory synaptic transmission both in dentate gyrus and in CA1 of the human hippocampus by the same NPY concentration in the aCSF6. One could speculate that an increasing NPY concentration in the perfusion medium may have more pronounced inhibitory effect on epileptiform activity.
In translationally designed animal experiments, reflecting possible future clinical scenarios, NPY/Y2 receptor based gene therapy has been shown to inhibit SRSs and therefore proposed as an alternative treatment strategy for focal epilepsies, particularly drug-resistant epilepsies, such as in patients undergoing resective surgery11. In this context, the present study takes a major decision-making step for a future gene therapy approach when demonstrating for the first time that NPY is able to suppress epileptiform activity in drug-resistant epileptic tissue from patients with TLE. Lastly, this study underscores the value of the unique possibility provided by epilepsy surgery to validate novel treatment strategies in human drug-resistant epileptic tissue, thereby potentially minimising the risks for failure when proceeding to costly and time-consuming clinical trials (Fig. 1).
Methods
Study design
The aim of this study was to test if NPY can have a seizure-suppressant effect in human hippocampal tissue, resected from patients with drug-resistant epilepsy. For the whole study, we collected hippocampal tissue from 10 patients (eight from Copenhagen and two from Lund) undergoing surgical treatment for drug-resistant epilepsy between years 2016 and 2017 (Table 1). The use of tissue from epilepsy surgery patients for research were approved by the local Ethical Committee in Copenhagen (H-2-2011-104) and Lund (#212/2007) and was performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from all subjects prior to each surgery. First, epileptiform activity was induced with [0Mg2+]/4-AP-aCSF and recorded with whole-cell patch-clamp technique on granule cells in the dentate gyrus. Subsequently, NPY was added to the [0Mg2+]/4-AP-aCSF, and changes in epileptiform activity were recorded and later analysed. To further investigate the mechanism behind the anti-seizure effect of NPY we focused on the Y2 receptor. In these experiments, epileptiform activity was induced as before but the Y2 receptor antagonist BIIE0246 was added together with NPY. Post-surgical morphological evaluation of the hippocampus was performed by a pathologist at the respective hospitals, and diagnosis for hippocampal sclerosis was determined according to ILAE guidelines39.
Slicing and incubation
A detailed description of the methods for slicing and incubation of human brain tissue in the interface chamber can be found in our previous publication40. In short, we transported the tissue from the operating room to the electrophysiology laboratory in an ice-cold sucrose-based slush containing in mM: 200 sucrose, 21 NaHCO3, 10 glucose, 3 KCl, 1.25 NaH2PO4, 1.6 CaCl2, 2 MgCl2, 2 MgSO4 (all from Sigma-Aldrich, Sweden), adjusted to 300–310 mOsm, pH 7.4. At the laboratory, the tissue was then transferred into the same type of sucrose slush, continuously bubbled with 95% O2 and 5% CO2. The tissue, as a whole or parts of the tissue, was then glued onto the cutting platform and placed in a vibratome (VT1200, Leica Microsystems). The hippocampus was sliced in the coronal direction with a slice thickness of 400 µm, while submerged in the sucrose-based slush. When cut, the slices rested fully submerged in a pre-incubation bath with 34 °C aCSF containing in mM: 129 NaCl, 21 NaHCO3, 10 glucose, 3 KCl, 1.25 NaH2PO4, 2 MgSO4, and 1.6 CaCl2, adjusted to 300–310 mOsm, pH 7.4 for 15–30 min before they were transferred to the interface incubation chamber. The closed interface incubation chamber contained humidified air, and the slices rested on cell culture insets (Millicell) fixed in a smaller open box with a constant flow of carbonated aCSF. The slices were incubated for approximately 24 hours in the interface chamber before electrophysiology recordings.
Electrophysiology
Slices were individually transferred to a dual-flow recording chamber and held in place by a horseshoe shaped flattened platinum wire. The slices were placed on a metal grid perfused with preheated 32 °C oxygenated aCSF with laminar flow directed above and below the slice at a flow rate of 2 ml/min/channel. Whole-cell recordings were performed from granule cells in the dentate gyrus with glass capillaries containing a AgCl-electrode, with a tip resistance between 2.5 and 6 MΩ when backfilled with a solution containing in mM: 122.5 K-gluconate, 12.5 KCl, 10 KOH-HEPES, 0.2 KOH-EGTA, 2 Mg-ATP, 0.3 Na3GTP, and 8 NaCl, pH 7.2–7.4 (mOsm 290–300). Pipettes were connected to a HEKA amplifier (HEKA, Germany) controlled with HEKA Patchmaster software. After formation of a Giga-seal, the patch was ruptured and whole-cell current-clamp recordings were performed. In some cases, field recordings in the granule cell layer, at least 500 µm apart from the patched granule cell, were simultaneously performed using glass capillaries with a tip resistance between 1 and 2 MΩ when backfilled with aCSF.
Epileptiform activity
In 31 hippocampal slices, 20 out of 39 whole-cell recorded dentate granule cells displayed stable epileptiform activity induced by [0Mg2+]/4-AP-aCSF. In these cases, application of NPY or NPY + BIIE0246 (a selective Y2 receptor blocker) was performed. In 12 of these cells, the recording was stable enough to also allow for their monitoring during the washout period. In eight of the 20 slices, field potentials and whole-cell recordings were simultaneously performed, with field electrodes positioned in the granule cell layer. Cells were recorded in current-clamp mode to monitor baseline activity for at least 15 min in normal aCSF, before switching to [0Mg2+]/4-AP-aCSF. The [0Mg2+]/4-AP-aCSF was prepared in the same way as normal aCSF but omitting Mg2+ and adding 4-AP, a blocker of the Kv1-family voltage-gated potassium channels, at a final concentration of 100 µM. A baseline recording with robust epileptiform activity in current clamp-mode was recorded for at least 10 min before applying NPY.
Administration of NPY and Y2 receptor antagonist
After baseline recordings in [0Mg2+]/4-AP-aCSF, the perfusion solution was switched to NPY-containing [0Mg2+]/4-AP-aCSF, in which C-terminally amidated NPY (dissolved in dH2O, batch: 16-05-2016, Schafer-N, Denmark) was added to a final concentration of 1 µM, all in a siliconized bottle to prevent NPY adhesion to the container walls. Continuous current-clamp recording was performed for 10 min, while perfusing slices with NPY-[0Mg2+]/4-AP-aCSF, followed by a washout period, in [0Mg2+]/4-AP-aCSF. In a subset of recordings, the Y2 receptor antagonist (BIIE0246, first dissolved in ethanol (0.25 mM) then dH2O, Tocris Bioscience, UK) was added to the NPY-[0Mg2+]/4-AP-aCSF to a final concentration of 0.6 µM.
Immunohistochemistry
Biocytin (Sigma-Aldrich) was added to the intracellular solution to label recorded cells and after completion of electrophysiological recordings, the slices were fixed in 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS) overnight and then stored submerged in Walter’s antifreeze solution (ethylene glycol and glycerol in PBS) at −20 °C. After rinsing the slices in potassium-PBS (KPBS), a 1-hour incubation in blocking solution containing 5% normal goat serum (NGS) and triton-KPBS (TKPBS) was followed by 48 hours incubation (at −4 °C) with mouse-MAP2 (1:200 Sigma, M2320) as primary antibody in 5% NGS. Slices were rinsed (3 × 10 min) with TKPBS followed by incubation with mouse-Cy3 (1:400 Jackson ImmunoResearch, 115-165-003) and streptavidin-Alexa488 (1:400, Invitrogen, S11223) as secondary antibodies in 5% NGS for 2 hours, rinsed (3 × 10 min) with KPBS and then mounted on glass slides and cover slipped with DABCO.
Data analysis and statistics
Electrophysiological recordings were analysed in IGOR Pro version 6.3 (Wavemetrics) and Minianalysis (Synaptosoft), while Prism (GraphPad) was used for further statistical analysis. The last 300 seconds of baseline, NPY, NPY + BIIE0246 and washout recordings were compared in the evaluation of epileptiform activity. In the first part of the analysis, Minianalysis and Prism were used to calculate the intervals between APs and to generate a cumulative plot on which the Kolmogorov-Smirnov test was applied to test if the plotted curves were significantly separated from each other. Further analysis of the recordings included counting of APs and PDSs. The APs were automatically detected by the Minianalysis software, using a detection threshold of 15 mV above baseline. The PDS were detected manually, defined by a depolarising ridge, which is not disrupted by repolarisation between APs, with the largest PDS in the baseline recording as a standard giving the inclusion parameter of 50% of largest PDS amplitude. All PDS with lower amplitude than 50% of the largest PDS in the baseline recording were excluded from analysis. The Ratio-based Paired t-test was used to compare the data from the baseline recording with the wash-in period of either NPY or NPY + BIIE0246. Differences between groups were considered statistically significant when p < 0.05. The correlation analysis between number field potentials and PDSs from the simultaneous whole-cell and field recordings were done with Spearman correction. The Deming regression line was calculated using the standard deviation from each group (number of field potentials, number of PDS). Cross-correlation analysis was performed in Clampfit software (Molecular Devices) using a 1-second sliding window across 10 minutes of recording. Traces from both the whole-cell and field recordings were filtered with two iterations of low-pass filtering at 40 Hz before the analysis, to reduce the contribution of action potentials to the shape of individual PDS.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Duncan, J. S., Sander, J. W., Sisodiya, S. M. & Walker, M. C. Adult epilepsy. Lancet 367, 1087–1100 (2006).
Perucca, P. & Gilliam, F. G. Adverse effects of antiepileptic drugs. Lancet Neurol. 11, 792–802 (2012).
Chen, Z., Brodie, M. J., Liew, D. & Kwan, P. Treatment outcomes in patients with newly diagnosed epilepsy treated with established and new antiepileptic drugs a 30-year longitudinal cohort study. JAMA Neurol. 75, 279–286 (2018).
Andersson, M. et al. Optogenetic control of human neurons in organotypic brain cultures. Sci. Rep. 6, 24818 (2016).
Avaliani, N., Andersson, M., Runegaard, A. H., Woldbye, D. & Kokaia, M. DREADDs suppress seizure-like activity in a mouse model of pharmacoresistant epileptic brain tissue. Gene Ther. 23, 760–766 (2016).
Ledri, M. et al. Differential effect of neuropeptides on excitatory synaptic transmission in human epileptic hippocampus. J. Neurosci. 35, 9622–9631 (2015).
Tatemoto, K., Carlquist, M. & Mutt, V. Neuropeptide Y-a novel brain peptide with structural similarities to peptide YY and pancreatic polypeptide. Nature 296, 659–660 (1982).
Marksteiner, J., Ortler, M., Bellmann, R. & Sperk, G. Neuropeptide Y biosynthesis is markedly induced in mossy fibers during temporal lobe epilepsy of the rat. Neurosci. Lett. 112, 143–148 (1990).
Vezzani, A. & Sperk, G. Overexpression of NPY and Y2 receptors in epileptic brain tissue: An endogenous neuroprotective mechanism in temporal lobe epilepsy? Neuropeptides 38, 245–252 (2004).
Sørensen, A. T. et al. Hippocampal NPY gene transfer attenuates seizures without affecting epilepsy-induced impairment of LTP. Exp. Neurol. 215, 328–333 (2009).
Nikitidou Ledri, L. et al. Translational approach for gene therapy in epilepsy: Model system and unilateral overexpression of neuropeptide Y and Y2 receptors. Neurobiol. Dis. 86, 52–61 (2016).
Powell, K. L. et al. Gene therapy mediated seizure suppression in genetic generalised epilepsy: neuropeptide Y overexpression in a rat model. Neurobiol. Dis. 113, 23–32 (2018).
Richichi, C. et al. Anticonvulsant and antiepileptogenic effects mediated by adeno-associated virus vector neuropeptide Y expression in the rat hippocampus. J. Neurosci. 24, 3051–3059 (2004).
Klapstein, G. J. & Colmers, W. F. Neuropeptide Y suppresses epileptiform activity in rat hippocampus in vitro. J Neurophysiol. 78, 1651–61 (1997).
El Bahh, B. et al. The anti-epileptic actions of neuropeptide Y in the hippocampus are mediated by Y2 and not Y5 receptors. Eur. J. Neurosci. 22, 1417–1430 (2005).
El Bahh, B., Cao, J. Q., Beck-Sickinger, A. G. & Colmers, W. F. Blockade of neuropeptide Y2receptors and suppression of NPY’s anti-epileptic actions in the rat hippocampal slice by BIIE0246. Br. J. Pharmacol. 136, 502–509 (2002).
Klapstein, G. J. & Colmers, W. F. 4-Aminopyridine and low Ca2+ differentiate presynaptic inhibition mediated by neuropeptide. October 470–474 (1992).
Greber, S., Schwarzer, C. & Sperk, G. Neuropeptide Y inhibits potassium-stimulated glutamate release through Y2 receptors in rat hippocampal slices in vitro. Br. J. Pharmacol. 113, 737–740 (1994).
Silva, A. P., Pinheiro, P. S., Carvalho, C. M. & Zimmer, J. Activation of neuropeptide Y receptors is neuroprotective against excitotoxicity in organotypic hippocampal slice cultures. FASEB J. 17, 1118–1120 (2003).
Patrylo, P. R., van den Pol, A. N., Spencer, D. D. & Williamson, A. NPY inhibits glutamatergic excitation in the epileptic human dentate gyrus. J. Neurophysiol. 82, 478–483 (1999).
Woldbye, D. P. D. et al. Adeno-associated viral vector-induced overexpression of neuropeptide Y Y2 receptors in the hippocampus suppresses seizures. Brain 133, 2778–2788 (2010).
Jandová, K. et al. Carbamazepine-resistance in the epileptic dentate gyrus of human hippocampal slices. Brain 129, 3290–3306 (2006).
Sandow, N. et al. Drug resistance in cortical and hippocampal slices from resected tissue of epilepsy patients: No significant impact of P-glycoprotein and multidrug resistance-associated proteins. Front. Neurol. 6, 1–18 (2015).
Hsiao, M. C. et al. An in vitro seizure model from human hippocampal slices using multi-electrode arrays. J. Neurosci. Methods 244, 154–163 (2015).
Jones, R. S. G., da Silva, A. B., Whittaker, R. G., Woodhall, G. L. & Cunningham, M. O. Human brain slices for epilepsy research: Pitfalls, solutions and future challenges. J. Neurosci. Methods 260, 221–232 (2016).
Traub, R. D., Colling, S. B. & Jefferys, J. G. Cellular mechanisms of 4-aminopyridine-induced synchronized after-discharges in the rat hippocampal slice. J. Physiol. 489, 127–140 (1995).
Avoli, M. et al. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog. Neurobiol. 68, 167–201 (2002).
Buckle, P. J. & Haas, H. L. Enhancement of synaptic transmission by 4-aminopyridine in hippocampal slices of the rat. The Journal of Physiology 326, 109–122 (1982).
Rutecki, P. A., Lebeda, F. J. & Johnston, D. 4-Aminopyridine produces epileptiform activity in hippocampus and enhances synaptic excitation and inhibition. J. Neurophysiol. 57, 1911–1924 (1987).
Gabriel, S. et al. Stimulus and potassium-induced epileptiform activity in the human dentate gyrus from patients with and without hippocampal sclerosis. J. Neurosci. 24, 10416–10430 (2004).
Schiller, Y. Activation of a Calcium-Activated Cation Current During Epileptiform Discharges and Its Possible Role in Sustaining Seizure-Like Events in Neocortical Slices. J. Neurophysiol. 92, 862–872 (2004).
Sanon, N. T., Pelletier, J. G., Carmant, L. & Lacaille, J. C. Interneuron subtype specific activation of mGluR15 during epileptiform activity in hippocampus. Epilepsia 51, 1607–1618 (2010).
Lin, C. H. et al. Effects of anti-epileptic drugs on spreading depolarization-induced epileptiform activity in mouse hippocampal slices. Sci. Rep. 7, 1–14 (2017).
Noè, F. et al. Neuropeptide Y gene therapy decreases chronic spontaneous seizures in a rat model of temporal lobe epilepsy. Brain 131, 1506–1515 (2008).
Huberfeld, G. et al. Glutamatergic pre-ictal discharges emerge at the transition to seizure in human epilepsy. Nat. Neurosci. 14, 627–634 (2011).
Misgeld, A. U., Deisz, R. A., Dodt, H. U. & Lux, H. D. The Role of Chloride Transport in Postsynaptic Inhibition of Hippocampal Neurons. 232, 1413–1415 (1986).
Andersen, P., Dingledine, R., Gjerstad, L., Langmoen, I. A. & Mosfeldt Laursen, A. Two different responses of hippocampal pyramidal cells to application of gamma-amino butyric acid. J. Physiol. 305, 279–296 (1980).
Avoli, M. & de Curtis, M. GABAergic synchronization in the limbic system and its role in the generation of epileptiform activity. Prog. Neurobiol. 95, 104–132 (2011).
Blümcke, I. et al. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: A Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia 54, 1315–1329 (2013).
Wickham, J. et al. Prolonged life of human acute hippocampal slices from temporal lobe epilepsy surgery. Sci. Rep. 8, 4158 (2018).
Acknowledgements
This study was supported by the Swedish Research Council (M.L., Grant No. 2015-00353, M.A., Grant No. 2016-02605; M.K. Grant No. K2013-61X-14603-11-5), The Swedish Brain Foundation (M.A. and M.K.), The Crafoord Foundation (M.A.), Marie Sklodowska Curie Actions (M.L., INCA 600398), ALF-grant (J.B.) and Region Skåne (J.B.), and EPITARGET: FP7-HEALTH project (602102). Open access funding provided by Lund University.
Author information
Authors and Affiliations
Contributions
J.W., M.A. and M.L. performed electrophysiological recordings. J.W. and M.A. performed immunohistochemical imaging. E.E. performed pathological evaluation and imaging. J.W. performed data analysis. J.B., B.J. and L.P. performed the surgery and provided the postoperative tissue with help from D.W. The main text was written by J.W., M.A., M.L. and M.K. All authors contributed to experimental design and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
M.K. and D.W. are co-founders and consultants of spin-off company COMBIGENE AB, Sweden. M.K. and D.W. are also inventors on awarded patent pertaining to the data presented (WO2008004972).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wickham, J., Ledri, M., Bengzon, J. et al. Inhibition of epileptiform activity by neuropeptide Y in brain tissue from drug-resistant temporal lobe epilepsy patients. Sci Rep 9, 19393 (2019). https://doi.org/10.1038/s41598-019-56062-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-56062-1
This article is cited by
-
Transcriptomic alterations in cortical astrocytes following the development of post-traumatic epilepsy
Scientific Reports (2024)
-
Combinatorial gene therapy for epilepsy: Gene sequence positioning and AAV serotype influence expression and inhibitory effect on seizures
Gene Therapy (2023)
-
Ligands of the Neuropeptide Y Y2 Receptors as a Potential Multitarget Therapeutic Approach for the Protection of the Neurovascular Unit Against Acute Ischemia/Reperfusion: View from the Perspective of the Laboratory Bench
Translational Stroke Research (2022)
-
Impact of predictive, preventive and precision medicine strategies in epilepsy
Nature Reviews Neurology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
