
Abstract
Air pollution resulted from fossil fuel burning has been an environmental issue in developing countries in Asia. Sulfur-bearing compounds, in particular, are species that are regulated and monitored routinely. To assess how the species affect at local and global scales, regional background level has to be defined. Here, we report analysis of sulfur isotopes in atmospheric sulfate, the oxidation end product of sulfur species, in particulate phase collected at the Lulin observatory located at 2862 m above mean sea level in 2010. The averaged sulfate concentration for 44 selected samples is 2.7 ± 2.3 (1-σ standard deviation) μg m−3, and the averaged δ34S is 2.2 ± 1.6‰, with respect to the international standard Vienna Canyon Diablo Troilite. Regardless of the origins of air masses, no noticeable difference between the low-altitude Pacific and high-altitude free troposphere sulfate aerosols is observed. Also, no identifiable seasonal cycle in seen. Correlation analysis with respect to coal burning tracers such as lead and oil industry tracers such as vanadium shows sulfate concentration is in better correlation with vanadium (R2 = 0.86, p-value < 0.001) than with lead (R2 = 0.45, p-value < 0.001) but no statistically significant correlation is found in δ34S with any of physical quantities measured. We suggest the sulfate collected at Lulin can best represent the regional background level in the Western Pacific, a quantity that is needed in order to quantitatively assess the budget of sulfur in local to country scales.
Similar content being viewed by others
Introduction
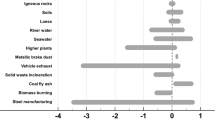
Sulfur is ubiquitous in natural environments and in the atmosphere is present primarily either as sulfate in aerosol/aqueous phases or as OCS and SO2 in gas phase. Sulfur isotopic compositions vary with sources and cycling pathways, thus have received tremendous attention as key tracers in geochemical, biological, and atmospheric processes1,2,3,4,5,6. Sulfur isotopes have also been used to investigate sources and chemical evolution processes of atmospheric aerosols7,8,9. The main source materials of atmospheric sulfur include volcanic sulfur, marine sulfate/sulfide, fossil fuel, and sulfide ores. Despite the wide distribution of stable sulfur isotope ratios (δ34S) in these materials (−50‰ to +50‰), the main sulfur emissions within a specific regional reservoir possess distinctive characteristics of sulfur isotopic values10,11,12,13,14. Table 1 summarize the range of δ34S values reported in the literature, along with the value determined in this work (see below).
Climatically identified to be the most significant aerosol species that gives negative radiative forcing15 (−0.4 ± 0.2 W m−2), atmospheric sulfate is produced primarily by aqueous phase chemical reactions (via oxidation chemistry with dissolved ozone and hydrogen peroxide) in cloud droplets and by dust particle-mediated gas phase chemistry (via first oxidation of SO2 with hydroxyl radicals followed by subsequent condensation and heterogeneous chemistry) on pre-existing particles (e.g.,16,17,18,19,20,21). In addition to anthropogenic inputs, volcanic eruptions also release a significant amount of sulfur-bearing gases into the atmosphere of the Earth. In a global perspective, major sources of SO2 include fossil fuel burning (~72%), biomass burning (~2%), marine dimethyl sulfide (~19%), and volcanic emissions (~7%)22. The last are the most relevant species concerning the climatic impact of volcanic activities. For example, Eyjafjallajökull, a volcano on southern Iceland, began to erupt on 14 April 2010. The volcanic ash and gases were ejected several kilometers into the atmosphere and transported over long distance. The ash was detected over the Netherland, Germany, Italy, and Greece23,24. The transport distance of volcanic aerosols is expected to be much longer. However, little observation has been made on the transport pathways. In particular, a recent new analysis showed volcanic emissions of SO2 during passive degassing are 23 ± 2 Tg/yr (ref. 25), comparable to the total SO2 emission from China26 (see below for anthropogenic emissions). In this paper, we present the concentration and sulfur isotopic composition in aerosol sulfate, in attempt to see how anthropogenic and natural emissions (such as the Eyjafjallajökull mentioned above) affect regional sulfate concentration in a regional scale in Asia.
In the last two decades or so, anthropogenic emissions have been shifted from the western countries like USA and Europe to China and southeast Asia, leading to significant regional shifts in radiative forcing and environmental impact (e.g.,26,27,28,29,30,31). Indeed, it has been documented that China alone contributes nearly a quarter of the global emission (e.g.,30), amounting to ~30 Tg SO2 per year, largely from coal burning (~90%)26; China shares slightly more than 50% of world coal consumption32. Though fuel gas desulfurization device in power plants is widely applied, the emission from industry remains, accounting for ~70% to total SO2 emission from China26. Total emission from other countries in Asia is about 20% that of China30. We then expect that the regional background sulfur emissions and sulfate concentrations are largely set by coal emissions from China. A recent study by Sakata et al.33, however, does not support this scenario; and instead they noted that sulfate concentrations in Japan (from the results derived from two coastal sites) are heavily influenced by oil industry in seasons other than the core winter (December, January, and February). The new isotope and concentration analysis presented in this work also shows that the China coal emission signals are not clearly seen (see below). See Table 1 for the isotopic values of sulfur (δ34S) for known emission sources.
Sampling and Extraction
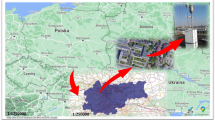
The aerosol samples were collected at Lulin Atmospheric Background Station (NOAA code: LLN; 120°52′25″E, 23°28′07″N, 2,862 m above mean sea level) during 2010. This site is located on the summit of Mt. Lulin in central Taiwan and considered as a clean air station with minimum influence of local pollution. At such high elevation in the free troposphere, the observatory is an ideal station for monitoring levels of pollutants and background traces in regional to global scales (e.g., see Hsu et al.34). The location of the site allows for studies of long-range transport of aerosols34. Aerosol samples were collected daily using high volume TSP collectors34 onto pre-baked (900 °C for ~5 h) PALL Pallflex tissue quartz filters (8″ × 10″), and stored at a temperature close to 0 °C during transportation. The average volume of air that passed through the TSP collectors in a day was 1700 ± 275 m3 (the quoted error bar refers to 1-σ standard deviation of the sampling volume variation). For isotopic sulfate analysis, 44 samples were selected, chosen based on their five-day back trajectories, to best represent air masses in the region. They were originated either over Pacific Ocean, continental low altitudes, or mid-troposphere. In addition, the selection was also based on the consideration of possible seasonal variations. As a result, about 2–4 samples per month were picked. The aerosol was then extracted by shredding 1/16 part of a filter paper placed in a sterilized centrifuge tube, containing 10 ml ultrapure Milli-Q water, kept in an ultrasonic bath for 60 min. The extracts were then filtered using syringe filters (Minisart 17 597-K, pore size 0.2 mm), and the sample stock solution was ready for subsequent sample preparation procedures. The selected samples and their analytical results are summarized in Table 2. Supporting data such as CO, O3, major ions, and metals are obtained and measured following the methods described by Ou-Yang et al.35 and Hsu et al.36; the data of CO and O3 are available in Guha et al.37
Sulfur isotope analysis
Acids used in this study for sample digestion are high purity ones procured from JT Baker. Acids were diluted using Milli-Q water (MQW; resistivity 18.2 MΩ cm). SPEX (aqueous NH4SO4), used as the bracketing standard, was from SPEX CertiPrep Group, Metuchen, USA. PFA vials used in this work were cleaned using sequential cleaning of hot HNO3, HCl and MQW for >12 hour durations. Anion exchange resin (AG1X8; Cl form; 200–400 mesh; BIORAD labs, Richmond, USA) has been used for separation of sulfate from other matrix elements. The sulfur separation procedures were adopted from Das et al.38. All operations (cleaning and sample preparation) were done in CLASS-1000 laboratory, and column chemistry was performed within CLASS-10 working bench maintained at positive airflow.
Dissolution of IAEA S-1 standard was made using protocols following Craddock et al.39. Aliquots of IAEA S-1 solution and aerosol stock solution were evaporated to dryness on a 65 °C hotplate contained within a homemade clean box equipped with the filtered influx air and a venting system to reduce possible contamination from the surroundings. Then, 2–4 mL of 0.3 N HNO3 were added to re-dissolve all the dried material and the sulfur concentrations were measured by ICP-OES. Known amount of this solution was dried and taken in 0.028 M HNO3 to yield sulfur stock concentration of 8 µg mL−1 for subsequent column chemistry. The recovered sulfur (2 µg) is finally taken in 1 mL of 0.3 M HNO3 for isotopic measurements. All measurements of δ34S were done using the Thermo Neptune MC-ICPMS (Thermo Fischer Scientific, Germany) facility at the Isotope Geochemistry Laboratory at the National Cheng Kung University, Taiwan. δ34S measurements were made in the high resolution mode, similar to that of Craddock et al.39, to separate sulfur from major molecular interferences. Isotopic measurements are made at masses 32S, 33S and 34S (monitored at L1, C and H1 faradays cups, respectively), and sulfur isotopic ratios are determined on the low mass shoulder to avoid heavier molecular interferences from O2. (In this work, we limit our discussion to 34S. Because of precision for 33S, no measurable mass-independent effect is found for the samples reported in this work.) Contributions of isobaric interference from 64Zn2+ and 68Zn2+ to 32S and 34S, respectively, were found to be negligible. This was assessed by scanning an ultra pure solution of 50 ng g−1 Zn and monitoring the signal intensities at 32S and 34S and was found to be similar to that of the HNO3 solution. Standard-sample-standard bracketing was used to correct for instrumental mass bias using the SPEX standard. Peak centering was done with respect to the 34S mass scan. All measurements were taken at an integration time of 4 seconds and data acquisition was made for 48 measurements. Mean isotopic ratios of bracketing standard (SPEX) and samples evaluated by the Neptune software were used for calculating δ34S. Two blank tests were performed during the analytical session, the overall procedural blanks vary between 12–18 ng. Since bracketing standard were processed through columns similar to that of a sample, no additional procedural blank correction was needed. Typical 2-σ external measurement precision (relative to SPEX) ranged from 0.24–0.34‰; however, the expanded (propagated) uncertainty increased to 0.45‰ because of two normalizations (sample and SPEX; IAEA S-1 and SPEX) involved in converting to VCDT (Vienna Canyon Diablo Troilite) scale. In the following, the value of IAEA S-1 has been assumed to have δ34S of −0.3‰ relative to VCDT40.
δ34S of a sample relative to the VCDT scale is calculated using the following relation:
Results and Discussion
Overall, the concentrations of major ions are highly variable, with [NH4+] = 1115 ± 988 ng m−3, [SO42−] = 2674 ± 2271 ng m−3, and [NO3−] = 1264 ± 1263 ng m−3. Largely affected by wet deposition, the concentrations are lower in summer time (June-September) than the rest of the time of the year. In summer, daily precipitation is 1.9 ± 2.6 mm (air relative humidity is 93 ± 7%); in winter and spring, the value is 0.2 ± 0.4 mm (air relative humidity is 78 ± 19%). [NH4+] is 474 ± 346 and 1383 ± 1048 ng m−3, respectively, for summer and the rest of the time; [SO42−] is 1196 ± 1048 and 3246 ± 2368 ng m−3; [NO3−] is 425 ± 298 and 1589 ± 1345 ng m−3. Strong temporal variability in winter and spring is closely associated with northeast Asia monsoon that significantly modifies the trajectories of air masses arriving at the sampling location. The phenomenon has been noted previously from the analysis of multiple isotope compositions of nitrate aerosols collected at the same location37.
In the region, there are three major sources of sulfur: ocean, oil industry (ship business), and coal burning. We examine them below. (The aforementioned natural sources such as volcanic emission from Eyjafjallajökull eruption as a major source of sulfate at LLN were not supported, because of good correlation between sulfate concentration and man-made trace metal levels such as vanadium and absence of correlation between δ34S and the other variables measured and analyzed in this work. See the analysis presented below for details.) Fig. 1 shows the time series of the observed major ions for the selected 44 aerosol samples. The single most important cation is NH4+, contributing 76 ± 8%, followed by Ca2+ (8 ± 4%), K+ (7 ± 5%), Na+ (6 ± 4%), and Mg2+ (3 ± 1%). The positively charged ions are balanced primarily by SO42−, NO3−, and Cl−, with the first two contributing 90 ± 8%. SO42− is about a factor of 3 more important than NO3−; the former accounts for 67 ± 11% and the latter is 24 ± 6%. Cl− contribution is variable at 10 ± 8%, with a maximal contribution of 40% appearing on July 26 when the highest δ34S value in sulfate is observed (see Table 2). Surprisingly the lowest sea salt anion contribution (the fraction of Cl−) to the selected occurring on October 3 corresponds to the second largest δ34S value measured. The data shows that the δ34S values of sulfates do not follow the fraction of sea salts in the collected aerosols, suggesting that oceanic sulfur contribution to the sulfate observed at LLN is variable but is not likely to be the major source. Further analysis for the other two sources follows.

Time series of the concentrations (ng/m3) of major ions and metals (Na+, Ca2+, and NH4+, nss-SO4/SO42−, NO3−, and Cl−. Pb and V). The data values are provided in Table 2.
To assess sulfate originated from anthropogenic emission only we first remove the sea salt component following Hsu et al.36 Sea salt sulfate contributes little to the aerosol sulfate collected at Mt. Lulin. The contribution ranges from 0.2% to 2.7% maximum by mass, with an average of 1.2 ± 0.7%, further verifying the proposition that oceanic dimethyl sulfide is not a major source of sulfate at LLN. Correlation analysis shown in Fig. 2 demonstrates that the collected non-sea salt sulfates (nss-SO42−) are largely affected anthropogenically. Tight correlation of [nss-SO42−] with [NO3−] (R2 = 0.70) or [NH4+] (R2 = 0.83) suggests human activities play a major role in the production of sulfate aerosols in the atmosphere; the correlation with nitrate is expected as a result of high temperature combustion and the correlation with ammonium is via NH3 slipped from power plants. Anthropogenic origin of sulfate aerosols is also supported by statistically good correlation with [CO], with R2 = 0.36 and p-value = 2 × 10–5. Complete regression analysis (not shown here but analysis results supporting the statement are available in Guha et al.37) shows that statistically significant correlation is found for the gaseous (CO and O3) and aerosol-phase (ions) species considered, demonstrating anthropogenic alteration is a major source in affecting their abundances. The collected sulfates covering all seasons with little sea salt contribution suggest one may take the values of sulfates collected at the site to represent a regional background anthropogenic level in east Asia.

Scatter plots of Cl−, NO3−, NH4+ versus SO42−. The respective R2 values are 0.47, 0.70, and 0.83, respectively. The p-values are all less than 0.001.
The δ34S values vary between −1.0 and 8.2‰ and are averaged to 2.2 ± 1.6‰ (Table 2). Unlike sulfate concentration (see also Guha et al.37), there is no observable seasonality (Fig. 3). The values in summer and the rest of the seasons are 2.1 ± 2.3‰ and 2.3 ± 1.3‰, respectively. We then analyze the data with aid from their air mass 5-day back trajectories obtained using NOAA ARL HYSPLIT4 model41 with the GDAS (Global Data Assimilation System) meteorological data provided by NCEP (National Center for Environmental Prediction) at a resolution of 6 hours in time and 190.5 km in horizontal spread (see Guha et al.37 for details) and divide the data into two categories (noted in Table 2): one tracks back to a lower region (lower than the sampling site altitude) of the atmosphere and near the surface (ocean surface exclusively) and the other one in regions higher than the sampling location; see Guha et al.37 for a thorough discussion and presentation on the origins of air masses. Similar to seasonal variations, no statistically significant difference is noted: the former is 2.5 ± 2.3‰ and the latter is 2.1 ± 1.0‰. Moreover, no statistically significant correlation is found for δ34S and other variables examined in this work, suggesting sulfur-bearing species have been processed physically and chemically many times attaining certain level of homogenization in space and time before turning into sulfate phase arriving at the sampling location. That is, the source characteristics have been lost, and the sulfur isotopes represent a regional average. The conclusion is supported by triple-oxygen isotope analysis made for sulfate at a background site in east Asia42.

Time series of δ34S and SO42−.
We then compare with major regional sources of sulfur from China. The values of δ34S in sulfate aerosols in PM2.5 reported for Beijing, China during 2015 China Victory Day (with strict pollution control) and non-control periods are 4.7 ± 0.8‰ and 5.0 ± 2.0‰, respectively43. The corresponding concentrations are 3560 ± 2050 ng m−3 and 9590 ± 10910 ng m−3. For comparison, the level of sulfate at another strict control period, the 2008 Olympic, is even higher than the non-control period in 2015 mentioned above7,44. Indeed, strategic regulation help reduce pollution level but the outcome heavily depends on local/regional meteorology7. Overall, the 2015 control period gives sulfate ~50% greater than the value of LLN. The seasonal variations are apparent in the concentrations of the species reported in this work, but δ34S is not. From their two-year (2004–2005) of study in Japan, Sakata et al.33 showed that both the δ34S and concentration of sulfate varied seasonally. Heavier sulfate (that is, higher δ34S value) reported in winter time tends to be less abundant in the concentration, and they suggested the aerosols were originated in northern China33,45. From their analysis, elevated abundance in sulfate in summer time is related to petroleum combustion and has little to do with coal burning. The argument is supported by the δ34S values and vanadium concentrations in the collected aerosols and air mass back trajectory for the samples.
The δ34S values measured in the aerosols collected at LLN are significantly lower than those reported in China7,45,46,47,48,49. Our values are in general close to the values obtained by Sakata et al. (2013) in summer time and to some degree, our results are in agreement with the values from a high mountain in southeast China, Mt. Wuyi in summer time48,49 when there is less influence from coal burning. Following the same analysis as Sakata et al.33, strong correlation between non-crustal vanadium (nc-V) and nss-SO42− concentrations is found (R2 = 0.86, p-value < 0.001; Fig. 4); crustal contribution is estimated using the V/Al ratios reported in Japan arc upper crust50. The overall crustal contribution is 12 ± 6%, with the summer time value (8 ± 6%) slightly less than the rest of the seasons (13 ± 6%). Both the current study and that of Sakata et al.33 suggest that a major source of sulfate in the east Asia is likely from oil industry, rather than coal burning. Evidence is also seen from the poorer correlation (R2 = 0.45, p-value < 0.001) between sulfate and lead. The core reason behind for the correlations is that emission from oil industry is enhanced in vanadium concentration and that from coal burning is lead-enriched (see Sakata et al.33 and references contained therein). Finally, we note that the non-seasonally varying sulfate δ34S values measured at LLN strongly suggests LLN can be a representative site for regional background sulfate. The regional contribution from coal industry, however, is yet to be determined and that is critically dependent on the source characteristics of sulfur-bearing compounds from oil industry which has not been quantified in east Asia.

Scatter plot of nss-SO42− versus nc-V (noncrustal vanadium). See text for the calculation of noncrustal fraction. The R2 and p-values are 0.86 and <0.001, respectively.
Concluding Remarks
We reported one-year sulfur isotope analysis for suspended sulfate aerosols collected at the high mountain station Lulin in the Western Pacific. Regardless of the origins of air masses, the δ34S values in the sulfates are averaged to 2.2 ± 1.6‰. No clear seasonality is seen, and the marine contribution for the sulfate loading is determined to be less than 3%. Time series analysis for the concentrations of lead and vanadium, however, does show significant enhancement in spring (March-June) and winter (September-December) time. The former is due clearly to biomass burning is southeast countries (e.g., see37,51,52). The latter is affected by winter monsoons that carry pollutants from China. Correlation analysis for sulfate with lead and vanadium shows that [SO42−] correlates with vanadium (R2 = 0.85, p-value < 0.001) better than lead (R2 = 0.45, p-value < 0.001), suggesting oil industry plays a critical role in affecting sulfate level at Mt. Lulin. The results indicate that coal burning is less significant than oil industry but its contribution is yet to be determined. Despites the correlations observed and noted above, no statistically significant correlation is observed for δ34S with any of the physical quantities measured. The results imply that the sulfur-bearing species might have been processed many times before converting into sulfate aerosols and reaching the sampling location, with their source isotopic information greatly diminished. As a result, we suggest the δ34S values of Lulin sulfates can represent the level of the background in the Western Pacific. The average is 2.2 ± 0.2‰ (1 standard error, n = 44). This regional value is essential to quantitatively estimate the budget of sulfur in a local and even to a country-sized scale in Asian countries where fossil fuel burning affected air quality has been an issue of public concerns in the past decade and will likely remain in the coming decade.
References
Brüchert, V. Early diagenesis of sulfur in estuarine sediments: the role of sedimentary humic and fulvic acids. Geochim. Cosmochim. Acta 62, 1567–1586 (1998).
Fry, B., Gest, H. & Hayes, J. M. 34S/32S fractionation in sulfur cycles catalyzed by anaerobic bacteria. Appl. Environ. Microbiol. 54, 250–256 (1988).
Canfield, D. E. & Teske, A. Late proterozoic rise in atmospheric oxygen concentration inferred from phylogenetic and sulphur-isotope studies. Nature 382, 127–132 (1996).
Canfield, D. E., Farquhar, J. & Zerkle, A. L. High isotope fractionations during sulfate reduction in a low-sulfate euxinic ocean analog. Geology 38, 415–418 (2010).
Farquhar, J., Savarino, J., Jackson, T. L. & Thiemens, M. H. Evidence of atmospheric sulphur in the martian regolith from sulphur isotopes in meteorites. Nature 404, 50–52 (2000).
Farquhar, J., Nanping, W. U., Canfield, D. E. & Oduro, H. Connections between sulfur cycle evolution, sulfur isotopes, sediments and base metal sulfide deposits. Econ. Geol. 105, 509–533 (2010).
Han, X. et al. Using stable isotopes to trace sources and formation processes of sulfate aerosols from Beijing, China. Sci. Rep. 6, 29958, https://doi.org/10.1038/srep29958 (2016).
Harris, E. et al. Enhanced role of transition metal ion catalysis during in-cloud oxidation of SO2. Science. 340, 727–730 (2013).
Norman, A. L. et al. Aerosol sulphate and its oxidation on the Pacific NW coast: S and O isotopes in PM2.5. Atmos. Environ. 40, 2676–2689 (2006).
Wei, L. et al. Stable sulfur isotope ratios and chemical compositions of fine aerosols (PM2.5) in Beijing, China. Sci. Total Environ. 633, 1156–1164 (2018).
Inomata, Y., Ohizumi, T., Take, N., Sato, K. & Nishikawa, M. Transboundary transport of anthropogenic sulfur in PM2.5 at a coastal site in the sea of japan as studied by sulfur isotopic ratio measurement. Sci. Total Environ. 553, 617–625 (2016).
Nakai, N. & Leroy Jensen, M. Sources of atmospheric sulfur compounds. Geochem. J. 1, 199–210 (1967).
Rees, C. E., Jenkins, W. J. & Monster, J. The sulphur isotopic composition of ocean water sulphate. Geochim. Cosmochim. Acta 42, 377–381 (1978).
Martin, E. Volcanic plume impact on the atmosphere and climate: O- and S-isotope insight into sulfate aerosol formation. Geosci. 8, 198 (2018).
Stocker, T. F. et al. Climate change 2013 the physical science basis: Working Group I contribution to the fifth assessment report of the intergovernmental panel on climate change. Climate Change 2013 the Physical Science Basis: Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change 9781107057 (2013).
Pham, M., Müller, J.-F., Brasseur, G. P., Granier, C. & Mégie, G. A three-dimensional study of the tropospheric sulfur cycle. J. Geophys. Res. 100, 26061–26092 (1995).
Andrea, M. et al. Aerosols, Their Direct and Indirect Effects. A1 - Penner,J.E. in Climate Change 2001: The Scientific Basis. Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change (IPCC) 291–336 (2001).
Sheng, J. X. et al. Global atmospheric sulfur budget under volcanically quiescent conditions: Aerosol-chemistry-climate model predictions and validation. J. Geophys. Res. 120, 256–276 (2015).
Dupart, Y. et al. Mineral dust photochemistry induces nucleation events in the presence of SO2. Proc. Natl. Acad. Sci. USA 109, 20842–20847 (2012).
Jacob, D. J. Chemistry of OH in remote clouds and its role in the production of formic acid and peroxymonosulfate. J. Geophys. Res. 91, 9807–9826 (1986).
J H. Seinfeld, S. N. P. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change. (Wiley, 2016).
Haywood, J. & Boucher, O. Estimates of the direct and indirect radiative forcing due to tropospheric aerosols: A review. Reviews of Geophysics 38, 513–543 (2000).
Balis, D. et al. EARLINET observations of the Eyjafjallajökull ash plume over Greece. in Lidar Technologies, Techniques, and Measurements for Atmospheric Remote Sensing VI 7832, 78320O (SPIE, 2010).
Madonna, F., Amodeo, A., D’Amico, G., Mona, L. & Pappalardo, G. Observation of non-spherical ultragiant aerosol using a microwave radar. Geophys. Res. Lett. 37, L21814 (2010).
Carn, S. A., Fioletov, V. E., Mclinden, C. A., Li, C. & Krotkov, N. A. A decade of global volcanic SO2 emissions measured from space. Sci. Rep. 7, 44095, https://doi.org/10.1038/srep44095 (2017).
Lu, Z., Zhang, Q. & Streets, D. G. Sulfur dioxide and primary carbonaceous aerosol emissions in China and India, 1996-2010. Atmospheric Chemistry and Physics 11, 9839–9864 (2011).
Boucher, O. & Pham, M. History of sulfate aerosol radiative forcings. Geophys. Res. Lett. 29, 22-1-22–4 (2002).
Smith, S. J. & Andres, R. Historical Sulfur Dioxide Emissions 1850–2000: Methods and Results. (Joint Global Change Research Institute, 2004).
Pham, M., Boucher, O. & Hauglustaine, D. Changes in atmospheric sulfur burdens and concentrations and resulting radiative forcings under IPCC SRES emission scenarios for 1990-2100. J. Geophys. Res. D Atmos. 110, 1–10 (2005).
Smith, S. J. et al. Anthropogenic sulfur dioxide emissions: 1850-2005. Atmos. Chem. Phys. 11, 1101–1116 (2011).
Wang, T. et al. Anthropogenic agent implicated as a prime driver of shift in precipitation in eastern China in the late 1970s. Atmos. Chem. Phys. 13, 12433–12450 (2013).
OCDE, O. Coal Information 2017. (OECD, 2017).
Sakata, M., Ishikawa, T. & Mitsunobu, S. Effectiveness of sulfur and boron isotopes in aerosols as tracers of emissions from coal burning in Asian continent. Atmos. Environ. 67, 296–303 (2013).
Hsu, S. C. et al. Dust transport from non-East Asian sources to the North Pacific. Geophys. Res. Lett. 39, L12804 (2012).
Ou Yang, C. F., Lin, N. H., Sheu, G. R., Lee, C. T. & Wang, J. L. Seasonal and diurnal variations of ozone at a high-altitude mountain baseline station in East Asia. Atmos. Environ. 46, 279–288 (2012).
Hsu, S. C. et al. Ammonium deficiency caused by heterogeneous reactions during a super Asian dust episode. J. Geophys. Res. 119, 6803–6817 (2014).
Guha, T. et al. Isotopic ratios of nitrate in aerosol samples from Mt. Lulin, a high-altitude station in Central Taiwan. Atmos. Environ. 154, 53–69 (2017).
Das, A., Chung, C.-H., You, C.-F. & Shen, M.-L. Application of an improved ion exchange technique for the measurement of δ34S values from microgram quantities of sulfur by MC-ICPMS. J. Anal. At. Spectrom. 27, 2088–2093 (2012).
Craddock, P. R., Rouxel, O. J., Ball, L. A. & Bach, W. Sulfur isotope measurement of sulfate and sulfide by high-resolution MC-ICP-MS. Chem. Geol. 253, 102–113 (2008).
Ding, T. et al. Calibrated sulfur isotope abundance ratios three IAEA sulfur isotope reference materials and V-CDT with a reassessment of the atomic weight of sulfur. Geochim. Cosmochim. Acta 65, 2433–2437 (2001).
Draxler, R. R., G. D. R. HYSPLIT (HYbrid Single-Particle Lagrangian Integrated Trajectory) Model Access via NOAA ARL READY Website. Available at, http://ready.arl.noaa.gov/HYSPLIT.php (2010).
Lin, M. et al. Vertically uniform formation pathways of tropospheric sulfate aerosols in East China detected from triple stable oxygen and radiogenic sulfur isotopes. Geophys. Res. Lett. 44, 5187–5196 (2017).
Han, X. et al. Effect of the pollution control measures on PM2.5 during the 2015 China Victory Day Parade: Implication from water-soluble ions and sulfur isotope. Environ. Pollut. 218, 230–241 (2016).
Okuda, T. et al. The impact of the pollution control measures for the 2008 Beijing Olympic Games on the chemical composition of aerosols. Atmos. Environ. 45, 2789–2794 (2011).
Mukai, H. et al. Regional characteristics of sulfur and lead isotope ratios in the atmosphere at several Chinese urban sites. Environ. Sci. Technol. 35, 1064–1071 (2001).
Hong, Y., Zhang, H. & Zhu, Y. Sulfur isotopic characteristics of coal in China and sulfur isotopic fractionation during coal-burning process. Chinese J. Geochemistry 12, 51–59 (1993).
Maruyama, T. et al. Sulfur isotope ratios of coals and oils used in China and Japan. Nippon Kagaku Kaishi / Chem. Soc. Japan - Chem. Ind. Chem. J. 2000, 45–52 (2000).
Lin, M. et al. Unexpected high35S concentration revealing strong downward transport of stratospheric air during the monsoon transitional period in East Asia. Geophys. Res. Lett. 43, 2315–2322 (2016).
Lin, M. et al. Five-S-isotope evidence of two distinct mass-independent sulfur isotope effects and implications for the modern and Archean atmospheres. Proc. Natl. Acad. Sci. 115, 8541 LP–8546 (2018).
Togashi, S. et al. Young upper crustal chemical composition of the orogenic Japan Arc. Geochemistry, Geophys. Geosystems 1, https://doi.org/10.1029/2000GC000083(2000).
Laskar, A. H., Lin, L. C., Jiang, X. & Liang, M. C. Distribution of CO2 in Western Pacific, Studied Using Isotope Data Made in Taiwan, OCO-2 Satellite Retrievals, and CarbonTracker Products. Earth Sp. Sci. 5, 827–842 (2018).
Ou Yang, C. F. et al. Impact of equatorial and continental airflow on primary greenhouse gases in the northern South China Sea. Environ. Res. Lett. 10, https://doi.org/10.1088/1748-9326/10/6/065005 (2015).
Acknowledgements
The work is supported in part by MOST grants (105-2111-M-001-006-MY3, 107-2111-M-001-008, and 105-2611-M-006-003) to Academia Sinica and National Cheng Kung University. We thank Chang-Feng Ou-Yang for helpful discussion on data analysis. All the new data used in the manuscript are provided in the Table.
Author information
Authors and Affiliations
Contributions
C.F.Y, M.C.L. and S.C.H. conceived and designed the study; C.H.C. performed the sulfur isotope analysis, prepared figures and tables, and wrote the first draft; C.F.Y, M.C.L. and C.H.C contributed to data interpretation and to the writing of the final version of this paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chung, CH., You, CF., Hsu, SC. et al. Sulfur isotope analysis for representative regional background atmospheric aerosols collected at Mt. Lulin, Taiwan. Sci Rep 9, 19707 (2019). https://doi.org/10.1038/s41598-019-56048-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-56048-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
