
Abstract
The observation that primitive arc magmas are more oxidized than mid-ocean-ridge basalts has led to the paradigm that slab-derived fluids carry SO2 and CO2 that metasomatize and oxidize the sub-arc mantle wedge. We combine petrography and thermodynamic modelling to quantify the oxygen fugacity (fO2) and speciation of the fluids generated by serpentinite dehydration during subduction. Silicate-magnetite assemblages maintain fO2 conditions similar to the quartz-fayalite-magnetite (QFM) buffer at fore-arc conditions. Sulphides are stable under such conditions and aqueous fluids contain minor S. At sub-arc depth, dehydration occurs under more reducing conditions producing aqueous fluids carrying H2S. This finding brings into question current models in which serpentinite-derived fluids are the cause of oxidized arc magmatism and has major implications for the global volatile cycle, as well as for redox processes controlling subduction zone geodynamics.
Similar content being viewed by others


Introduction
Extensive studies on arc lavas have found that arc basalts are more oxidized than mid-ocean ridge basalts1,2,3. Most of these works discount magmatic low-pressure differentiation as an oxidation mechanism and attribute the oxidized nature of arc lavas to the oxidation of the mantle wedge magma source by slab-derived fluids. Intense hydrothermal interaction of oceanic lithosphere with seawater results in the precipitation of carbonates, sulphates and ferric iron oxides4,5,6 producing a km-wide zone on top of subducted slabs that is comparatively oxidized with respect to the mantle wedge (Fig. 1). Hydrothermally altered oceanic lithosphere contains oxidized species (Fe3+, C4+, S6+) and these elements are present in different concentrations in sediments, mafic crust and mantle lithologies that are introduced into subduction zones7,8,9. The potential release of such oxidized species during dehydration reactions may be coupled with a change in the rock redox state and thus explain the oxidation of the mantle wedge by infiltrating fluids10.

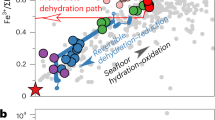
Cartoon of a subduction zone illustrating the subduction of hydrated, relatively oxidized, oceanic lithosphere and where fluids releasing reactions from ultramafic rocks occur at the slab surface, along with calculated fluid fO2 expressed as ∆logQFM (in square bracket) and composition. Thermal model for Central Honshu from Syracuse et al.58 and temperatures are given in degrees Celsius. MORB and arc lava oxygen fugacity values are from Christie et al.59, Bézos and Humler60, Lee et al.40; abyssal serpentinite from Deschamps et al.61; island arc basalt (IAB) source from Ballhaus37 and Parkinson and Arculus39.
Hydrous ultramafic rocks (i.e. serpentinites) are generally considered as the main water carriers and a major source of fluids in subducting oceanic lithosphere at fore-arc to subarc depth11. Mantle peridotites, exposed to the seafloor on slow/ultraslow spreading ridges during tectonic extension, are serpentinized and represent a sink for water, and redox sensitive elements such as carbon and sulphur6,12 (Fig. 1). In particular sea-floor oxidation leads to the precipitation of abundant magnetite and serpentine containing ferric iron12,13,14,15,16. Consequently, serpentinites have been regarded by several researchers as principle carriers of excess oxygen into subduction zones9,17,18,19, with oxidised sulphur released in the fluid phase as the main means of transport of redox budget from the slab to the locus of partial melting. This has led to the current paradigm that oceanic serpentinization and oxidation control the redox potential of fluids released by dehydration9,20.
We investigated the silicate-oxide-sulphide relationships in subducted serpentinites that document the effects of the three key dehydration reactions that occur between fore-arc and sub-arc conditions. This information is combined with thermodynamic modelling along a prograde P-T path in order to constrain the evolution of rock-buffered fO2 during these dehydration reactions. Our findings show that in the presence of magnetite, silica activity imposed by the different silicate mineral assemblages controls oxygen fugacity in hydrous peridotitic systems. We argue that in hydrated ultramafic rocks dehydration reactions take place at rock-buffered conditions and that this buffering has important consequences for the solubility and speciation of sulphur. Thermodynamic calculations show that sulphur occurs predominantly as reduced rather than oxidized fluid species when liberated from hydrous mantle rocks at sub-arc depth and thus serpentinite-derived fluids are unlikely to be responsible for the oxidized character of subduction zone magmas, contrary to claims in previous works.
Silicate-Oxide-Sulphide Relations in High-Pressure Serpentinites, Chlorite-Harzburgites, and Garnet Metaperidotite
The first major dehydration reaction in subducted antigorite serpentinites is
and occurs at 500–550 °C and fore-arc depth for typical slab geotherms, liberating up to ~2.2 wt.% H2O21. This reaction is well-documented in the Zermatt-Saas unit, North-Western Alps, Switzerland, which experienced peak metamorphic conditions of 550 °C, 2.5 GPa22,23. The newly formed olivine is in textural equilibrium with antigorite, chlorite, magnetite23,24 and Fe-Ni sulphides (pentlandite, ±pyrrhotite) (Fig. 2a).

Backscatter images of hydrated ultramafic rocks from Zermatt (a), Cerro del Almirez (b) and Cima di Gagnone (c) showing equilibrium texture between silicates, magnetite and pentlandite. Ol = olivine; Atg = antigorite; Pn = pentlandite; Mt = magnetite; Cr-Mt = chromium-magnetite; Opx = orthopyroxene; Chl = chlorite; Cpx = clinopyroxene.
The antigorite breakdown reaction
is the most important dehydration reaction in serpentinites and occurs at 650–700 °C and 1.6–1.8 GPa, liberating between 5–12 wt.% H2O11. This reaction is well recorded at Cerro del Almirez, Spain25,26 where the reaction products olivine, orthopyroxene and chlorite (chlorite-harzburgite hereafter) coexist with magnetite and pentlandite (±pyrrhotite; ±ilmenite). Previous studies have suggested that hematite is also stable in the chlorite-harzburgite27. However, our examination revealed that hematite occurs only along cracks in association with retrograde talc and chrysotile/lizardite mixtures and as exsolution lamellae from high-T ilmenite. These textures indicate that hematite is most likely a product of retrogression and does not constrain prograde fO2 conditions.
The final dehydration reaction liberates 2.5–3 wt.% H2O and is related to the consumption of chlorite at 750–800 °C and 2.8 GPa at subarc depth by the reaction:
This reaction is documented in lenses of chlorite-harzburgite and garnet metaperidotite at Cima di Gagnone, Central Alps, Switzerland28,29. The peak paragenesis of the chlorite-harzburgite comprises olivine, orthopyroxene, clinopyroxene, chlorite, with accessory Cr-rich magnetite, pentlandite and pyrrhotite (this study30,31,). In garnet peridotite Cr-Al-spinel, Cu-sulphide, Fe-Ni-Cu and Fe-Cu sulphide, pentlandite, Fe-Ni arsenide, and ilmenite are in equilibrium with the peak silicate assemblage composed of olivine, orthopyroxene, clinopyroxene and garnet (this study30,31,). The presence of arsenides and di-sulphides such as chalcopyrite in garnet peridotite indicates higher S fugacity conditions compared to chlorite-harzburgite32.
Calculated Oxygen Fugacities
The equilibrium of Fe-bearing silicates with magnetite reflects the fO2 in metaperidotite33. Iron is the most abundant redox sensitive element in the subducted serpentinites. Previous studies have focused on calculating the redox budget (amount of moles of electrons that need to be added to the rock to reach the reference state34) of the silicate assemblage of serpentinites32 and on the antigorite breakdown reaction (2)20. However, redox conditions prevailing upon reaction (1) (Br-out) and reaction (3) (Chl-out) have remained largely unconstrained. In this study we investigated a suite of samples of progressively dehydrating ultramafic rocks and show how the silicate-oxide assemblages can be used to reconstruct the entire prograde P-T-fO2 evolution. We modelled conditions from 450 to 850 °C and from 2 to 3 GPa along a linear subduction geotherm of 15 °C/km (details and modelling parameters in Supplementary Information, Fig. S1). The results are reported with reference to the commonly used oxygen fugacity buffer quartz-fayalite-magnetite (QFM) in delta log notation to eliminate the P-T dependence of the absolute oxygen fugacity value (Fig. 3). The QFM equilibrium is given by:

Pressure-Temperature-fO2 evolution of hydrated ultramafic rocks. Dehydration reactions occur along a divariant field (yellow highlighted areas). Stars indicate the end of the stability field of hydrated phases: brucite, antigorite and chlorite. On the y-axis, fO2 is expressed as ∆logQFM to eliminate the P-T dependence of the fO2 absolute value (see also Fig. S1). Ol = olivine; Br = brucite; Opx = orthopyroxene; Atg = antigorite; Gt = garnet; Sp = spinel; Chl = chlorite; Hem = hematite; Mt = magnetite; Fa = fayalite; Q = quartz; Fs = ferrosilite; Herc = hercynite; Alm = almandine; FeS = troilite.
Metamorphic olivine is present from the onset of reaction (1) and the fayalite component is decreasing with increasing temperature (Fig. S2). We have shown that magnetite is the stable oxide upon dehydration reaction (1) and (2). Free quartz is not present but a(SiO2) is buffered by coexisting silicates such as brucite and antigorite or olivine and orthopyroxene. This means that knowing the P-T conditions for reaction (1) and (2), and with magnetite being present, we can constrain the oxygen fugacity by modelling the change in Si activity (i.e. change in silicate assemblage). Olivine-free antigorite serpentinite from Zermatt-Saas has the lowest Si activity, with the Si buffering assemblage being brucite + antigorite (~log(aSiO2) = −2.3). Such a low Si activity indicates that these samples are the most reduced (4 log unit below the QFM; Fig. 3). With increasing T and olivine crystallization, the fayalite component in olivine decreases and Si activity increases up to ~log(aSiO2) > −2 (Fig. S2) driving the fO2 to higher values, close to QFM. This corresponds to an increase in fO2 by 4 log units (Fig. 3).
With continued subduction, antigorite breaks down by a continuous reaction to form orthopyroxene (reaction 2) and Si-activity is buffered by orthopyroxene + olivine (Fig. S3). Samples from Cerro del Almirez show that dehydrating antigorite serpentinites have a redox solid buffer (magnetite + olivine + orthopyroxene). The fayalite-ferrosilite-magnetite buffer is one log unit below QFM. However, because of the high Mg# of olivine and orthopyroxene (i.e., molar Mg/(Fe + Mg) ∼ 0.90, measured by electron microprobe), fO2 is ~0.5 log units higher than the QFM buffer (Figs. 3 and S1). Consequently, the antigorite-out reaction (2) does not cause an increase in fO2. It is worth noting that antigorite dehydration at lower pressure, such as in Cerro del Almirez (1.6–1.8 GPa), will occur at identical fO2 conditions because the Si activity is still buffered by olivine + orthopyroxene, with the only difference being that the peak assemblage includes tremolite26,35.
The last dehydration reaction occurs at higher P-T conditions (3 GPa − 770 °C), where chlorite + orthopyroxene react to produce garnet + olivine (reaction 3, Fig. 2c). The oxide assemblage at HP-HT conditions includes magnetite together with spinel. Magnetite + spinel in equilibrium with olivine + garnet is a solid buffer and indicates that reaction (3) occurs at buffered oxygen fugacity conditions. As the reaction proceeds magnetite is progressively replaced by Cr-Al bearing spinel with low ferric iron content (this study30,). This depletion in ferric iron requires that during chlorite dehydration, oxygen fugacity decreases by 2–3 log units below the QFM (Fig. 3). At the end of the reaction, Fe3+ is mainly hosted in clinopyroxene and garnet36. This implies that at higher temperature, when magnetite is completely consumed, redox conditions in the residual anhydrous metaperidotite are governed by equilibria involving Fe3+ bearing silicates36,37 and thus will be sensitive to the bulk rock Fe3+/Fetot.
Fluid Speciation
As the P-T-fO2 conditions for each dehydration reaction have been established, we can estimate the H-O-S molecular speciation of the fluid. Here we provide the predicted speciation of a sulphur bearing fluid liberated from serpentinite and chlorite-harzburgite. At the conditions prevailing at the brucite and antigorite-out reactions (fO2 increasing from −2 to 0 ΔQFM and QFM buffered, respectively; Fig. 4a,b) a HOS-fluid will be situated close to the water maximum (oxygen molar ratio XO = 1/3) with very minor H2S, and H2. In both cases, total sulphur mobilized by the fluid is extremely low (Fig. 4a,b). This result is also supported by the petrographic observations on samples from Zermatt and Cerro del Almirez, where the stability of Fe-Ni sulphide across dehydration reactions (1) and (2) documents subordinate mobilization of sulphur.

Isobaric-isothermal diagrams for H-O-S fluid in equilibrium with sulphide (pyrrhotite buffer) illustrating the mole fraction (Yi) of fluid species as a function of X(O) (oxygen molar ratio: O/(O + H) in the fluid phase). The oxygen fugacity (black line) and the position of mineral buffers (red marks) are also reported (lower boxes). The dashed line indicates the water maximum (X(O) = 0.33) and the grey field indicates the rock buffered oxygen fugacity.
Reaction 3 occurs close to the spinel + olivine = garnet + magnetite buffer (Fig. 3). Our calculations show that at these P-T- fO2 conditions fluids are reduced. Sulphide dissolution in such a reduced environment will produce H2S bearing fluids (Fig. 4c). It is noteworthy that a comparatively large amount of S is released at these P-T-fO2 conditions (Fig. 4c). This result is consistent with the de-sulphidation observed in garnet peridotite (i.e., presence of Fe-Ni arsenides replacing pentlandite).
Absolute S solubility in high-pressure fluids are strongly dependent on the chosen models: molecular species or electrolytes (Table S1). Here we report the results for molecular species fluids (see also Fig. S4). These values are to be considered as conservative estimates. We modelled sulphur solubility at rock buffered conditions using S bulk content ranging from 0.05 to 0.2 wt.%. These values are in the range of reported values for abyssal serpentinites (ranging from 320 to 2300 ppm)6. Both the obtained phase diagrams predict the stability of magnetite with pyrrhotite and/or pyrite, thus indicating that the initial sulphur content does not affect the silicate-magnetite solid buffer, in agreement with the large fO2 stability field predicted for the assemblage magnetite + pentlandite by Evans et al.32. In such a case, solubility does not depend on bulk concentration. Our model predicts that H2S in fluid at brucite-out and antigorite-out conditions does not exceed tens of ppm (20 and 70 ppm, respectively). At chlorite-out condition, S solubility increase by one order of magnitude and H2S content reaches hundreds of ppm (300 ppm). Importantly, both models (molecular species and electrolytes) indicate that S solubility in fluids equilibrated with ultramafic rocks increases prominently from fore-arc to subarc conditions.
Implications for Redox Processes and The Global S Cycle
It is widely accepted that the sub-arc mantle is oxidized19,38,39. This has led to the paradigm that oxidation of the mantle wedge is related to the oxidizing properties of slab derived fluids1,2,19,40. We have demonstrated that in subducting ultramafic rocks magnetite-silicate equilibria buffer the fO2 and that during the major dehydration reactions reduced, rather than oxidised, fluids are liberated. Interestingly, if magnetite forms during seafloor hydration, the bulk rock Fe3+ content does not influence the excess oxygen-content of the fluids that evolve during subsequent dehydration since the system is buffered by the silicate + magnetite assemblage. Ocean floor-inherited Fe3+ content only becomes relevant after the chlorite dehydration reaction (3) when redox conditions are governed by equilibria involving ferric iron in clinopyroxene and garnet36,41. The reduced nature of aqueous fluids released from subducting ultramafic rocks affects the recycling capacity of redox-sensitive elements to arc magmas and the convecting mantle. The interaction of seawater with peridotites leads to an enrichment of U in serpentinites42. Recent findings on the trace element composition of subducted serpentinite have reported no mobilization of U during the brucite and antigorite dehydration reactions43, consistent with our results that indicate fO2 conditions close to QFM for reactions (1) and (2). Reducing conditions during chlorite dehydration (reaction 3) enable near complete retention of U beyond subarc depths in subducting, fully dehydrated, garnet metaperidotite. We therefore suggest that serpentinite-derived fluids cannot account for the observed elevated U/Th ratio in arc lavas44,45,46. Moreover, recent mass balance modelling has revealed that excess U relative to Th and Pb in ocean floor serpentinites recycled to the deep mantle offers a solution to the second terrestrial Pb-isotope paradox47, also known as the kappa conundrum48. Our findings support this hypothesis and emphasize the relevance of redox controls on element recycling in subduction zones.
The antigorite dehydration (reaction 2) generates fluids at fore-arc depth if serpentinites are located at the top of the slab. However, this reaction will occur at subarc depth in partially serpentinized peridotites within the cooler slab interior (Fig. 1). To date no information is available on the sulphide-oxide systematics of subducted oceanic mantle. Our study sets an upper bound on fO2 conditions for a potentially magnetite-free partially serpentinized peridotite as increasing pressure has a negligible effect on the position of the Fe-silicates - oxide mineral buffer.
Chlorite dehydration in serpentinites at the top of the slab occurs below the volcanic arc and may trigger partial melting of the mantle wedge (Fig. 1). Our model indicates that fluids produced upon the chlorite-breakdown reaction can be the source of S to sub-arc magmatism and may thus be relevant to the formation of magmatic-hydrothermal porphyry-type ore deposits. However, reduced conditions imply that S is present as H2S. Reduced mantle wedge conditions are indicated by studies on orogenic garnet peridotites that report subduction metasomatism with fO2 below the QFM buffer49,50,51. Aqueous fluids with reduced S species are in equilibrium with the mantle wedge mineral assemblage and thus could percolate until the magma source region.
Our new findings have important consequences for arc magma genesis. Four main processes have been proposed to explain why arc lavas are more oxidised than MORB: (1) transfer of oxidised species, especially S from the slab to the locus of partial melting in the mantle wedge2,9,19,20; (2) oxidation in the mantle during the ascent of hydrous magmas by dissociation of H2O in olivine and orthopyroxene52,53; (3) Fe3+ enrichment during differentiation of hydrous magmas at lower crustal conditions54,55; and (4) oxidation during degassing at the late stage of eruption/emplacement of magmas40,56. The results of this study indicate that the largely accepted view that oxidised slab fluids are the main cause for producing oxidised arc lavas has to be reconsidered. Our results could also explain why V/Sc and Zn/Fe, ratios used to determine the oxidation conditions of mantle partial melting, are indistinguishable in MORB and arc sources40.
The results we present in this work refer to slow/ultraslow spreading oceanic lithosphere. Nevertheless, the interaction of reduced serpentinite-derived fluids with thick altered oceanic crust and/or sediments that are present in fast-spreading lithosphere likely leads to the precipitation of sulphides, transformation of ferric to ferrous iron, and potentially to the formation of graphite/diamond during subsequent subduction. Thermodynamic calculations have shown that aqueous fluids equilibrated with altered oceanic crust are dominated by CO257. However, we speculate that when such fluids are leaving the slab, interaction with a reduced garnet-peridotite mantle wedge might lead to a change in fluid speciation from CO2 to CH4 dominated aqueous fluids along with a concomitant oxidation of the mantle wedge only immediately above the slab41. Further studies are needed to better quantify the nature of these multiple interaction processes.
References
Carmichael, I. S. The redox states of basic and silicic magmas: a reflection of their source regions? Contributions to Mineralogy and Petrology 106, 129–141 (1991).
Kelley, K. A. & Cottrell, E. Water and the oxidation state of subduction zone magmas. Science 325, 605–607 (2009).
Brounce, M., Kelley, K. & Cottrell, E. Variations in Fe3+/∑ Fe of Mariana Arc basalts and mantle wedge f O2. Journal of Petrology 55, 2513–2536 (2014).
Alt, J. C. Sulfur isotopic profile through the oceanic crust: Sulfur mobility and seawater-crustal sulfur exchange during hydrothermal alteration. Geology 23, 585–588 (1995).
Delacour, A., Früh-Green, G. L., Bernasconi, S. M. & Kelley, D. S. Sulfur in peridotites and gabbros at Lost City (30°N, MAR): Implications for hydrothermal alteration and microbial activity during serpentinization. Geochimica et Cosmochimica Acta 72, 5090–5110 (2008).
Alt, J. C. et al. The role of serpentinites in cycling of carbon and sulfur: seafloor serpentinization and subduction metamorphism. Lithos 178, 40–54 (2013).
Cannaò, E. & Malaspina, N. From oceanic to continental subduction: Implications for the geochemical and redox evolution of the supra-subduction mantle. Geosphere 14, 2311–2336 (2018).
Tumiati, S., Godard, G., Martin, S., Malaspina, N. & Poli, S. Ultra-oxidized rocks in subduction mélanges? Decoupling between oxygen fugacity and oxygen availability in a Mn-rich metasomatic environment. Lithos 226, 116–130 (2015).
Schwarzenbach, E. M. et al. Sulphur and carbon cycling in the subduction zone mélange. Scientific reports 8, 15517 (2018).
Evans, K. The redox budget of subduction zones. Earth-Science Reviews 113, 11–32 (2012).
Ulmer, P. & Trommsdorff, V. Serpentine stability to mantle depths and subduction-related magmatism. Science 268, 858–861 (1995).
Cannat, M., Fontaine, F. & Escartin, J. Serpentinization and associated hydrogen and methane fluxes at slow spreading ridges. Diversity of hydrothermal systems on slow spreading ocean ridges 241–264 (2010).
Malvoisin, B., Carlut, J. & Brunet, F. Serpentinization of oceanic peridotites: 1. A high‐sensitivity method to monitor magnetite production in hydrothermal experiments. Journal of Geophysical Research: Solid Earth 117 (2012).
Andreani, M., Munoz, M., Marcaillou, C. & Delacour, A. μXANES study of iron redox state in serpentine during oceanic serpentinization. Lithos 178, 70–83 (2013).
Schwarzenbach, E. M., Caddick, M. J., Beard, J. S. & Bodnar, R. J. Serpentinization, element transfer, and the progressive development of zoning in veins: evidence from a partially serpentinized harzburgite. Contributions to Mineralogy and Petrology 171, 5 (2016).
Charlou, J., Bougault, H., Appriou, P., Nelsen, T. & Rona, P. Different TDM/CH4 hydrothermal plume signatures: TAG site at 26 N and serpentinized ultrabasic diapir at 15 05′ N on the Mid-Atlantic Ridge. Geochimica et Cosmochimica Acta 55, 3209–3222 (1991).
Evans, K.-A. & Tomkins, A.-G. The relationship between subduction zone redox budget and arc magma fertility. Earth and Planetary Science Letters 308, 401–409 (2011).
Evans, K. & Powell, R. The effect of subduction on the sulphur, carbon and redox budget of lithospheric mantle. Journal of Metamorphic Geology 33, 649–670 (2015).
Bénard, A. et al. Oxidising agents in sub-arc mantle melts link slab devolatilisation and arc magmas. Nature communications 9, 3500 (2018).
Debret, B. & Sverjensky, D. Highly oxidising fluids generated during serpentinite breakdown in subduction zones. Scientific reports 7, 10351 (2017).
Padrón-Navarta, J. A. et al. Tschermak’s substitution in antigorite and consequences for phase relations and water liberation in high-grade serpentinites. Lithos 178, 186–196 (2013).
Angiboust, S., Agard, P., Jolivet, L. & Beyssac, O. The Zermatt‐Saas ophiolite: the largest (60‐km wide) and deepest (c. 70–80 km) continuous slice of oceanic lithosphere detached from a subduction zone? Terra Nova 21, 171–180 (2009).
Li, X., Rahn, M. & Bucher, K. Serpentinites of the Zermatt‐Saas ophiolite complex and their texture evolution. Journal of Metamorphic Geology 22, 159–177 (2004).
Kempf, E. D. & Hermann, J. Hydrogen incorporation and retention in metamorphic olivine during subduction: Implications for the deep water cycle. Geology 46, 571–574 (2018).
Trommsdorff, V., Sánchez-Vizcaíno, V. L., Gomez-Pugnaire, M. & Müntener, O. High pressure breakdown of antigorite to spinifex-textured olivine and orthopyroxene, SE Spain. Contributions to Mineralogy and Petrology 132, 139–148 (1998).
Padrón-Navarta, J. A., Lopez Sanchez-Vizcaino, V., Garrido, C. J. & Gómez-Pugnaire, M. T. Metamorphic record of high-pressure dehydration of antigorite serpentinite to chlorite harzburgite in a subduction setting (Cerro del Almirez, Nevado–Filábride Complex, Southern Spain). Journal of Petrology 52, 2047–2078 (2011).
Debret, B. et al. Redox state of iron during high-pressure serpentinite dehydration. Contributions to Mineralogy and Petrology 169, 36 (2015).
Evans, B. W. & Trommsdorff, V. Petrogenesis of garnet lherzolite, Cima di Gagnone, Lepontine Alps. Earth and Planetary Science Letters 40, 333–348 (1978).
Trommsdorff, V., Hermann, J., Müntener, O., Pfiffner, M. & Risold, A.-C. Geodynamic cycles of subcontinental lithosphere in the Central Alps and the Arami enigma. Journal of Geodynamics 30, 77–92 (2000).
Pfiffner, M. A. Genese der hochdruckmetamorphen ozeanischen Abfolge der Cima Lunga-Einheit (Zentralalpen) (1999).
Scambelluri, M., Pettke, T. & Cannaò, E. Fluid-related inclusions in Alpine high-pressure peridotite reveal trace element recycling during subduction-zone dehydration of serpentinized mantle (Cima di Gagnone, Swiss Alps). Earth and Planetary Science Letters 429, 45–59 (2015).
Evans, K., Reddy, S., Tomkins, A., Crossley, R. & Frost, B. Effects of geodynamic setting on the redox state of fluids released by subducted mantle lithosphere. Lithos 278, 26–42 (2017).
Frost, B. R. On the stability of sulfides, oxides, and native metals in serpentinite. Journal of Petrology 26, 31–63 (1984).
Evans, K. Redox decoupling and redox budgets: Conceptual tools for the study of earth systems. Geology 34, 489–492 (2006).
Bretscher, A., Hermann, J. & Pettke, T. The influence of oceanic oxidation on serpentinite dehydration during subduction. Earth and planetary science letters 499, 173–184 (2018).
Malaspina, N., Langenhorst, F., Fumagalli, P., Tumiati, S. & Poli, S. Fe3+ distribution between garnet and pyroxenes in mantle wedge carbonate-bearing garnet peridotites (Sulu, China) and implications for their oxidation state. Lithos 146, 11–17 (2012).
Ballhaus, C. Redox states of lithospheric and asthenospheric upper mantle. Contributions to Mineralogy and Petrology 114, 331–348 (1993).
Rielli, A. et al. Evidence of sub-arc mantle oxidation by sulphur and carbon. Geochemical Perspectives Letters 3, 124–132 (2017).
Parkinson, I. J. & Arculus, R. J. The redox state of subduction zones: insights from arc-peridotites. Chemical Geology 160, 409–423 (1999).
Lee, C.-T. A., Leeman, W. P., Canil, D. & Li, Z.-X. A. Similar V/Sc systematics in MORB and arc basalts: implications for the oxygen fugacities of their mantle source regions. Journal of Petrology 46, 2313–2336 (2005).
Malaspina, N., Poli, S. & Fumagalli, P. The oxidation state of metasomatized mantle wedge: insights from C–O–H-bearing garnet peridotite. Journal of Petrology 50, 1533–1552 (2009).
Niu, Y. Bulk-rock major and trace element compositions of abyssal peridotites: implications for mantle melting, melt extraction and post-melting processes beneath mid-ocean ridges. Journal of Petrology 45, 2423–2458 (2004).
Peters, D., Bretscher, A., John, T., Scambelluri, M. & Pettke, T. Fluid-mobile elements in serpentinites: Constraints on serpentinisation environments and element cycling in subduction zones. Chemical geology 466, 654–666 (2017).
Kelley, K. A., Plank, T., Farr, L., Ludden, J. & Staudigel, H. Subduction cycling of U, Th, and Pb. Earth and Planetary Science Letters 234, 369–383 (2005).
Hawkesworth, C., Turner, S., Peate, D., McDermott, F. & van Calsteren, P. Elemental U and Th variations in island arc rocks: implications for U-series isotopes. Chemical Geology 139, 207–221 (1997).
Allègre, C. J. & Condomines, M. Basalt genesis and mantle structure studied through Th-isotopic geochemistry. Nature 299, 21 (1982).
Pettke, T., Kodolányi, J. & Kamber, B. S. From ocean to mantle: new evidence for U-cycling with implications for the HIMU source and the secular Pb isotope evolution of Earth’s mantle. Lithos 316, 66–76 (2018).
Elliott, T., Zindler, A. & Bourdon, B. Exploring the kappa conundrum: the role of recycling in the lead isotope evolution of the mantle. Earth and Planetary Science Letters 169, 129–145 (1999).
Wang, J., Hattori, K. H., Kilian, R. & Stern, C. R. Metasomatism of sub-arc mantle peridotites below southernmost South America: reduction of fO 2 by slab-melt. Contributions to Mineralogy and Petrology 153, 607–624 (2007).
Ishimaru, S., Arai, S. & Shukuno, H. Metal-saturated peridotite in the mantle wedge inferred from metal-bearing peridotite xenoliths from Avacha volcano, Kamchatka. Earth and Planetary Science Letters 284, 352–360 (2009).
Song, S., Su, L., Niu, Y., Lai, Y. & Zhang, L. CH4 inclusions in orogenic harzburgite: evidence for reduced slab fluids and implication for redox melting in mantle wedge. Geochimica et Cosmochimica Acta 73, 1737–1754 (2009).
Brandon, A. D. & Draper, D. S. Constraints on the origin of the oxidation state of mantle overlying subduction zones: an example from Simcoe, Washington, USA. Geochimica et Cosmochimica Acta 60, 1739–1749 (1996).
Tollan, P. & Hermann, J. Arc magmas oxidized by water dissociation and hydrogen incorporation in orthopyroxene. Nature geoscience 12, 667 (2019).
Burgisser, A. & Scaillet, B. Redox evolution of a degassing magma rising to the surface. Nature 445, 194 (2007).
Humphreys, M. C. et al. Coupled interactions between volatile activity and Fe oxidation state during arc crustal processes. Journal of Petrology 56, 795–814 (2015).
Li, J.-L., Gao, J., Klemd, R., John, T. & Wang, X.-S. Redox processes in subducting oceanic crust recorded by sulfide-bearing high-pressure rocks and veins (SW Tianshan, China). Contributions to Mineralogy and Petrology 171, 72 (2016).
Kerrick, D. M. & Connolly, J. A. D. Metamorphic devolatilization of subducted oceanic metabasalts: implications for seismicity, arc magmatism and volatile recycling. Earth and Planetary Science Letters 189, 19–29 (2001).
Syracuse, E. M., van Keken, P. E. & Abers, G. A. The global range of subduction zone thermal models. Physics of the Earth and Planetary Interiors 183, 73–90 (2010).
Christie, D. M., Carmichael, I. S. & Langmuir, C. H. Oxidation states of mid-ocean ridge basalt glasses. Earth and Planetary Science Letters 79, 397–411 (1986).
Bézos, A. & Humler, E. The Fe3+/ΣFe ratios of MORB glasses and their implications for mantle melting. Geochimica et Cosmochimica Acta 69, 711–725 (2005).
Deschamps, F., Godard, M., Guillot, S. & Hattori, K. Geochemistry of subduction zone serpentinites: A review. Lithos 178, 96–127 (2013).
Acknowledgements
This work was supported by the Swiss National Science Foundation grants No. 200021_172688 to T.P. and No. 200021_169062 to J.H. We thank Editor Takeshi Kuritani and four anonymous reviewers for their constructive comments.
Author information
Authors and Affiliations
Contributions
F.P. performed the petrographic study, thermodynamic modelling, and wrote the manuscript with input from J.H., T.P. and J.A.D.C.; E.D.K. and J.F.V.D. contributed to the petrographic study of samples from Zermatt-Saas and Cerro del Almirez, respectively.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Piccoli, F., Hermann, J., Pettke, T. et al. Subducting serpentinites release reduced, not oxidized, aqueous fluids. Sci Rep 9, 19573 (2019). https://doi.org/10.1038/s41598-019-55944-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-55944-8
This article is cited by
-
Mantle wedge oxidation from deserpentinization modulated by sediment-derived fluids
Nature Geoscience (2023)
-
Oxide-silicate petrology and geochemistry of subducted hydrous ultramafic rocks beyond antigorite dehydration (Central Alps, Switzerland)
Contributions to Mineralogy and Petrology (2023)
-
Early release of H2O during subduction of carbonated ultramafic lithologies
Contributions to Mineralogy and Petrology (2023)
-
Trace-element distribution and ore-forming processes in Au–Ag-rich hydrothermal chimneys and mounds in the TA25 West vent field of the Tonga Arc
Mineralium Deposita (2023)
-
Slab-derived devolatilization fluids oxidized by subducted metasedimentary rocks
Nature Geoscience (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
