
Abstract
CRISPR/Cas9 editing efficacies in tetraploid potato were highly improved through the use of endogenous potato U6 promoters. Highly increased editing efficiencies in the Granular Bound Starch Synthase gene at the protoplast level were obtained by replacement of the Arabidopsis U6 promotor, driving expression of the CRISPR component, with endogenous potato U6 promotors. This translated at the ex-plant level into 35% full allelic gene editing. Indel Detection Amplicon Analysis was established as an efficient tool for fast assessment of gene editing in complex genomes, such as potato. Together, this warrants significant reduction of laborious cell culturing, ex-plant regeneration and screening procedures of plants with high complexity genomes.
Similar content being viewed by others


Introduction
Genome editing provides an efficient route of translating genetic knowledge into improved crop varieties in the field. In contrast to many breeding techniques, CRISPR-Cas genome editing minimizes introduction of undesired mutations1, which requires backcrossing and/or extensive selection to identify vigorous offspring. This becomes important in outbreeding crops and especially in those with complex genomes, where genome editing enables breeding of desired traits in existing elite cultivars, without compromising existing good agronomic performance2. Elite cultivars of tetraploid potato, Solanum tuberosum, are known to be extremely genetically diverse with a very high Small Nucleotide Polymorphism (SNP) frequency3,4; and are thus propagated clonally to maintain agronomic traits that are the result of balancing such overwhelming genetic diversity. Many traits can be improved by loss of function alleles, for example generated through CRISPR/Cas9 induction of the Non Homologous End Joining (NHEJ) pathway5. The most famous example is mlo-based resistance to powdery mildew6,7.
High efficient CRISPR/Cas9 editing are in many plants complicated by the presence of complex and high ploidy genomes and inefficient or poorly controlled delivery of the CRISPR/Cas9 components to cells with regenerative potential. Fluorescence Activated Cell Sorting (FACS) of cells expressing GFP tagged CRISPR/Cas9 is regularly used for enrichment of edited cell populations in mammalian cell systems8 and more recently also expanded to plant protoplast cells9. The Indel Amplicon Analysis (IDAA)10 technique allows for fast and direct assessment of insertions/deletions (indels), with a sensitivity down to + /−1 bp, without the need for in depth Sanger sequencing11. IDAA was recently used for editing scoring of protoplast cell populations9, and in the present study the use of IDAA was expanded and adapted to plants with complex genomes, such as potato, where it proved an efficient and fast tool for editing assessment.
Genome editing has been applied to several gene targets in potato albeit with moderate editing frequencies in protoplasts and subsequent regenerated ex-plant shoots. In one study TALENs were targeted to the acetolactate synthase gene resulting in 7–8%, 11–13%, 10% editing at the protoplast, calli, and regenerated shoot/ex-plant levels, respectively; but full allelic edited ex-plants were not obtained12. In another study an vacuolar invertase gene was targeted by CRISPR/Cas9. Here 18 of 600 shoots displayed editing and 5 (0.8%) had editing in all four alleles13. A recent study where the potato granule bound starch synthase (GBSS) gene was targeted by CRISPR/Cas9, showed that replacement of the standard Arabidopsis thaliana U6-1 (AtU6-1) promoter, driving expression of the guide RNA, with a endogenous potato U6 promoter resulted in a doubling in editing frequency from ca. 5% to 10% in regenerated ex-plant lines of which 2% displayed full allelic editing14. The presence of a single wild-type allele was sufficient for conferring significant amounts of the GBSS gene product amylose thus demonstrating that KO of all four alleles is necessary for producing amylopectin (waxy) potato starch devoid of amylose14. Similarly, the endogenous cotton (Gossypium hirsutum) GhU6.3 promoter also resulted in increased CRISPR/Cas9 editing as compared to the AtU6-29 promoter in a transient reporter expression system in cotton15.
In this study, we significantly improved CRISPR/Cas9 editing efficacy by applying endogenous potato StU6 promoters for driving the CRISPR component of the CRISPR/Cas system, and demonstrate that this optimization has a dramatic effect on editing frequencies at both the protoplast and shoot/ex-plant level.
Results
Retrieval of endogenous U6 promotors and editing analysis at the protoplast level
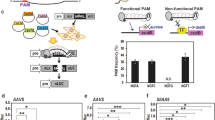
To identify and retrieve StU6 promoters, we used sequences described in16,17,18 to search the potato genome. Upstream 5′ flanking sequences of the StU6 promoters were retrieved and used to PCR amplify the promoter regions from the cultivars Desirée and Wotan. Four promoter sequences were identified of which two were hitherto unannotated (StU6-2 and StU6-3) and one (StU6-4) displayed size polymorphism (StU6-4a and StU6-4b). An alignment of the retrieved U6 promoter sequences displays an overall high heterogeneity with respect to length and composition (Supplementary Fig. 1). The four potato StU6-1-4 promoters, defined as 350 bp upstream of the U6 gene start, were cloned to replace the Arabidopsis thaliana AtU6-1 promoter, thus driving expression of the target GBSS gRNA1 and gRNA2 in constructs also expressing the SpCas9 enzyme (Fig. 1A, Supplementary Fig. 2). In our initial designs we selected gRNAs targeting exon 1 of GBSS with diagnostic restriction sites for scoring editing. Potato leaf-derived protoplasts were isolated and the CRISPR/Cas9 expressing constructs (Supplementary Fig. 2) were delivered by polyethylene glycol (PEG) transformation as earlier described12. Indels were initially scored by PCR amplification of the targeted region from pools of protoplasts harvested 24 hrs after transformation followed by restriction enzyme digestion. This revealed a significant increase in editing for the endogenous StU6-1-4 promoters when compared to the AtU6-1 promoter as judged by restriction enzyme resistant band intensities (Fig. 1B). The experiment was repeated three times with overall similar results (data not shown). High resolution assessment of indel formation and distribution at the cell pool level was carried out through the use of the Indel Amplicon Analysis (IDAA) technique (Fig. 1C, Supplementary Fig. 3) and analyzed in detail by sequencing (Fig. 1D). When applied on the cultivar Wotan similar increases in editing were observed albeit at lower absolute editing frequencies (Supplementary Fig. 3). We speculate that subtle differences in chromatin structure, such as methylation status or packaging, or other factors between the two cultivars may explain these differences.

U6 promoter efficacy analysis at the protoplast level. StU6 promoters were here defined as 350 nt upstream of the transcription start site (Supplementary Fig. 1 and Supplementary Fig. 2D), and the 257 bp AtU6-1 promoter of Arabidopsis thaliana was obtained from the vector pHBT-pcoCas927. (A) Structure of the Granular Bound Starch Synthase (GBSS) gene3 with the guide RNA (gRNA1) targeting part of the conserved motif and proposed active site KTGGL23. gRNA1 includes a diagnostic BsrI restriction enzyme site spanning the SpCas9 cleavage site28, −3 bp upstream of the Photospacer Adjacent Motif (PAM) (red), which upon digestion yields the allele specific fragments: allele III (286, 140), allele I and II (289, 140) and allele IV (289, 144). The outer most primer set includes the SNPs (+SNPs) and gives rise to PCR amplicons of 426 (allele III), 429 (alleles I and II) and 433 bp (allele IV), and the innermost (-SNPs) to a PCR amplicon of 218 bp (all alleles). These length SNPs were conserved between the cultivars Desirée and Wotan. (B) Construct design and StU6-1-4 versus AtU6-1 promoter analysis at the cell pool (protoplast) level as evidenced by the presence of indel mediated destruction of the BsrI site (BsrI resistant band). (C) Indel Detection by Amplicon Analysis (IDAA) chromatograms for the + SNP and –SNP PCR amplicons of WT and AtU6-1 and StU6-1 derived indels. + SNP IDAA reveals allele complexity, while –SNP and BsrI digested + SNP amplicons permit estimation of editing efficacy. (D) Sequence analysis of 34 individual clones of the BsrI resistant band of StU6-1 (B) isolated and cloned into the pJet vector, confirmed the indel distribution of the IDAA (Fig. 1C, panel: StU6-1, -SNP) also peaking at −4 bp deletions. WT peak positions are indicated by dotted lines. PPDK and NOS designate Pyruvate phosphate dikinase promoter and nopaline synthase terminator, respectively. All experiments were done in Desirée, except for panel C (upper right side) and D, which were done in the cultivar Wotan.
We expanded IDAA by restriction enzyme digestion of the IDAA samples, thus separating WT and indel peaks and allowing for direct assessment and quantification of only the indel derived peaks. Peak quantification revealed complete removal of the WT peaks in the Ctrl sample and a ca. 9 fold increase, from 8% to 71%, of edited alleles upon replacement of the AtU6-1 promoter with the StU6-1 (Fig. 1C) or StU6-2 (data not shown) promoter in protoplasts of the cultivar Desirée. The restriction enzyme resistant band of the StU6-1 experiment (Fig. 1B) was cloned, sequenced and the indels scored, showing a distribution of deletions peaking at −4 bp, a few 1 nt additions (Fig. 1D) as well as large insertions (data not shown). IDAA and sequence analysis were consistent. The same analysis was applied on a second target in exon 1 of GBSS, gRNA2, which also showed higher efficacy using the StU6 promoters compared to the AtU6-1 promoter, albeit with GBSS-gRNA2 having lower general editing efficacy (Supplementary Fig. 4). The indel distribution of these two gRNA target sites, i.e. the high prevalence of −4 and −1 deletions, respectively, is in agreement with a recent study, where Cas9 editing of a vast number of gRNAs/targets showed that each gRNA conferred individual cell-line-dependent bias toward particular Cas9 editing outcomes19. When exon 1 of GBSS was subjected to five selected in silico gRNA prediction servers19, the highly in vivo efficient GBSS-gRNA1 (Fig. 1) was ranked highest by the efficiency score in three of the predictors, but with a specificity score ranking it as #23 or #24 of 36 possible gRNA’s in the two predictors that allowed off-target assessment (Supplementary Fig. 5). The non-trivial weighting of gRNA efficiency and specificity ranking when selecting gRNA targets may be augmented by unreliable or inadequate genome sequence information. While traditional breeding techniques that use mutagenic chemicals or irradiation result in mutation frequencies of 0.2–5 × 10–320, only four off-target mutations could be attributed to CRISPR/Cas9 activity in tetraploid cotton analysed by whole genome sequencing1. This is less than 1% of the 466 SNPs and 77 indels identified as spontaneous mutations and in agreement with the recently reported none or very low off-target mutation frequencies in mammalian cells21.
Ex-plant molecular and phenotypic characterization
Protoplast isolation, transformation, alginate embedment, shoot and ex-plant generation and transfer to soil for phenotypic scoring of starch composition in the tubers is outlined in Supplementary Fig. 6. Twenty three shoots/ex-plants were randomly selected and analyzed and out of these eight (35%) displayed full allelic editing, six appeared to have editing in 1–3 alleles, and nine were un-edited as evidenced by restriction enzyme analysis. Eleven out of the 23 shoots/ex-plants had plasmid derived insertions (Fig. 2B, left panel) and one displayed a dwarfed phenotype (Fig. 2B, right panel, plantlet K8). The high frequency of insertion mutants is in agreement with a recent study in potato, where CRISPR/Cas nucleoproteins were assembled in vitro. Here, an unexpected high frequency of insertions of both potato genomic DNA and fragments derived from the plasmid used for in vitro transcription of the gRNA was observed22. Molecular and phenotypic analyses of shoots/ex-plants initially selected for further analysis are outlined in Supplementary Fig. 7.

Amylopectin only potato: Geno- and phenotypic analysis of GBSS loss of function ex-plants. (A) Regenerated ex-plants from tissue culture with resulting potato tubers.(B) In total, 23 randomly selected shoots/ex-plants were screened to identify indels using the BsrI restriction enzyme digestion. K8, K10, K18, K19, K33 & K52 appeared to have editing in all four alleles (fully BsrI resistant), K11, K12, K26, K32 & K42 displayed a mixture of WT type and edited alleles, while K1, K2, K7, K9, K13, K34, K47, K50 & K51 appeared to be un-edited. * denotes insertions. K5 and K6 appear at a first glance to be mixed, but sequence analysis revealed that the BsrI digestion bands were derived from plasmid insertions comprising the gRNA1 sequence, which reintroduced the BsrI site and that K5 and K6 in fact had full allelic editing. (C) WT and the full allelic edited ex-plants, K10 and K33, were corroborated by IDAA of the larger (+SNP) region and the inner smaller (-SNP) region without length SNPs (Fig. 1C). WT peak positions are indicated by dotted lines. Lugol staining was used to analyse tuber starch from greenhouse grown plants (Supplementary Fig. 6). The presence of amylose gives rise to the dark blue colour (WT) tubers, while amylose free/’amylopectin only’ yields the red-brownish color. Additional shoots/ex-plants selected for IDAA analysis of which a subset was propagated to set tubers were also stained with lugol29 (Supplementary Fig. 7). (D) Sequence analysis confirmed that both K10 and K33 were full allelic edited. K10 had a 3 bp deletion in allele III, IV and in one of the alleles I or II and a 990 bp insertion (denoted ‘*’ left to the sequence, see also arrow on gel insertion (Fig. 2C)) in the other as also evident in the IDAA analysis displaying 1:1:1 ratio of the three chromatographic peaks. K33 had 6 bp and 5 bp deletions in allele I and II, a 4 bp deletion in allele IV and a 1 bp insertion in allele III.
It was previously shown that full allelic knock-out of the GBSS gene is necessary for obtaining amylose-free starch14. Interestingly, two of the plants with four allele editing and amylose free starch, K10 and K33 (Fig. 2C), displayed three or six nucleotide in frame deletions in three or one allele, respectively (Fig. 2C,D), which resulted in elimination of the K-codon of the proposed ADPG binding site in the KTGGL motif23. The lack of amylose in K10 and K33 thus confirmed the importance of this amino acid for GBSS activity.
Noteworthy, our initial sequencing based characterization of the target region of the single copy GBSS gene having an expected maximum of four alleles, revealed a seemingly implausible complexity of up to 15 alleles. More detailed analysis, using combinations of different PCR amplifications to increase robustness of the analysis revealed that the observed hyper diversity was likely due to chimera formation caused by priming with incompletely extended PCR products from previous cycles (Supplementary Fig. 8) as has been previously observed for simultaneous amplification of highly homologous templates24.
Discussion
Most agronomic traits are influenced by multiple loci and obtaining individuals harboring the desired alleles at these loci are very unlikely. In hybrid breeding crops, such as most cereals, genetic diversity at important loci is frequently fixed in inbreds that are then combined to provide homogenous and vigorous offspring thus blocking further breeding improvement. Outbreeding crops, such as tetraploid potato, harbor numerous allelic variants at important loci and agronomic performance of single individuals, propagated as clones (seed potato), is the result of a complex balance of alleles at a multitude of loci, which is perturbed by further crossing. In both instances, crop development may be accelerated by targeted genetic mutations in already existing elite varieties25 using genome editing technology, such as CRISPR/Cas9. In outbreeding crops, the opportunity for altering specific traits in existing elite cultivars holds the promise of increasing performance of specific traits, without compromising good agronomic performance. Potato is tetraploid and elite germplasms are extremely genetically diverse with observed SNP frequency between two individuals of 1 per 29 bp3. Thus, genome editing is particularly attractive in potato where good agronomic performance is the result of balancing such overwhelming genetic diversity.
In the present study, we devised schemes for efficient editing and editing assessment of high ploidy complex genomes, here in potato, which are characterised by a high SNP prevalence between alleles but also between different cultivars.
We performed a survey for assessing the endogenous U6 promotor repertoire in potato and retrieved both published and hitherto unpublished potato U6 promotor sequences, and assessed their conferred editing performance in protoplasts as a first readout. Replacement of the regularly used Arabidopsis AtU6-1 promotor with endogenous potato StU6 promotors resulted in a dramatic increased editing efficiency of the target GBSS gene. Editing frequencies of 30–70% in protoplasts, corresponding to an up to a ca. 9 fold increased editing, were obtained as evidenced by IDAA. Theoretically 50% editing in protoplasts would translate into 6.25% (0.54) ex-plants with full allelic editing. However, in agreement with earlier CRISPR/Cas editing in potato13,14, we observed full allelic editing in 35% of the ex-plants/shoots analysed, thus suggesting that in transformed protoplasts there is an increased likelihood that multiple alleles will undergo editing.
Our implementation of the IDAA technique permits fast and high throughput assessment of editing in organisms with high ploidy and complex genomes, such as potato, as evidenced here where all peak positions of the WT chromatogram were shifted in full allelic knock out of explants (Fig. 2). Editing may be scored at both the protoplast and ex-plant level, without the need for comprehensive sequence analysis, which, however, is still needed for full characterization at the ex-plant level.
Somaclonal variation has been reported to be a concern in relation to ex-plant regeneration from callus/shoots, including potato26. The present study did not include a comprehensive survey in regard hereto, but the regenerated first generation ex-plants displayed vigorous growth in vitro when scored ca 2 month after first visible shoot formation and later in pots in the greenhouse (Fig. 2A). Both the full allele GBSS edited ex-plants, such K10 and K33, and un-edited GBSS but regenerated plants, such as K34, for example, displayed the same vigorous growth under the growth and culturing conditions applied here. One ex-plant, K8 (depicted in insert to Fig. 2A), displayed a major growth phenotype, but further genotyping of this ex-plant was not pursued. Although somaclonal/phenotypic variation was generally not encountered in the present limited explant material, additional explant lines and propagation generations are needed for proper assessment of the extent of genetic as well as non-genetic derived somaclonal/penotypic variation.
In conclusion, this study demonstrates that i) use of endogenous U6 promoters has an great impact on editing efficiencies, ii) in complex tetraploids an exhaustive molecular characterization of WT alleles is required to avoid SNP’s at gRNA targets and recombinant PCR derived erroneous allele assignment, iii) plasmid derived insertions were prevalent prompting for the use of purified DNA free nucleoprotein (RNP) to at least reduce the insertion prevalence, and iv) IDAA provides a simple qualitative and quantitative means of editing analysis at the cell pool and ex-plant level in the complex potato genome. The obtained highly improved editing efficacy may significantly reduce downstream cell culturing and ex-plant re-generation in particular where full allelic and heritable transmitted gene editing is desired.
Methods
Methods and any associated references are available in the online version of the paper. Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
Online methods
Plant material
Sterile in vitro grown plantlets of Desirée and Wotan were obtained from Vitroform (Årslev, Denmark) and propagated in a Fitotron growth cabinet with 16/8 h, 24 °C/20 °C, 70% humidity, at 65μE light intensity as described in12. Regenerated plants were transferred to soil by rooting on peat under a white plastic cover. After rooting, plants were transferred to 25 cm pots and grown in the greenhouse to set tubers.
StU6 promoter sequence and cloning
StU6 promoter sequences were retrieved by BLAST searches in the reference genome (DM1–3 516 R44)3 with the sequences Z17290, Z17292, Z17293, Z17301 from the cultivar Record18 as baits. Z17290, Z17292, Z17293 and Z17301 were mapped to the reference genome using CLC Genomics Workbench (Qiagen). 5′ flanking sequences AGCAAGATGCAATGTATCAACTCA (Z17290), ACCACTTAAACTGAGAACAGTCAA (Z17292), TTCACTTAGTTCAGTTGCATTATGTC (Z17293), GATAAATTCTTAAAGTTGAGTAACC (Z17301) were used to amplify and sequence U6 upstream regions from cultivars Desirée and Wotan of 421 bp (Z17290), 433 bp (Z17292), 372 bp (Z17293) and 374 bp, 321 bp and 318 bp (Z17301) using the common RV primer GCCATGCTAATCTTCTCTGTATCG that anneals to nt 33–55 of the U6 sequence. These sequences were used to define StU6-1 (Z17290), StU6-2 (Z17292), StU6-3 (Z17293) and StU6-4, StU6-4a and StU6-4b (Z17301), which were cloned to drive the gRNA expression using Nebuilder assisted Gibson assembly (Supplementary Fig. 2C).
In silico prediction analysis of GBSS-gRNA1 and 2 targets
Exon 1 of GBSS, which includes 36 potential gRNA targets, was used as input for evaluation of the ranking of gRNA1 and gRNA2. The in silico assisted gRNA selection tools30 amenable for plants and included in the survey were CHOPCHOP v2 (http://chopchop.cbu.uib.no/)31, CRISPR-P 2.0 (http://crispr.hzau.edu.cn/CRISPR2/)32, SSC (http://crispr.dfci.harvard.edu/SSC/)33, CRISPRater (https://crispr.cos.uni-heidelberg.de/)34,35, and CRISPOR (http://crispor.tefor.net/)36.
Protoplast isolation, transformation, editing, ex-plant regeneration
Protoplast isolation, transformation, editing and ex-plant regeneration were essentially done as described in12.
DNA extraction
Plant gDNA template for PCR and IDAA was purified with GenElute Plant genomic DNA miniprep kit from Sigma and quantified using nanodrop. gDNA from harvested protoplasts was obtained by suspending the cells in H2O, snap freezing in liquid nitrogen followed incubation at 96 °C for 15′.
Genotyping
To characterize the target region, different combinations of FW primers tgtagaccacacatcacATG, tgtagaccacacatcacATGG, GCAAGCATCACAGCTTCACACC, AGCATCACAGCTTCACACCACT and, + SNP FW, and RV primers atcatttagGCCCGCGGACA, AAACGTGGGGTTGATCGTGT, ATGGCCCCAAAGCTGGACTAG and + SNP RV, were used for PCR amplification with CloneAmp™ HiFi PCR Premix (Clontech) in total reaction volumes of 25 µl with 0.25 µM primers and 0.4–0.8 ng/µl gDNA according to the manufacturers recommendations with the PCR parameters of 35 cycles: 98 °C 10″, 64 °C 15″, 72 °C (60″ per 1000 bases, extension). In the attempts to reduce PCR derived chimeras, extension time was doubled and the primer concentration increased twofold. For genotyping of edited plants and protoplasts, the edited region was amplified with + SNP FW and + SNP RV. Gel purified PCR products were cloned using the CloneJet PCR cloning Kit (Thermo Scientific), sequenced by Sanger sequencing at Macrogen and analyzed on the CLC Main Workbench.
Screening by Indel Detection by Amplicon Analysis (IDAA)
Tri-primer PCR amplicons for IDAA10 were obtained using the primers + SNP FW, IDAA + SNP RV and Fluorescein Amidite (FAM) labelled FAMF, while primers –SNP FW and FAM-labelled -SNP RV were used for di-primer PCR. PCR reactions of 25 µl using CloneAmp™ HiFi PCR Premix (Clontech) with 0.25 µM primers except IDAA + SNP RV at 0.025 µM on a template of 0.4–0.8 ng/ µl gDNA or 2.5 µl protoplast cell pool suspension were run with PCR parameters: 98 °C 10″, 64 °C 15″, 72 °C (60″ per 1000 bases, extension) 35 cycles for gDNA, 40 cycles for protoplast suspensions. The PCR product was analysed on a 3500xL Genetic analyzer (Applied Biosystems) as previously described10.
Phenotypic analysis
Lugol staining was done as described in29.
References
Li, J. et al. Whole genome sequencing reveals rare off-target mutations and considerable inherent genetic or/and somaclonal variations in CRISPR/Cas9-edited cotton plants. Plant Biotechnol J (2018).
Nadakuduti, S. S., Buell, C. R., Voytas, D. F., Starker, C. G. & Douches, D. S. Genome Editing for Crop Improvement - Applications in Clonally Propagated Polyploids With a Focus on Potato (Solanum tuberosum L.). Front Plant Sci 9, 1607 (2018).
Potato Genome Sequencing, C. et al. Genome sequence and analysis of the tuber crop potato. Nature 475, 189-195 (2011).
D’Hoop B, B. et al. Identification of agronomically important QTL in tetraploid potato cultivars using a marker-trait association analysis. Theor Appl Genet 127, 731–748 (2014).
Puchta, H. The repair of double-strand breaks in plants: mechanisms and consequences for genome evolution. J Exp Bot 56, 1–14 (2005).
Wang, Y. et al. Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol 32, 947–951 (2014).
Kusch, S. & Panstruga, R. mlo-Based Resistance: An Apparently Universal “Weapon” to Defeat Powdery Mildew Disease. Mol Plant Microbe Interact 30, 179–189 (2017).
Lonowski, L. A. et al. Genome editing using FACS enrichment of nuclease-expressing cells and indel detection by amplicon analysis. Nat Protoc 12, 581–603 (2017).
Petersen, B. L. et al. Improved CRISPR/Cas9 gene editing by fluorescence activated cell sorting of green fluorescence protein tagged protoplasts. BMC Biotechnol 19, 36 (2019).
Yang, Z. et al. Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res. 43, 8 (2015).
Yang, Z. et al. Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res 43, e59 (2015).
Nicolia, A. et al. Targeted gene mutation in tetraploid potato through transient TALEN expression in protoplasts. J Biotechnol 204, 17–24 (2015).
Clasen, B. M. et al. Improving cold storage and processing traits in potato through targeted gene knockout. Plant Biotechnol J 14, 169–176 (2016).
Andersson, M. et al. Efficient targeted multiallelic mutagenesis in tetraploid potato (Solanum tuberosum) by transient CRISPR-Cas9 expression in protoplasts. Plant Cell Rep 36, 117–128 (2017).
Long, L. et al. Optimization of CRISPR/Cas9 genome editing in cotton by improved sgRNA expression. Plant Methods 14, 85 (2018).
Wang, S. et al. Efficient targeted mutagenesis in potato by the CRISPR/Cas9 system. Plant Cell Rep 34, 1473–1476 (2015).
Hu, Y. Q., Brown, J. W., Waugh, R. & Turner, P. C. Cloning and characterisation of a U6 small nuclear RNA gene from potato. Biochim Biophys Acta 1129, 90–92 (1991).
Guerineau, F. & Waugh, R. The U6 small nuclear RNA gene family of potato. Plant Mol Biol 22, 807–818 (1993).
Allen, F. et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat Biotechnol (2018).
Koornneef, M., Dellaert, L. W. & van der Veen, J. H. EMS- and radiation-induced mutation frequencies at individual loci in Arabidopsis thaliana (L.) Heynh. Mutat Res 93, 109–123 (1982).
Wang, S. et al. No off-target mutations in functional genome regions of a CRISPR/Cas9-generated monkey model of muscular dystrophy. The Journal of biological chemistry 293, 11654–11658 (2018).
Andersson, M. et al. Genome editing in potato via CRISPR-Cas9 ribonucleoprotein delivery. Physiol Plant 164, 378–384 (2018).
Ainsworth, C., Clark, J. & Balsdon, J. Expression, organisation and structure of the genes encoding the waxy protein (granule-bound starch synthase) in wheat. Plant Mol Biol 22, 67–82 (1993).
Smyth, R. P. et al. Reducing chimera formation during PCR amplification to ensure accurate genotyping. Gene 469, 45–51 (2010).
Gao, C. The future of CRISPR technologies in agriculture. Nat Rev Mol Cell Biol 19, 275–276 (2018).
Barrell, P. J., Meiyalaghan, S., Jacobs, J. M. & Conner, A. J. Applications of biotechnology and genomics in potato improvement. Plant Biotechnol J 11, 907–920 (2013).
Li, J. F. et al. Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol 31, 688–691 (2013).
Nishimasu, H. et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949 (2014).
Heilersig, H. J., Loonen, A., Bergervoet, M., Wolters, A. M. & Visser, R. G. Post-transcriptional gene silencing of GBSSI in potato: effects of size and sequence of the inverted repeats. Plant Mol Biol 60, 647–662 (2006).
Chuai, G. H., Wang, Q. L. & Liu, Q. In Silico Meets In Vivo: Towards Computational CRISPR-Based sgRNA Design. Trends Biotechnol 35, 12–21 (2017).
Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B. & Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res 44, W272–276 (2016).
Liu, H. et al. CRISPR-P 2.0: An Improved CRISPR-Cas9 Tool for Genome Editing in Plants. Mol Plant 10, 530–532 (2017).
Xu, H. et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res 25, 1147–1157 (2015).
Stemmer, M., Thumberger, T., Del Sol Keyer, M., Wittbrodt, J. & Mateo, J. L. CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLoS One 10, e0124633 (2015).
Labuhn, M. et al. Refined sgRNA efficacy prediction improves large- and small-scale CRISPR-Cas9 applications. Nucleic Acids Res 46, 1375–1385 (2018).
Prykhozhij, S. V., Rajan, V., Gaston, D. & Berman, J. N. CRISPR multitargeter: a web tool to find common and unique CRISPR single guide RNA targets in a set of similar sequences. PLoS One 10, e0119372 (2015).
Acknowledgements
This work was supported by Kartoffelafgiftsfonden, The Danish Councils for Strategic and Independent Research (12–125709, 12–131859), The Danish National Research Foundation (DNRF107), the Copenhagen University Excellence Program for Interdisciplinary Research (CDO2016) and National Science Foundation (NSF) (1755482).
Author information
Authors and Affiliations
Contributions
I.E.J., B.L.P. and E.A. designed the experiments; K.L.N. performed U6 sequence retrieval and associated analysis, I.E.J. performed the experiments and Y.L. IDAA analysis; B.J. advised on potato tissue culture work; A.B. advised on starch analyses; E.P.B. advised on IDAA analysis. B.L.P., I.E.J., K.L. and A.B. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Johansen, I.E., Liu, Y., Jørgensen, B. et al. High efficacy full allelic CRISPR/Cas9 gene editing in tetraploid potato. Sci Rep 9, 17715 (2019). https://doi.org/10.1038/s41598-019-54126-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-54126-w
This article is cited by
-
CRISPR/Cas system for the traits enhancement in potato (Solanum tuberosum L.): present status and future prospectives
Journal of Plant Biochemistry and Biotechnology (2024)
-
Genome editing advancements in potato (Solanum tuberosum L.): operational challenges and solutions
Journal of Plant Biochemistry and Biotechnology (2023)
-
Agrobacterium tumefaciens-Mediated Plant Transformation: A Review
Molecular Biotechnology (2023)
-
CRISPR/Cas9-mediated multiple guide RNA-targeted mutagenesis in the potato
Transgenic Research (2023)
-
Recent advances and challenges in potato improvement using CRISPR/Cas genome editing
Planta (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
