
Abstract
Sponges (Phylum Porifera) are among the oldest Metazoa and considered critical to understanding animal evolution and development. They are also the most prolific source of marine-derived chemicals with pharmaceutical relevance. Cell lines are important tools for research in many disciplines, and have been established for many organisms, including freshwater and terrestrial invertebrates. Despite many efforts over multiple decades, there are still no cell lines for marine invertebrates. In this study, we report a breakthrough: we demonstrate that an amino acid-optimized nutrient medium stimulates rapid cell division in 9 sponge species. The fastest dividing cells doubled in less than 1 hour. Cultures of 3 species were subcultured from 3 to 5 times, with an average of 5.99 population doublings after subculturing, and a lifespan from 21 to 35 days. Our results form the basis for developing marine invertebrate cell models to better understand early animal evolution, determine the role of secondary metabolites, and predict the impact of climate change to coral reef community ecology. Furthermore, sponge cell lines can be used to scale-up production of sponge-derived chemicals for clinical trials and develop new drugs to combat cancer and other diseases.
Similar content being viewed by others


Introduction
Sponges (Phylum Porifera) are key components of many benthic marine ecosystems. There are more than 9,000 described species that occur worldwide, from the intertidal to the deep sea1. Among the oldest metazoans, sponges have evolved a variety of strategies to adapt to different environments. Because they are sessile as adults, they have evolved sophisticated chemical systems for communication, defense from predators, antifoulants to prevent other organisms from growing over them, and to prevent infection from microbes filtered out of the water2,3. These chemicals interact with molecules that have been conserved throughout evolutionary history and are involved in human disease processes, for example, cell cycling4, immune and inflammatory responses5, and calcium and sodium regulation6,7.
Vertebrate, insect, and plant cell lines are important tools for research in many disciplines, including human health, evolutionary and developmental biology, agriculture, and toxicology. Although cell lines have been established for freshwater and terrestrial invertebrates (e.g., Hydra, Caenorhabditis), and long-term (>1 month) primary cultures have been reported for cells derived from tissues of the cnidarian Anemonia viridis and the shrimp Penaeus8,9, attempts to establish cell lines from marine invertebrates have been unsuccessful9,10,11.
Marine sponges, including some of the species in this study, are the source of thousands of novel chemicals with pharmaceutically relevant properties12,13,14. Supply of these chemicals is a bottleneck to development of sponge-derived drug leads: wild harvest is not ecologically sustainable, and chemical synthesis is challenging due to the complexity of many of the bioactive chemical compounds. In vitro production has been proposed as an option, but the lack of permanent sponge cell lines makes this option unfeasible.
Sponge cell lines could be used as models to understand the role of secondary metabolites in sponges, to use this information to develop new models for drug discovery, and to scale-up production of sponge-derived bioactive compounds for novel medicines. Cell lines of common reef sponges could also be used to quantify the effects of climate change (ocean warming and acidification) on uptake of dissolved organic material (DOM), a major component of the “sponge loop hypothesis” of carbon cycling and to test the hypothesis that coral reefs could become sponge reefs as climate changes15.
To date, primary cultures have been established from dissociated and cryopreserved cells of several sponge species4,16,17,18; optimized nutrient media have been developed16,19,20,21,22; cell division has been stimulated with growth factors and mitogens23,24 (Munroe et al. in prep); transient expression of immortalizing genes has been obtained25; somatic cell hybridization has been demonstrated26; and methods for three-dimensional culture in hydrogels have been established27,28 (Munroe et al. in prep). These improvements in sponge cell culture were sporadic and incremental, and resulted only in a limited number of cell divisions. In this study, we report a breakthrough in marine invertebrate (sponge) cell culture: using an optimized nutrient medium22, a substantial increase in both the rate and number of cell divisions has been accomplished for the first time.
Results
An amino acid-optimized nutrient medium stimulates rapid cell division in primary cell cultures of marine sponges
We cultured cells of 12 sponge species in three different media: artificial seawater (ASW), Medium 199 (M199), and M1 (Fig. 1). As predicted from prior research4,16,17, the number of cells cultured in ASW either remained the same or decreased, except for an unidentified species of Spongiidae (Fig. 1i). Although the pattern of cell number increase and decrease in ASW for Amphimedon erina parallels the pattern for M199 and M1 after the initial decrease in ASW (Fig. 1d), there was only a small increase in cell number in ASW (~25%) compared with nearly a three-fold increase in cell number in M199 and M1 after day 1. Within two days of incubation in M1, cell numbers increased for each of the following species: Geodia barretti, Geodia sp., G. neptuni, A. erina, Amphimedon compressa, Niphates erecta, Aplysina fulva, the unidentified species of Spongiidae, and Tedania ignis (Fig. 1a–g,i,l). Of these, G. barretti, Geodia sp., G. neptuni, A. erina, A. compressa, and A. fulva had the largest increase in cell number in M1, with between 1.5 and 3 population doublings (Fig. 1a–e,g). Cell numbers also increased in M199 for the same six species, although either lower than (G. barretti, Geodia sp., G. neptuni, and A. compressa) or equal to (A. erina and A. fulva) the increase in cell numbers in M1. For three species (D. etheria, A. corrugata, and C. varians), there was no increase in cell number in any medium (Fig. 1h,j,k). The medium in cultures of each individual of each species with increases in cell number changed color from pale orange to dark grey, and the cells appeared microscopically to have dark inclusions (unpublished data). These changes were observed as soon as cultures increased in cell number and became increasingly darker as the cell density increased. The color change is not associated with a change in pH of the culture medium (spent medium pH: 7.8). Research is in progress to determine the cause of the color change and to characterize the cell inclusions.

Primary cultures of 12 sponge species in three different media: artificial seawater (ASW), Medium 199 (M199) and M1 medium. (a) Geodia barretti, (b). Geodia sp., (c). Geodia neptuni, (d). Amphimedon erina, (e). Amphimedon compressa, (f). Niphates erecta, (g). Aplysina fulva, (h). Dysidea etheria, (i). Spongiidae, (j). Axinella corrugata, (k). Cliona varians, (l). Tedania ignis. Only 1 individual of each species was tested, however, the results are the average of 3 technical replicates (n = 3) ± standard deviation.
Individual (intraspecific) variation must be factored into selection of source material
To evaluate individual (intraspecific) variation, we focused subsequent studies on the six species with the largest increase in cell density in M1: G. barretti, Geodia sp., G. neptuni, A. compressa, A. erina, and A. fulva (Fig. 1). For these species, cells from multiple individuals were cultivated in triplicate for 48 hours in M1 medium. There was little individual (intraspecific) variation in final cell density for each of the three species of Geodia cultured for 48 hours (Fig. 2a–c). Conversely, individuals of A. erina, A. compressa, and A. fulva had individual (intraspecific) variation in final cell density (Fig. 2d–f).

Primary cultures of six sponge species selected for further studies in M1 medium. (a) Geodia barretti (n = 4), (b). Geodia sp. (n = 6), (c). Geodia neptuni (n = 7), (d). Amphimedon erina (n = 9), (e). Amphimedon compressa (n = 4), (f). Aplysina fulva (n = 5). Each line represents an individual. Results are the average of 3 technical replicates (n = 3) ± standard deviation.
Marine sponge cells are capable of rapid cell division
In this study, we selected three species of Geodia because there was little intraspecific variation when cultured in M1 medium. In addition, G. barretti is the source of compounds with anti-inflammatory activity29. Cell density of G. barretti, Geodia sp., and G. neptuni was measured with a finer time resolution to understand the dynamics of cell division for each species (Fig. 3) and to identify when the cultures were near the end of exponential growth and, therefore, ready to subculture/passage. Growth curves were analyzed for all three species at both 22 °C (Fig. 3a–c), the temperature at which M1 medium was optimized, and 4 °C (Fig. 3d–f). These temperatures were chosen because Geodia sp. occurs on shallow grass flats (~2 meters) and G. neptuni occurs on shallow reefs (~20 meters) off Summerland Key, FL, USA, with sea surface temperatures ranging from 21.5 °C to 30.5 °C, and G. barretti occurs in deeper water (~500 meters) in Norwegian fjords, with sea surface temperatures ranging from 5.5 °C to 15.5 °C. Rapid cell division was observed in all Geodia species (Fig. 3a–f), although the number of population doublings (Nd) varied between species and incubation temperatures (Table 1).

Growth curves of three species of Geodia. (a–c) 22 °C, (d–f) 4 °C. (a,d) Geodia barretti, (b,e) Geodia sp., (c,f) Geodia neptuni. Each line represents an individual. The results are the average of 3 technical replicates (n = 3) ± standard deviation.
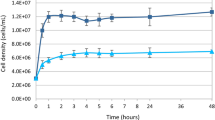
Geodia barretti cultures did not reach the same density as the other two species. There was little individual variation in peak cell density for G. barretti when cultured at 4 °C, with an average of 8.54E + 06 cells/mL (Fig. 3a). When G. barretti cells were cultured at 22 °C, two individuals reached peak densities of 1.67E + 07 cells/mL and 1.73E + 07 cells/mL (Fig. 3a). One individual at 22 °C had a lower peak density of 1.11E + 07 cells/mL (Fig. 3a).
Two individuals of Geodia sp. cultured at 22 °C reached a peak density of 6.03E + 07 cells/mL and 6.08E + 07 cells/mL within 12 hours (Fig. 3b). The third individual cultured at 22 °C reached a significantly higher peak density (8.38E + 07 cells/mL) within 9 hours (Fig. 3b). Different individual responses were observed for Geodia sp. cells cultured at 4 °C. Two individuals cultured at 4 °C reached a peak density of 4.29E + 07 cells/mL and 4.71e + 07 cell/mL (Fig. 3e). At 4 °C, one individual reached a higher peak density of 5.61E + 07 cells/mL within 24 hours (Fig. 3e); this was not the same individual with the highest density at 22 °C.
Cultures of all 3 individuals of G. neptuni incubated at 22 °C reached an average peak density of 5.54E + 07 cells/mL within 12 hours (Fig. 3c). The average peak cell density of G. neptuni cells incubated at 4 °C was lower (4.97E + 07 cells/mL) (Fig. 3f).
Cultures can be subcultured and maintained for up to several weeks
Figure 4 shows the results of subculture experiments for G. barretti, Geodia sp., and G. neptuni at both 22 °C and 4 °C. Passaging times and total number of population doublings varied among the three species. As in the growth curve experiments, G. barretti cells did not reach the same density as the other two species (~9.82E + 06 cells/mL at 4 °C and ~1.35E + 07 cells/mL at 22 °C) (Fig. 4a,d), however, G. barretti cells continued to divide until the 5th passage, ultimately reaching a total of 6.95 population doublings at 4 °C and 5.54 population doublings at 22 °C (Nd) (Table 1). Cell cultures of Geodia sp. had individual variations in peak cell densities when cultured at 22 °C, from 3.45E + 07 to 5.77E + 07 cells/mL (Fig. 4b). Similarly, the peak cell density of Geodia sp. cells cultured at 4 °C varied between individuals, from 3.79E + 07 to 5.13E + 07 cells/mL (Fig. 4e). Geodia sp. cells cultured at 4 °C reached a higher number of population doublings (Nd = 5.50) after 28 days of culture compared to cultures at 22 °C after 120 hours (Nd = 4.67) (Table 1). Geodia neptuni cultures reached 4.20 population doublings after 120 hours (5 days) (Table 1) but had an average peak cell density of 5.77E + 07 cell/mL within 12 hours (Fig. 4c). On the other hand, G. neptuni cultured at 4 °C had 5.53 population doublings over a course of 28 days (Table 1) and a lower average peak cell density, only reaching 4.38E + 07 cells/mL (Fig. 4f).

Passaging of three species of Geodia in M1 medium. (a–c) 22 °C, (d–f) 4 °C. (a,d) Geodia barretti, (b,e). Geodia sp., (c,f) Geodia neptuni. Each line represents an individual. The results are the average of 3 technical replicates (n = 3) ± standard deviation.
Cell identity was verified by 18S rRNA gene sequence analyses
Cultures were routinely monitored microscopically, however, verification that the subcultures were the three Geodia species was confirmed by 18 S rRNA gene amplicon sequencing. For all samples sequenced, at least 99.96% of the reads had a 100% match with published sequence data for Geodia species (Table 2).
Discussion
We established finite cell lines30,31 for G. barretti, Geodia sp., and G. neptuni. The cultures were monitored microscopically and were not axenic. However, sponges are holobionts with a diverse community of microbes32 that may be obligate symbionts: the microbiome of G. barretti is species-specific and stable33. Continued development of sponge cell lines, and specifically, the establishment of an axenic cell line, will provide a sponge model to test hypotheses related to the functional role(s) of the sponge microbiome.
As noted, the medium in cultures with dividing cells changed color, from pale orange to dark grey, and the sponge cells appeared microscopically to have dark inclusions (~0.5 µm). The change in medium color and appearance of dark inclusions were present in each individual of each species that had increases in cell number. These changes were observed as soon as cultures increased in cell number and became increasingly darker as the cell density increased. We hypothesize that the color change is associated with the production of melanin, a photo-protective pigment that has been reported from the ectosome of marine sponges34. Sponge-associated bacteria also produce melanin, causing color changes to media35. Research is in progress in our group to determine the exact cause of the color change in the medium and to characterize the inclusions present in the cells.
Both interspecific and intraspecific (individual) variation have been observed in metabolic responses of sponges18,22. Over the course of this study, inter- and intra-species responses were observed for peak cell densities, number of population doublings, passaging times and culture lifespan. Cells remained in stationary phase for up to 1 week, depending on the species and the culture temperature. Our results demonstrate the necessity of testing multiple individuals of the target species to identify the appropriate individuals for continued development of cell lines. The choice of which sponge species and even which individuals to use can have a significant impact on the outcome of the study. Selecting the appropriate species and individual source material to establish marine sponge cell lines cannot be overemphasized.
The establishment of sponge cell lines requires the optimization of several variables, including nutrient media, incubation temperature, inoculation/seeding density, duration between passages, and the use of antibiotics, to name a few. Optimization of nutrient media is especially important: A genetic algorithm approach was used to optimize the amino acid composition of M199 to improve metabolic activity in primary sponge cell cultures. M1 medium was optimized from M199, based on 48-hour cultures of one sponge species, D. etheria22. Even though M1 was developed for and stimulated metabolic activity in D. etheria, cells from this species did not divide22. Nevertheless, we hypothesized that the optimized medium would stimulate cell division in other sponge species, and our results demonstrate that M1 stimulated rapid cell division in 9 other species. M199 also stimulated cell division, but M1 was better for most species. Since M1 contains extra amino acids, for some sponges the amino acid content of M199 is suboptimal. Research is in progress to optimize other medium components (e.g., lipids, vitamins, trace metals, growth factors) (Munroe et al. in prep) and to develop cell lines from additional species of sponges. We hypothesize that, not unlike optimization of other eukaryotic cell lines, medium optimization will be required for each species and for the intended application of the cell line.
Conclusion
Our demonstration of exceptionally fast cell division for marine invertebrates (sponges), as well as our ability to subculture the cells, is a breakthrough in marine biotechnology. From this study, we conclude that optimization needs to be species-specific and may depend on the intended use of the cell lines. Our results form the basis for developing marine invertebrate (sponge) cell models to better understand early animal evolution and to test hypotheses related to the effects of higher temperature and lower pH on sponges. Furthermore, sponge cell lines may be used to scale-up production of sponge-derived chemicals with pharmaceutical relevance, and to gain more insight into the role of secondary metabolites in sponges to develop new models for marine natural products drug discovery.
Methods
Sample collection
Individuals of twelve sponge species (Class Demospongiae) from seven orders and eight families were collected for this study. Amphimedon erina (Order Haplosclerida, Family Niphatidae), Cliona varians (Order Clionaida, Family Clionaidae), Dysidea etheria (Order Dictyoceratida, Family Dysideidae), Geodia sp. (Order Tetractinellida, Family Geodiidae), an unidentified species of Spongiidae (Order Dictyoceratida), and Tedania ignis (Order Poecilosclerida, Family Tedaniidae) were collected from Atlantic coastal waters off Summerland Key, Florida (24°39′36.9″N 81°27′18.0″W) at a depth of approximately one to two meters, from sandy bottom grass flats and mangrove roots. Amphimedon compressa (Order Haplosclerida, Family Niphatidae), Aplysina fulva (Order Verongiida, Family Aplysinidae), Axinella corrugata (Order Axinellida, Family Axinellidae), Geodia neptuni (Order Tetractinellida, Family Geodiidae) and Niphates erecta (Order Haplosclerida, Family Niphatidae) were collected from a deeper (~20 meters) reef site off Looe Key, Florida (24°32′44.6″N 81°24′21.4″W), characterized by hard bottom with dense sponge, coral and algae cover. Geodia barretti (Order Tetractinellida, Family Geodiidae) individuals were collected in a single trawl at a depth of ~500 meters in a fjord (59°58.8″N 5°22.4″E) close to Bergen, Norway. Collections from Florida were authorized under permit #FKNMS-2014-070 from the National Oceanic and Atmospheric Administration, Office of National Marine Sanctuaries, and Special Activity License SAL-14-1588-SR from the Florida Fish and Wildlife Conservation Commission.
Sample identification
Taxonomic identification of the sponges was confirmed by evaluation of morphological characters: morphology, color, surface texture, and microscopic analysis of the skeleton. Geodia sp. (reported as Geodia vosmaeri36) is in the process of being re-described (Cardenas, personal communication), but it is an easily recognized species in shallow water grass flats off the coast of south Florida.
Dissociation and cryopreservation
Cells from all studied species were dissociated and cryopreserved immediately after sampling using previously established methods18,24,37. Sponges were cleaned of debris and rinsed in seawater filtered through a sterile 0.22 µm filter (FSW). Cells were dissociated by squeezing fragments of sponge through sterile gauze (grade 16 mesh size for G. barretti [B. Braun Medical] and grade 10 mesh size for all other species [Fisherbrand]) and filtering the cell suspension through a cell strainer (40 μm [Greiner Bio-One] for G. barretti and 70 μm [Fisherbrand]) for all other species) to eliminate debris, cell aggregates and spicules. Cells were washed twice by centrifugation at 300 × g for 5 minutes and resuspended in FSW. Cell concentrations of G. barretti were microscopically counted using disposable hemocytometers (C-chip™, Neubauer improved); cells from all other species were automatically counted using the Countess II FL Automated Cell Counter (Thermo Fisher). Dissociated cells from all species were cryopreserved at a cell concentration of approximately 1.00E + 08 cells/mL in a cryoprotectant solution (10% dimethyl sulfoxide (DMSO) and 10% fetal bovine serum (FBS) in FSW)18,24,38. Cells were pipetted (1 mL) into cryogenic vials (Fisherbrand), the vials were placed in Nalgene® Mr. Frosty freezing containers and cooled to −80 °C at a steady rate of 1 °C per minute.
Media preparation
Artificial seawater (ASW), modified from Zhang et al. (2004), was prepared by dissolving salts into filter sterilized distilled water (DIW) and then autoclaving at 121 °C for 25 min18,22. Medium 199 (Sigma Aldrich, M3769) was prepared according to the manufacturer’s protocol. M1 medium was prepared by dissolving Medium 199 powder (Sigma Aldrich, M3769) in distilled water (DIW). Salts were added in concentrations to approximate the pH (medium: 7.9; ocean: 8.1) and salinity (medium: 33.5 ppt; ocean: 35 ppt) of seawater (Table 3). Next, amino acids were added in the concentrations that were optimized for in vitro culture of the sponge D. etheria22 (Table 3). Both M1 and M199 were supplemented with rifampicin (Sigma Aldrich, R3501) and amphotericin B (Sigma Aldrich, A2411) to control bacterial and fungal contamination, respectively.
Establishment of primary cultures
For the first set of experiments, primary cultures of twelve species (one individual of each species) were evaluated to determine which species would proliferate in three different media (Table 3): artificial seawater (ASW), Medium 199 (M199)24 and M1 medium22. Cryopreserved cells were thawed rapidly in a 50 °C water bath to minimize ice crystal damage to the cells24,37. The cell suspension was rinsed twice by centrifugation at 4000 × rpm for 5 minutes and resuspended in ASW. Cell number was measured automatically using the Countess II FL Automated Cell Counter (Thermo Fisher) for all time points for all species except G. barretti, for which cell concentrations were counted microscopically using a disposable hemocytometer (C-chip, Neubauer improved). Prior to counting the cells, the cell suspension was gently pipetted to disperse aggregates. Cell aggregation was generally not an issue, however, if aggregation prevented accurate cell counts, the cells were resuspended in calcium- and magnesium-free artificial seawater (CMF)4 prior to counting. Cell concentrations at time point zero were calculated by counting the cell suspension and then resuspending in either ASW, M199, or M1 to the desired concentration. Samples were cultured in 24-well plates (Falcon): for the first two experiments (to determine if cells were dividing and to further evaluate cell proliferation in the 6 selected species), cells were incubated for four to six days at ~22 °C for all species except G. barretti, which was incubated at 4 °C. These temperatures are within the range of ambient seawater temperatures for the Florida and Norway sites, respectively. To further characterize cell division and to determine the number of times the cells could be subcultured, the three Geodia species were incubated at both ~22 °C and 4 °C.
Growth characterization
Growth curves in M1 were determined for G. barretti, Geodia sp., and G. neptuni (n = 3 specimens for each species). Specimens (=individuals) were prepared in triplicate, in 96-well plates (Falcon), for each individual and time point. All samples were incubated at ~22 °C and at 4 °C. Cell number was measured hourly for 12 hours, every 12 hours for 48 hours and every 24 hours for 120 hours (5 days) for all cultures at 22 °C, as well as G. barretti cultures at 4 °C. Cultures of Geodia sp. and G. neptuni at 4 °C were measured hourly for 24 hours and every 24 hours for 120 hours. Three seeding densities (5.00E + 05, 1.00E + 06, and 5.00E + 06 cells/mL) were evaluated to determine the effect of inoculation density on final cell densities (unpublished data). The optimal seeding density (i.e., the inoculation density that resulted in the greatest increase in cell number) was 3.00E + 06 cells/mL for G. barretti and 5.00E + 06 cells/mL for Geodia sp. and G. neptuni. Cells were automatically counted using the Countess II FL Automated Cell Counter (Thermo Fisher) for Geodia sp. and G. neptuni, and manually counted for G. barretti using disposable hemocytometers (C-chip, Neubauer improved) for all time points except for G. barretti at time point zero, which was calculated by counting the cell suspension, and then resuspending in M1 at the desired concentration.
Passaging cells
Cultures were passaged in M1 using three individuals per species, in triplicate, in 24-well plates (Falcon). All three species were cultured at both ~22 °C and 4 °C. The seeding density was 3.00E + 06 cells/mL for G. barretti, and 5.00E + 06 cells/mL for Geodia sp. and G. neptuni. Passaging was performed by resuspending the cells by pipetting, determining the cell concentration and subsequently diluting the cells, by either splitting the culture (e.g., 1:2 ratio) or diluting back to the seeding density. Cultures were passaged until the cells stopped dividing in order to determine the lifespan of the cell lines for each species. Cell number was measured automatically using the Countess II FL Automated Cell Counter (Thermo Fisher) for Geodia sp. and G. neptuni. Geodia barretti cells were counted microscopically using disposable hemocytometers (C-chip, Neubauer improved) for all time points except for time point zero which was calculated by counting the cell suspension and then diluting to the desired concentration. Cultures of all three species were monitored microscopically (EVOS FL Auto imaging system, Invitrogen).
Sponge cell verification
To confirm that the cells in culture were from the three species of Geodia, 18 S rRNA gene amplicon sequencing and eukaryotic community profiling was performed on cultures of all three species, both before (time point zero for all three species) and after passaging (one passage, time point 48 hours, T = 22 °C for G. neptuni and Geodia sp.; four passages, time point 28 days, T = 4 °C for G. barretti). Cell pellets (approximately 1.00E + 08 cell/mL) of each species were stored at −20 °C. Genomic DNA extraction, 18 S rRNA gene amplification through polymerase chain reaction (PCR) and Illumina MiSeq sequencing and sequence analysis were performed by RTL Genomics (Lubbock, Texas, USA). A High Pure PCR Template Preparation Kit (Roche Life Science, Basel, Switzerland) was used to extract genomic DNA following the manufacturer’s protocol, with one exception: after addition of binding buffer and proteinase K, the samples were incubated at 70 °C for a prolonged period (35 minutes) to increase DNA yield. Fungal primers that were previously used for eukaryotic community analysis of a sponge holobiont39 were used to amplify an approximately 350 base pair long region of the eukaryotic small-subunit rRNA gene, including the V7 and V8 hypervariable regions in a two-step process. The forward primer was constructed with (5′-3′) Illumina i5 sequencing primer (TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG) and the FF390 primer (CGATAACGAACGAGACCT)40. The reverse primer was constructed with (5′-3′) Illumina i7 sequencing primer (GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG) and the FR1 primer (ANCCATTCAATCGGTANT)40. Amplifications were performed in 25 µL reactions with Qiagen HotStarTaq master mix (Qiagen Inc., Valencia, California), 1 µL of each 5 µM primer, and 1 µL of template. Reactions were performed on ABI Veriti thermocyclers (Applied Biosystems, Carlsbad, California) under the following thermal profile: 95 °C for 5 minutes, then 30 cycles of 95 °C for 30 seconds, 50 °C for 45 seconds, and 72 °C for 1 minute, followed by a final extension of 72 °C for 10 minutes, and a 4 °C hold. Products from the first stage amplification were added to a second PCR based on qualitatively determined concentrations. Primers for the second PCR were designed based on the Illumina Nextera PCR primers:
Forward-AATGATACGGCGACCACCGAGATCTACAC[i5index]TCGTCGGCAGCGTC and Reverse-CAAGCAGAAGACGGCATACGAGAT[i7index]GTCTCGTGGGCTCGG. The second stage amplification was run with the following thermal profile: 95 °C for 5 minutes, then 10 cycles of 94 °C for 30 seconds, 54 °C for 40 seconds, and 72 °C for 1 minute, followed by a final extension of 72 °C for 10 minutes and a 4 °C hold. Sequence data were analyzed using the in-house data analysis pipeline of RTL Genomics (version 2.3.1).
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Van Soest, R. W. M. et al. Global diversity of sponges (Porifera). PLoS one 7, 1–23 (2012).
Loh, T. & Pawlik, J. R. Chemical defenses and resource trade-offs structure sponge communities on Caribbean coral reefs. PNAS 111, 4151–4156 (2014).
Carstens, B. B. et al. Isolation, characterization, and synthesis of the barrettides: disulfide- containing peptides from the marine sponge Geodia barretti. J. Nat. Prod. 78, 1886–1893 (2015).
Schippers, K. J., Martens, D. E., Pomponi, S. A. & Wijffels, R. H. Cell cycle analysis of primary sponge cell cultures. In Vitro Cell. Dev. Biol. Anim. 47, 302–311 (2011).
Keyzers, R. A. & Davies-Coleman, M. T. Anti-inflammatory metabolites from marine sponges. Chem. Soc. Rev. 34, 355–365 (2005).
Hilge, M., Aelen, J. & Vuister, G. W. Ca2+ regulation in the Na+/Ca2+ exchanger involves two markedly different Ca2+ sensors. Mol. Cell 22, 15–25 (2006).
Martin, V. et al. Differential effects of crambescins and crambescidin 816 in voltage-gated sodium, potassium and calcium channels in neurons. Chem. Res. Toxicol. 26, 169–178 (2013).
Ventura, P. et al. Cnidarian primary cell culture as a tool to investigate the effect of thermal stress at cellular level. Mar Biotechnol 20, 144–154 (2018).
Rinkevich, B. Marine invertebrate cell cultures: new millennium trends. Mar. Biotechnol. 7, 429–439 (2005).
Grasela, J. J., Pomponi, S. A., Rinkevich, B. & Grima, J. Efforts to develop a cultured sponge cell line: revisiting an intractable problem. In Vitro Cell. Dev. Biol. Anim. 48, 12–20 (2012).
Cai, X. & Zhang, Y. Marine invertebrate cell culture: a decade of development. J. Oceanogr. 70, 405–414 (2014).
Sipkema, D. et al. Large‐scale production of pharmaceuticals by marine sponges: Sea, cell, or synthesis? Biotechnol. Bioeng. 90, 201–222 (2005).
Laport, M. S., Santos, O. C. S. & Muricy, G. Marine sponges: potential sources of new antimicrobial drugs. Curr. Pharm. Biotechnol. 10, 86–105 (2009).
Perdicaris, S., Vlachogianni, T. & Valavanidis, A. Bioactive natural substances from marine sponges: new developments and prospects for future pharmaceuticals. Nat. Prod. Chem. Res. 1, 2329–6836 (2013).
de Goeij, J. M., Lesser, M. P. & Pawlik, J. R. Nutrient fluxes and ecological functions of coral reef sponges in a changing ocean in Climate Change, Ocean Acidification and Sponges: Impacts Across Multiple Levels of Organization (ed. Carballo, J. L. & Bell, J. J) 373–410 (Springer, 2017).
De Rosa, S. et al. Development in primary cell culture of demosponges. J. Biotechnol. 100, 119–125 (2003).
Richelle-Maurer, E. et al. Primary cultures from the marine sponge Xestospongia muta (Petrosiidae, Haplosclerida). J. Biotechnol. 100, 169–176 (2003).
Munroe, S., Martens, D. E., Sipkema, D. & Pomponi, S. A. Comparison of cryopreservation techniques for cells of the marine sponge Dysidea etheria. Cryoletters 39, 269–278 (2018).
Willoughby, R. & Pomponi, S. A. Quantitative assessment of marine sponge cells in vitro: development of improved growth medium. In Vitro Cell. Dev. Biol. Anim. 36, 194–200 (2000).
Zhang, X., Pennec, G. L., Steffen, R., Muller, W. E. G. & Zhang, W. Application of a MTT assay for screening nutritional factors in growth media of primary sponge cell culture. Biotechnol. Prog. 20, 151–155 (2004).
Zhao, Q., Zhang, W., Jin, M., Yu, X. & Deng, M. Formulation of a basal medium for primary cell culture of the marine sponge. Biotechnol. Prog. 21, 1008–1012 (2005).
Munroe, S., Sandoval, K., Martens, D. E., Sipkema, D. & Pomponi, S. A. Genetic algorithm as an optimization tool for the development of sponge cell culture media. In Vitro Cell. Dev. Biol. Anim. 55, 149–158 (2019).
Pomponi, S. A. & Willoughby, R. Sponge cell culture for production of bioactive metabolites. Proceedings of the 4th International Porifera Congress: Sponges in Time and Space (A. A. Balkema, Rotterdam, 1994).
Pomponi, S. A., Willoughby, R., Kaighn, M. E. & Wright, A. E. Development of techniques for in vitro production of bioactive natural products from marine sponges. Invertebrate Cell Culture: Novel Directions and Biotechnology Applications 231–237 (Science Publishers Inc., Enfield, New Hampshire, 1997).
De Caralt, S., Uriz, M. J. & Wijffels, R. H. Cell culture from sponges: pluripotency and immortality. Trends Biotechnol. 25, 467–471 (2007).
Pomponi, S. A., Jevitt, A., Patel, J. & Diaz, M. C. Sponge hybridomas: applications and implications. Integr. Comp. Biol. 53, 524–530 (2013).
Pozzolini, M. et al. Influence of rocky substrata on three-dimensional sponge cells model development. In Vitro Cell. Dev. Biol. Anim. 46, 140–147 (2010).
Wijffels, R. H., Osinga, R., Pomponi, S. A. & Tramper, J. Marine sponges as biocatalysts. Multiphase Bioreactor Design (Taylor & Francis, London and New York, 2001).
Di, X. et al. 6-bromoindole derivatives from the Icelandic marine sponge Geodia barretti: isolation and anti-Inflammatory activity. Mar. Drugs 16, 437 (2018).
Freshney, R. I. Culture of Animal Cells: A Manual of Basic Technique and Specialized Applications Ch. 12 (John Wiley & Sons, Inc., Hoboken, New Jersey, 2016).
Schaeffer, W. I. Terminology associated with cell, tissue, and organ culture, molecular biology and molecular genetics. In Vitro Cell. Dev. Biol. Anim. 26, 97–101 (1990).
Pita, L., Rix, L., Slaby, B. M., Franke, A. & Hentschel, U. The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome 6, 1–18 (2018).
Steinert, G., Rohde, S., Janussen, D. & Blaurock, S. P. J. Host-specific assembly of sponge- associated prokaryotes at high taxonomic ranks. Sci. Rep. 7, 2542 (2017).
Araujo, M., Xavier, J. R., Nunes, C. D., Vaz, P. D. & Humanes, M. Marine sponge melanin: a new source of an old biopolymer. Struct. Chem. 23, 115–122 (2012).
Vijayan, V. et al. Sponge-associated bacteria produce non-cytotoxic melanin which protects animal cells from photo-toxicity. Appl. Biochem. Biotechnol. 183, 369–411 (2017).
Ramsby, B., Massaro, A., Marshall, E., Wilcox, T. & Hill, M. Epibiont-basibiont interactions: examination of ecological factors that influence specialization in a two-sponge association between Geodia vosmaeri (Sollas, 1886) and Amphimedon erina (de Laubenfels, 1936). Hydrobiologia 687, 331–340 (2012).
Pomponi, S. A. & Willoughby, R. Development of sponge cell cultures for biomedical applications. Aquatic Invertebrate Cell Culture 36, 194–200 (2000).
Mussino, F. et al. Primmorphs cryopreservation: a new method for long-time storage of sponge cells. Mar. Biotechnol. 15, 357–367 (2013).
Naim, M. A., Smidtm, H. & Sipkema, D. Fungi found in Mediterranean and North Sea sponges: how specific are they? PeerJ 5, e3722 (2017).
Vainio, E. J. & Hantula, J. Direct analysis of wood-inhabiting fungi using denaturing gradient gel electrophoresis of amplified ribosomal DNA. Mycol. Res. 104, 927–936 (2000).
Acknowledgements
This research was supported by the European Union Marie Curie Grant (ITN-2013-BluePharmTrain-607786) (to D.S.), the European Union Horizon 2020 Project SponGES (grant agreement No. 679848) (to D.S., S.P., D.M.), the Harbor Branch Oceanographic Institute Foundation, Aquaculture and Save Our Seas Specialty License Program (to S.P.), and the National Oceanic and Atmospheric Administration, Cooperative Institute for Ocean Exploration, Research, and Technology (award number NA14OAR43202600 (to S.P.). This document reflects only the authors’ views; sponsors are not responsible for any use that may be made of the information it contains. We thank Dr. M. Cristina Diaz (HBOI-FAU) and Dr. Patricia Blackwelder (Nova Southeastern University) for microscopic analyses and discussions regarding the color change in nutrient media. We thank Dr. Hans Tore Rapp (University of Bergen) for collecting Geodia barretti samples. S.P. also acknowledges the many collaborators, undergraduate and graduate students (especially Dr. Robin Willoughby and Dr. Klaske Schippers), post-doctoral fellows, and research assistants whose contributions to the sponge biotechnology programs at Florida Atlantic University-Harbor Branch Oceanographic Institute and Wageningen University provided the foundation for the success achieved in this study.
Author information
Authors and Affiliations
Contributions
M.C. conceived, designed, and conducted experiments for all species except G. barretti, analyzed data for all species except G. barretti, assisted with preparation of figures for the paper, and wrote sections of the paper. K.H. conceived, designed, and conducted experiments for G. barretti, analyzed data for G. barretti, prepared figures for the paper, and wrote sections of the paper. S.M. and K.S. conceived, designed, and conducted experiments for nutrient media optimization and the development of medium M1. D.M. conceived and designed experiments, analyzed data, contributed reagents/materials/analysis tools, and reviewed drafts of the paper. D.S. conceived and designed experiments, analyzed data, contributed reagents/materials/analysis tools, wrote sections of the paper, and reviewed drafts of the paper. R.W. conceived and designed experiments, contributed reagents/materials/analysis tools, and reviewed drafts of the paper. S.P. conceived, designed, and conducted experiments, collected the sponges, supervised the research, analyzed data, contributed reagents/materials/analysis tools and field expenses, and wrote sections of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Conkling, M., Hesp, K., Munroe, S. et al. Breakthrough in Marine Invertebrate Cell Culture: Sponge Cells Divide Rapidly in Improved Nutrient Medium. Sci Rep 9, 17321 (2019). https://doi.org/10.1038/s41598-019-53643-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53643-y
This article is cited by
-
First continuous marine sponge cell line established
Scientific Reports (2023)
-
Development of an in vitro tissue culture system for hammer coral (Fimbriaphyllia ancora) ovaries
Scientific Reports (2021)
-
Cultivation of fractionated cells from a bioactive-alkaloid-bearing marine sponge Axinella sp.
In Vitro Cellular & Developmental Biology - Animal (2021)
-
Establishing Sustainable Cell Lines of a Coral, Acropora tenuis
Marine Biotechnology (2021)
-
Integrating novel tools to elucidate the metabolic basis of microbial symbiosis in reef holobionts
Marine Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
