
Abstract
The most common histological subtypes of cutaneous melanoma include superficial spreading and nodular melanoma. However, the spectrum of somatic mutations developed in those lesions and all potential druggable targets have not yet been fully elucidated. We present the results of a sequence capture NGS analysis of 114 primary nodular and superficial spreading melanomas identifying driver mutations using biostatistical, immunohistochemical and/or functional approach. The spectrum and frequency of pathogenic or likely pathogenic variants were identified across 54 evaluated genes, including 59 novel mutations, and the newly identified TP53 loss-of-function mutations p.(L194P) and p.(R280K). Frequently mutated genes most commonly affected the MAPK pathway, followed by chromatin remodeling, and cell cycle regulation. Frequent aberrations were also detected in the genes coding for proteins involved in DNA repair and the regulation and modification of cellular tight junctions. Furthermore, relatively frequent mutations were described in KDR and MET, which represent potential clinically important targets. Those results suggest that with the development of new therapeutic possibilities, not only BRAF testing, but complex molecular testing of cutaneous melanoma may become an integral part of the decision process concerning the treatment of patients with melanoma.
Similar content being viewed by others


Introduction
Cutaneous melanoma (CM) is a malignant tumor with an increasing incidence worldwide. The most common cutaneous melanoma subtypes include superficial spreading melanoma (SSM) and nodular melanoma (NM)1.
Germline mutations in a number of genes have already been associated with an increased risk of familial CM (including BAP1, CDKN2A, CDK4, MITF, TERT, POT1). Several studies have also analyzed a spectrum of somatic mutations in distinct melanoma subtypes and identified the key CM drivers that comprised mutations in BRAF, RAS (mainly NRAS, but mutations in HRAS and KRAS are responsible for <1%), NF1, TP53, CDKN2A, PTEN, IDH1, ARID2, RAC1, RB1, and PPP6C2,3,4,5,6,7. Systematical analyses of genomic data retrieved from publicly available datasets, especially from the largest available dataset at the TCGA Research Network (https://www.cancer.gov/tcga), identified other genes associated either with melanoma susceptibility, development or progression8,9,10,11. However, a majority of the previously published studies dealing with the identification of frequently-mutated melanoma genes lacked a detailed description of how the evaluation of pathogenicity of the detected mutations was performed (especially when it comes to novel, non-curated variants, and single nucleotide variants with a conflicting interpretation). Therefore, data on the prevalence of pathogenic and likely pathogenic (class 4 and 5) mutations and their contribution to melanomagenesis has not yet been analyzed systematically.
In this study, targeted next generation sequencing (NGS) analysis was performed using a custom-designed panel (spanning 219 kbp) in a sample set of 114 primary CMs from Czech patients, all with complete clinico-pathological data. The goal of the study was (i) to perform NGS analysis and a comprehensive biostatistical evaluation of the pathogenicity of the detected variants, (ii) to describe the spectrum and frequency of somatic class 4/5 mutations in primary CMs, (iii) to validate the biostatistical algorithm for pathogenicity prediction, and (iv) to analyze the relationship between frequently mutated genes and clinico-pathological variables or disease outcome, including disease-free survival (DFS), local recurrence-free survival (LFS), and metastasis-free survival (MFS). The results should help in further uncovering of the spectrum and frequency of the clinically important variants in CM.
Results
Variants detected in primary CMs
NGS analyses of primary CMs from 114 patients (Table 1) revealed on average 288 variants detected per sample, including all the detected synonymous and non-synonymous variants with variant allele frequency (VAF) >10%. Implementation of high-stringent probe design (chosen to eliminate non-specific probe binding) led to the exclusion of 18 genes from further evaluation due to low coverage in a majority of samples (Supplementary Data 1). The prioritized variants in 54 evaluated genes were subsequently processed (for detailed information see Methods and Supplementary Table 1). The biostatistical algorithm filtered out 142 different benign or likely benign variants in 41 genes (33 VUS and 109 novel missense variants, mainly novel variants detected in ATM, BRCA2, ESR1, KDR, PPM1D, SF3B1, and TJP1). Altogether, we found 144 different class 4/5 mutations in 42 out of 54 evaluated genes (Table 2). The spectrum of mutations included 59 novel mutations in 31 genes (underlined in Table 2). Among these were 16 protein truncation mutations in ARID1A, ARID2, ATM, CCND2, ESR1, KDR, MET, PDGFRA, PTEN, and TJP1, 8 splice mutations in the consensus splice site in AKT3, ARID2, HNF1B, PPM1D, RB1, and TP53, one in-frame mutation (ZEB2), one frameshift mutation leading to a loss of the termination codon (PARD3) and 33 missense class 4/5 mutations in 23 genes. The most prevalent pathogenic missense BRAF and NRAS mutations were mutually exclusive, except for one case with NRAS p.(G13C) and BRAF p.(G466E) variants, which are both outside the hot-spot codons and of uncertain clinical impact. The BRAF mutations (BRAFmut) were present in 62/114 (54.4%) cases, of those 57 (91.9%) harbored a mutation in the V600 codon. The NRAS mutations (NRASmut) were detected in 35/114 (30.7%) cases, and the alteration of the Q61 codon was present in 34 (97.1%) cases. Other genes frequently affected by class 4/5 mutations were ARID1A, ARID2, ATM, KDR, MAP2K1, MET, PARD3, and TP53 (>5% of cases), and BRCA2, ESR1, IDH1, NBN, PPP6C, PTEN, SF3B1, TJP1 and ZEB2 (3–5% of cases). At least one class 4/5 variant was identified in 108/114 (95%) of the samples. In 12 non-BRAFmut/non-NRASmut samples, class 4/5 mutations affected genes ARID1A, ARID2, AKT3, ATM, BRCA2, CCND2, CYP19A1, ESR1, ESR2, F11R, IDH1, KDR, KIT, MAP2K1, MET, MSH6, NBN, PARD3, SF3B1, TJP1, TP53, and ZEB2. In 6/114 (5%) patients no class 4/5 mutation was found (Fig. 1). The number of mutations/mutated genes per sample ranged between 0 to 6 (mean = 2.4/2.3, median = 2). We identified multiple mutations (>1) in 69/114 (60.5%) samples (Fig. 1). In samples with mutually-exclusive BRAF and NRAS mutations, we identified an additional mutation in 34/62 (54.8%) BRAFmut and in 26/35 (74.3%) NRASmut cases, respectively. Interestingly, one melanoma which developed a novel NRAS non-hot-spot mutation p.(P140S) predicted by in silico approach to be benign also carried a pathogenic hot-spot KRAS mutation p.(G12C). Furthermore, the in silico analyses algorithm suggested the pathogenicity of 15 missense variants in 10 genes (ATM, BRCA2, BRIP1, CDH1, IDH1, MET, NBN, RB1, SF3B1, and TP53) which are catalogued in the ClinVar with conflicting interpretation or uncertain significance. The pathogenicity of ARID1A and TP53 mutations was also confirmed by immunohistochemical and functional assays. We did not find any class 4/5 mutation in 12 of 54 evaluated genes, namely in BARD1, JAM2, JAM3, MAP2K1, MDM2, MITF, MSH2, MYC, PALB2, POT1, RAD51C, and RAD51D.

Spectrum of genes with detected pathogenic or possible pathogenic variants in 114 primary cutaneous melanomas with respect to their clinico-pathological characteristics. Cells in red indicate pathogenic mutation, cells in gray indicate likely pathogenic mutation.
Germline mutations
We identified germline mutation in three melanoma patients. The first one was a germline ATM c.8228C > T, p.(T2743M) variant (found alongside the somatic BRAF p.(V600E), and CDK4 p.(R24H) mutations) in a male patient diagnosed at the age of 32 years with pT3 primary NM located at forearm. The second one was a germline MLH3 mutation c.958T > G, p.(C320G) (found together with somatic pathogenic mutation in BRAF p.(G469E), and a likely pathogenic mutation in ZEB2 p.(V463A)) which was identified in a female patient diagnosed with primary pT4 NM with ulceration on the back at the age of 84. Finally, there was a germline, likely pathogenic IDH1 mutation c.245G > A, p.(R82K) (found alongside somatic, likely pathogenic mutations in BRAF p.(V600E), ATM p.(E2014K), ESR2 p.(T299I), and ZEB2 p.(G1068D)) detected in one female patient diagnosed with pT2 SSM on a lower extremity at the age of 76.
Primary melanoma pathways
The majority of the affected genes codes for proteins which are involved in RAS signaling (BRAF, NRAS, ESR1, ESR2, MAP2K1, MET, KDR, and PTEN), DNA damage response and cell cycle regulation (ATM, BRCA1, BRCA2, BRIP1, KDR, NBN, PPM1D, PPP6C, PTEN, SMARCB1, and TP53), chromatin remodeling (ARID1A, ARID2, SMARCB1), or tight junction regulation (PARD3, TJP1, ZEB2). Another frequently affected genes not assigned into main functional pathways were CYP19A1 (cytochrome P450 family monooxygenase), ESR1 (nuclear hormone receptor signaling pathway), IDH1 (enzyme in citrate cycle), or SF3B1 (splicing factor 3B subunit, RNA splicing).
Validation of the in silico prediction
Only the genes affected by mutations with an already known impact and those where an optimized functional and/or IHC analysis was available were chosen for this validation. We performed immunohistochemical (IHC) analysis of ARID1A and p53 protein expression in tissue sections from samples with mutations. Functional in vitro assessment of the detected TP53 variants was also performed in order to validate the utility of the in silico prediction of mutation’s pathogenicity. The comparison of the currently known impact of the detected ARID1A and TP53 mutations (databases) with our in silico evaluation and IHC/functional analyses is summarized in Table 3.
When comparing the impact of the detected ARID1A mutations, one sample with a missense ARID1A p.(E1779G) mutation (which had been previously classified as VUS and predicted as benign using our in silico pipeline) showed a strong expression of ARID1A in 80% of the tumor nuclei, which is a similar extent of expression to that found in tissues with wild-type ARID1A (Fig. 2A). Interestingly, a different sample with a nonsense mutation p.(R1721X) with VAF 19% showed immunohistochemical expression of ARID1A in <1% of tumor nuclei (Fig. 2B). Moreover, significantly reduced ARID1A expression (the nuclear ARID1A positivity ranged between 1 and 30%) was observed in all cases possessing class 4/5 mutations.

Representative examples for the ARID1A and p53 staining. (A) weak and focally strong ARID1A positivity in a case with a novel benign p.(E1779G) missense mutation, 200x magnification; (B) absence of ARID1A staining in a melanoma with a novel nonsense pathogenic mutation p.(R1721X), 200×; (C) wild-type p53 staining with an aberrant clone with nuclear p53 overexpression in a melanoma with detected novel splice TP53 mutation c.75-1G>T, affecting the consensus splice site in intron 2, 100×; (D) aberrant p53 staining with diffuse strong nuclear expression in a melanoma that developed pathogenic missense TP53 mutation p.(G245D), 100×.
We observed aberrant p53 expression (Fig. 2D) in four of the five melanomas with pathogenic TP53 mutation. The wild-type p53 expression was observed in one case with a novel variant (predicted as likely benign by in silico and functional evaluation), and in two cases with variants affecting the canonical splice-donor sites. However, there was one case with a transversion affecting the canonical splice-acceptor site in which we observed two different types of expression within a single tumor lesion – there was a wild-type p53 expression, found together with a focus of clonal-like aberrant p53 expression (Fig. 2C).
The impact of the missense TP53 mutations on cellular functions was studied by reconstitution assay, in which we expressed either the wild-type or mutant p53 in TP53 knock-out cells (Fig. 3A–C). Immunoblotting and flow cytometry showed that the TP53 mutation p.(P80S) was able to induce comparable levels of p21 expression as the wild-type p53. Conversely, p53 mutants p.(L194P), p.(R273P), p.(G245D) and p.(R280K) failed to induce p21 and scored similarly as the control loss-of-function TP53 mutation p.(L252P). In addition, the p.(L194P), p.(R273P), p.(G245D) mutants showed defects in acetylation of K382 and phosphorylation of S15 and S46. The cell growth suppression was only slightly reduced in the p.(P80S) mutant, which is comparable with wild-type p53, whereas p.(L194P), p.(R273P), p.(G245D) and p.(R280K) mutants showed an impaired ability to suppress cell growth, which is comparable with the loss-of function control RPE-p53-KO cells (Fig. 3D).

Functional analysis of the detected TP53 mutations. (A) RPE-TP53-KO cells were transiently transfected with a plasmid (expressing either wild-type or mutant p53) and a GFP reporter and were collected 20 hours after exposure to ionizing radiation. Parental RPE cells and mock transfected RPE-TP53-KO cells were used as controls. Cell lysates were probed with indicated antibodies by immunoblotting. All the samples were run in the same experiment, same blotting, on multiple gels. TFIIH was used as a loading control, GFP as a control of transfection. Grouped blots are cropped from different exposures (depending on the antibody’s signal strength), full length blots are included in the Supplementary Information File. (B,C) RPE-TP53-KO cells were processed as in (A) and analyzed by flow cytometry. A mean intensity of p21 and p53 was determined in the GFP positive cells. (D) RPE-TP53-KO cells were stably reconstituted with the wild type or mutant p53 and were grown in the presence of nutlin-3 for 7 days. The level of cell growth suppression was determined by resazurin assay and was normalized to the cells reconstituted with the wild type p53 (n = 3).
Occurrence of mutations in relation to clinico-pathological variables
The relationship between the occurrence of mutations in frequently mutated genes and the age, histological subtype, tumor stage, sex or tumor localization was studied (Table 4). The melanomas with ARID2 mutation were significantly associated with a higher age at diagnosis, both when the age was dichotomously categorized according to the mean age (χ2 = 5.07, p = 0.024) and when age was evaluated as a continuous predictor (logistic regression, Wald statistics = 4.03, p = 0.044). The PARD3 mutated melanomas were also associated with a higher age at diagnosis, but this result was only marginally significant (χ2 = 4.00, p = 0.046). This effect was not significant when age was treated as a continuous variable (logistic regression, Wald statistics = 3.52, p = 0.061). The ARID1A mutations were more frequently detected on the head + upper + lower extremities, compared to the trunk (χ2 = 5.10, p = 0.024).
Furthermore, we also analyzed the relationship between the number of mutations/mutated genes and the clinico-pathological variables (Fig. 4). The average number of pathogenic, likely pathogenic mutations and/or mutated genes per sample was significantly associated with particular histological subtypes. Both mutations and mutated genes were enriched in NM in comparison with SSM (ANOVA, F (2, 111) = 4.919, p = 0.009; Fig. 4C), and also in NRAS mutated patients when compared with BRAFmut or non-BRAFmut/non-NRASmut (ANOVA, F (4, 218) = 3.001, p = 0.019; Fig. 4D). Non-significant correlations were observed for tumor stage and location (Fig. 4A,B).

The average number of mutations/mutated genes in relation to the selected clinico-pathological variables in the cohort of 114 patients (A–C) and 113 patients (D – one case with concurrently detected both BRAF and NRAS mutations was excluded from this kind of analysis). P-values are based on ANOVA, significant p-values are indicated in bold. Vertical bars denote 0.95 confidence intervals.
The most prominent substitutions were the C>T (19%; 6193/32848) and G>A (19%; 6124/32848) transitions. The C>T substitutions constituting 5.7–39.3% (mean 16.8%, median 15.8%) of all the detected mutations with VAF >10% per patient were more common in NM when compared with the SSM cases (ANOVA, F (1,112) = 7.085, p = 0.009). They were also more frequently localized on the head than on other parts of the body (ANOVA, F (3,110) = 5.103, p = 0.002). There were no significant differences in the distribution of C>T substitutions among the tumor stage groups.
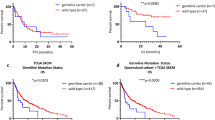
Prognostic value of BRAF and NRAS mutation status
Nine patients treated by targeted therapy (including BRAF inhibitors, anti-CTLA-4, and anti-PD-1) were excluded from the analyses to minimize the potential beneficial influence of the treatment on the length of survival. Out of those 105 patients 37 patients underwent adjuvant interferon alpha (IFN-α) therapy. There was no significant difference in the survival rates among BRAFmut, NRASmut and/or non-BRAFmut/non-NRASmut in any of the evaluated outcomes (DFS: χ2 = 0.767, p = 0.681, LFS: χ2 = 1.239, p = 0.538, MFS: χ2 = 2.582, p = 0.275).
Discussion
Melanoma is a complex and heterogeneous disease with multiple signaling pathways implicated in its pathogenesis. In our cohort of primary NM and SSM, the spectrum of the genes affected by somatic class 4/5 mutations included predominant BRAF and NRAS mutations which were mutually exclusive, with the exception of one case. However, concomitant BRAF and NRAS alterations, both located outside the hot-spots BRAFV600/NRASQ61, were also described in another study12. We identified somatic mutations with VAF >3% in the following genes: ARID1A, ARID2, ATM, BRCA2, BRIP1, ESR1, IDH1, KDR, MAP2K1, MET, NBN, PARD3, PPP6C, PTEN, SF3B1, TJP1, TP53 and ZEB2. Many of these have already been described in CM3,4,5,13,14. Interestingly, a majority of the detected class 4/5 mutations in ARID2, KDR, MET, PARD3 and ZEB2 were novel (Table 2). The ATM and PPP6C mutations have been previously described in a cohort of sun-exposed melanomas5,13. Somatic BRCA2 mutations are rare events and, additionally, germline mutations in BRCA2 have also already been described in melanoma in a few studies, although its reported association with an increased risk of melanoma is questionable15. Mutations in the IDH1 gene (codes for isocitrate dehydrogenase 1) have been associated with the development of multiple malignances. Relatively frequent IDH1 mutations (4.9%) were detected also in metastatic melanoma associated with NRAS mutation16. We found a co-occurrence of IDH1 with NRAS mutations in 3/5 primary melanomas (Fig. 1). In the SF3B1 gene we identified six class 4 mutations in 5/114 (4.4%) samples. Somatic SF3B1 mutations have been described in several malignancies, including mucosal3 and uveal melanoma, but not in CM2,17.
In 6 patients (6/114, 5.3%) there were no class 4/5 mutations detected in any of the 54 evaluated genes, which suggests the involvement of other genes which were not covered by our sequencing panel, or other alternative molecular mechanisms affecting the genes, such as promoter mutations, copy number variations, translocations or the involvement of the non-coding genome in melanoma pathogenesis9.
CM is a heterogeneous disease, where the interplay of several factors, including environmental exposures, genetic and other factors, leads to the formation of melanomas with different biological and clinical characteristics2,3. UV radiation has been suggested as a major mutagenic factor in melanoma4,13,18. The C>T transitions are typically associated with ultraviolet radiation-induced DNA damage18,19,20,21. Melanomas frequently show UV signature mutations, but not all melanomas are associated with UV exposure19. In our cohort, prominent C>T substitutions accounted for 6–39% (mean 16.8%) of all the mutations per case. None of the cases passed the previously set criteria for UV signature (>60% C>T substitutions)8, which suggests that middle-sized target panels (about 200 kb) are not suitable for the assessment of UV signature. Different mutation spectra between melanomas of the skin from locations with a high degree of cumulative sun damage (high-CSD melanomas) and low-CSD melanomas have also been described19, including increased frequencies of mutations of TP53 and ARID2 in high-CSD melanomas4. In our cohort, which is limited in its size, no correlation was found between the frequency of class 4/5 mutations in those genes and the location of the tumor. A higher mutation rate was observed in NM compared to SSM (p = 0.009). The same pattern was also observed by others in the metastatic NM cohort when compared with SSM22. The mutation rate in melanomas occurring on chronically sun exposed skin was found to be approximately five times higher than on skin without chronic sun exposure8,23. In our study, in which we only considered class 4/5 mutations, there was a clear trend where more mutations were detected in the melanomas located on the head (Fig. 4B). Nevertheless, the correlations are limited as only 10 cases were considered (5 NM and 5 SSM). The results are also influenced by the relatively small size of our panel, which does not allow for the assessment of the exact number of mutations or tumor mutation burden.
Germline, likely pathogenic mutations were identified in ATM c.8228C>T, p.(T2743M), MLH3 c.958T>G, p.(C320G), and IDH1 c.245G>A, p.(R82K) in three independent patients. The ATM gene (coding for apical double strand break repair24 kinase) was shown to be a rare susceptibility gene in familial melanoma25. However, the germline MLH3 (involved in mismatch repair - MMR) mutation does not seem to be involved in tumorigenesis, considering the high age at diagnosis and microsatellite stable phenotype of the affected tumors. It was shown that MMR complex MutLγ, which is formed by the MLH3 and MLH1 protein, is a less efficient MMR complex than MutLα. That demonstrates that the MLH3 mutations alone do not interfere with MMR26. The clinical impact of a novel, likely pathogenic germline IDH1 mutation p.(R82K) is unclear.
It has been described that most melanomas possess potentially actionable mutations in the components of the MAPK and PI3K-AKT pathways, cell cycle regulation and chromatin remodeling3,4,7,8,27,28. In this study, the majority of identified mutations affected genes coding for components of the RAS signaling, including mitogen-activated protein kinase (MAPK) or phosphatidylinositol 3-kinase (PI3K)-AKT/PKB signaling pathway. Other recurrently altered intracellular signaling pathways in our patients included DNA damage response, cell cycle regulation, and chromatin remodeling. There were also several genes frequently affected by somatic mutations, such as the genes coding nuclear hormone receptor (ESR1), RAS signaling pathway members (MET, KDR), DNA repair genes (BRIP1, and NBN), and genes coding for proteins involved in tight junction assembly or regulation of epithelial-to mesenchymal transition (PARD3, TJP1, and ZEB2), which have not yet been described as significantly mutated genes in primary CM. The TJP1 and PARD3 protein have not been described to play a role in melanomagenesis at all so far. The epithelial-to-mesenchymal transition transcription factors ZEB2 and SNAI2 have been shown to control melanocyte differentiation29,30. Loss of ZEB2 expression was already reported to be associated with poor melanoma-specific survival30.
The in silico prediction of the pathogenicity of the TP53 and ARID1A mutations was compared with the protein expression and function. Pathogenic mutations showed aberrant protein expression by immunohistochemical analysis and/or impaired function, as confirmed by the functional assay developed in our study. All nonsense ARID1A mutations showed a loss of ARID1A expression by IHC. Interestingly, one case with a TP53 nonsense mutation (VAF <10%) and two TP53 variants affecting the consensus splice-acceptor site showed a wild-type pattern of p53 expression when evaluated by IHC (Table 3). The wild-type expression of p53 in those cases could be explained by the low frequency of nonsense TP53 mutations present (suggesting limited impact of variants detected with low frequency) and/or by nonsense-mediated RNA decay in case of all variants leading to premature terminal codon31. All the detected TP53 mutations (except for the one splice mutation c.75–1G>A) have already been described in the IARC TP53 database (somatic mutations - release R19, July 2018;32), and are considered by in silico prediction to be pathogenic (except the benign variant p.P80S). We developed cell/based functional assays, and we identified that p.(L194P), p.(R273P), p.(G245D) and p.(R280K) are loss-of-function mutations of the TP53.
The consistent results obtained by different approaches support the accuracy of the used in silico analysis algorithm. However, we did observe conflicting impact interpretation results when comparing the classification in databases, the in silico predictions and literature in case of the CDK4. Its mutation p.(Arg24His), rs104894340 was predicted by our in silico prediction pipeline to be likely benign, however, in the ClinVar database this variant is classified as likely pathogenic somatic, albeit without strong assertion criteria. The same variant was also detected in a large Norwegian family with multiple atypical nevi and malignant melanomas33. Another mutation in the same codon p.(Arg24Cys), rs11547328 was classified as class 4/5 in the ClinVar. Therefore, we concluded that this variant is likely pathogenic. Similarly, the BRCA2 mutation p.(P2532L) was evaluated in the ClinVar database as likely benign by a single submitter, with its clinical significance being unknown according to the BIC and BRCA Exchange databases. However, the Cosmic database (including only FATHMM prediction), and our in silico algorithm suggested it to be pathogenic, and therefore we evaluated this variant as likely pathogenic. The pathogenicity of this variant was also supported by Align-GVGD34.
Several studies have identified clinical associations with BRAFV600 mutations, including melanoma subtype, primary tumor location, and prognosis35,36,37,38. Another study observed a higher rate of NRAS mutations in NM compared to SSM22. On the contrary, we did not find any significant correlations of the frequently mutated genes, including BRAFmut or NRASmut with the histological subtype (NM versus SSM), stage, location of the primary melanoma or the outcome. Nevertheless, our analysis suggested a significant association of the ARID2 or PARD3 mutations with a higher age at diagnosis (p < 0.05), and the loss-of-function ARID1A mutations were associated with sun exposed skin (p = 0.024). We did not observe any significant correlations of the frequent mutations with any of the studied outcomes (DFS, LFS or MFS). The strongest prognostic marker remains Breslow thickness of the primary melanoma and SLN positivity39. Considering other morphological parameters, the melanization of metastatic melanoma also seems to be an important prognostic and predictive marker40,41.
Modern targeted treatment strategies (including BRAF inhibitors and immunotherapy by immune checkpoint inhibitors) are currently widely used42,43. New potential therapy targets, such as inhibitors of CDK4/6, MDM2/p53, c-KIT, PI3K/Akt/mTOR, ERK, or IDH1, have also been considered for melanoma treatment27,28,44. The mutations BRAFV600 and NRASQ61 account for a large proportion of actionable mutations in CM and represent logical targets for downstream (MEK) inhibitors4,13,37,45,46. In this study we demonstrated relatively common pathogenic mutations in the KDR and MET genes, which code for tyrosine kinase growth factor receptors VEGFR2 and HGFR, respectively. Both these genes activate several signaling cascades, including the MAPK, RAS-ERK or PIK3-AKT signaling pathway. The KDR and MET mutations could represent new therapeutic options. The MET is currently studied as a potentially druggable target in non-small cell lung cancer47.
Limitations
Several genes were not covered by our analysis due to the high stringency of the designed probes, and therefore the mutation analysis does not cover some of the already known rare (e.g. AKT12,3 and SMARCA44) or frequently mutated genes (e.g. MAP2K214) in melanoma. From a clinical point of view, it would certainly be interesting to explore the correlations between TIL and tumor mutation burden (TMB). Unfortunately, the evaluation of TMB was impossible due to the small size of the sequenced targets (about 2Kbp) and the fact that the sample set is limited.
Conclusion
To the best of our knowledge, our analysis is the first to include the determination of pathogenicity of the detected mutations using biostatistical analyses and immunohistochemical and/or functional assays. We describe the spectrum and prevalence of class 4/5 mutations in primary NM and SSM, including several novel variants. Additionally, the functional analysis identified two loss-of-function TP53 missense mutations, which were previously classified as VUS. As well as the commonly described RAS-, cell cycle regulation- and chromatin remodeling pathways in CM, we revealed frequently mutated genes coding for proteins involved also in DNA damage response and cellular tight junction regulation. The newly identified significantly mutated genes included ATM, KDR, MET, PARD3, SF3B1 and ZEB2, which could all be of clinical importance. Our results suggest that with the development of new therapeutic possibilities, not only BRAF testing but complex molecular testing of CM may become an integral part of the decision process concerning the treatment of patients with melanoma.
Material and Methods
Patients and samples
Formalin-fixed paraffin-embedded (FFPE) tissue blocks of primary melanomas collected between the years 2006–2017 were obtained from the archive files processed at the Institute of Pathology and the Department of Dermatology and Venereology, First Faculty of Medicine, Charles University and General University Hospital in Prague. A review of the hematoxylin and eosin stained slides was performed in all cases and areas of non-tumor and tumor tissues were marked for macrodissection (with estimation of tumor cell percentage in the selected area; ranging between 60–99%). For this project, the DNA was isolated from more than 200 samples, but only about 50% of those passed the qualitative and quantitative requirements for NGS (massive parallel sequencing) analysis and biostatistical evaluation. The clinical and pathological characteristics of the 114 successfully analyzed patients and tumors are summarized in Table 1. The available patient information with respect to the follow-up data was updated at the end of this project in April 2019. The mean age of patients at diagnosis was 62 years (median 66, range 24–93 years). The melanoma samples were divided according to the TNM classification (7th Edition) into four groups (pT1 ≤1 mm, pT2 >1 and ≤2 mm, pT3 >2 and ≤4 mm, and pT4 >4 mm). Tumor infiltrating lymphocytes (TILs) were evaluated within our previous study48 which included a subset of samples analyzed also in this present study. In our sample set, mainly melanomas with a larger thickness of the primary tumor were selected in order to get enough material for DNA isolation. A corresponding non-tumor tissue from the selected patients was used for DNA isolation to determine germline or somatic origin of the identified mutations.
Ethics statement
In compliance with the Helsinki Declaration, the project has been approved by The Ethics Committee of General University Hospital in Prague (ethical approval number č.j. 56/15 Grant VES 2016 AZV 1. LFUK). The ethics committee which approved this study waived the need for informed consent. The methods were carried out according to the approved ethical guidelines.
DNA isolation and quality control
DNA from formalin-fixed paraffin-embedded (FFPE) tissue blocks was isolated using standard procedures implementing cobas® DNA Sample Preparation kit (Roche; Germany) or an automated MagCore® nucleic Acid Extractor using MagCore Genomic DNA FFPE One-step kit, (RBC Bioscience). Every sample underwent a quality control test of amplification efficacy by qPCR (5 ng DNA of sample was amplified using 5x HOT FIREPol® EvaGreen® HRM Mix NO ROX (Solis Biodyne), only the samples which passed the quality criteria (Ct <35 for a 180 bp product amplification) were used for the library preparation.
NGS analysis and biostatistical processing of data
Samples for sequence capture NGS were prepared using the KAPA HyperPlus kit according to Seq Cap EZ protocol (Roche NimbleGen). The target sequences were enriched using a panel of hybridization probes against multiple targets (72 genes or gene parts; 219 kbp; NimbleGen, Roche). High stringent probes were designed to match only one target to avoid the enrichment of homologous sequences. The prepared sample libraries were pair-end sequenced by MiSeq instrument (Illumina) using MiSeq Reagent Kit v3 (150 cycles; Illumina). Initial data sorting and demultiplexing to fastq files was carried out with the MiSeq software. NextGENe software (Softgenetics, State College, PA) was used for main biostatistical analysis of the sequencing data. PCR duplicate reads were removed using NextGENe Sequence Operation tool. The remaining unique paired reads in fastq file format were converted to fasta file format using NextGENe Format Conversion tool with low quality reads removal (median score <25; number of uncalled bases ≥3; called bases number <40, reads trimmed or rejected when ≥3 bases with score ≤20). The remaining unique high-quality paired reads in fasta format were mapped on human GRCh37 genome version by NextGENe using Sequence Alignment tool (allowable mismatched bases – 0; allowable ambiguous alignments – 10; seeds – 21 bases, move step 1 base, allowable alignments – 80; overall matching base percentage ≥95%) with a mutation filter VAF ≥5%. The alignment results were exported as a Mutation report (txt file), and the mapped reads were exported in a BAM format. The nonsynonymous variants in exons and splice variants in adjacent intronic regions with minimal average coverage 100x and VAF >10% were evaluated, and manually inspected in a BAM file using an IGV viewer (Broad Institute) in order to avoid false positive variations. The copy number variations (CNVs) were not evaluated due to the limited quality of the FFPE samples. A majority of the samples had significantly fragmented DNA which leads to non-homogenous length of the reads and coverage of target areas in the NGS panel.
Variant prioritization
Variants of each sample included in the Mutation Report obtained from the NextGENe software NGS data analysis were prioritized as follows: (i) all pathogenic and likely pathogenic variants based on Clinical Significance from ClinVar database, (ii) all nonsense, frameshift, in-frame, no-start and no-stop variants, and (iii) all splice variants in consensus splice sites. In the next step, (iv) all missense variants with a population frequency ≤0.01 and with a non-evaluated frequency based on dbNSFP 1000G_AF, and (v) potential splice variants with a rank score ≥0.5 based on dbNSFP_dbscSNV database were prioritized.
Databases and in silico prediction pipeline
In order to assess the impact of the prioritized variants we searched several databases and/or performed comprehensive analysis by 14 different in silico prediction tools implemented in the Variant Effect Predictor (VEP, Ensembl, http://grch37.ensembl.org/Homo_sapiens/Tools/VEP/)49. In VEP, the predictors were selected based on (i) evaluation of amino-acid change and protein structure (SIFT, PolyPhen-2, FATHM, PROVEAN and VEST3), (ii) evolutionary conservation (GERP++, MutationAssessor and SiPhy), (iii) evaluation of multiple databases information (CADD, MutationTaster and LRT) and (iv) based on combined algorithms (M-CAP, MetaLR, MetaSVM). In all the selected predictors, the rankscore which ranged from 0 (=likely benign) to 1 (=likely pathogenic), was evaluated. We considered a variant as “likely pathogenic” if at least 7/14 of selected predictors scored the variant as ≥0.8. The remainder of the variants was considered as likely benign. (Supplementary Table 1).
Variant characterization
Somatic mutations which were not found in the literature, the Single Nucleotide Polymorphism Database (dbSNP, http://www.ncbi.nlm.nih.gov/SNP/), the ClinVar Database (https://www.ncbi.nlm.nih.gov/clinvar/), or in the Catalogue of Somatic Mutations in Cancer (COSMIC, http://www.sanger.ac.uk/cosmic; databases accessed March 28, 2019) were considered as novel. Gene-specific databases were searched in the case of TP53 (IARC TP53 Database, http://p53.iarc.fr/), BRCA1 and BRCA2 (the BIC database, https://research.nhgri.nih.gov/bic/ and BRCA exchange, https://brcaexchange.org).
The protein truncating mutations, including nonsense, frameshift and any type of variants classified pathogenic in the ClinVar database were considered as “pathogenic”, also referred to as the class 5. The category of “likely pathogenic” mutations, also referred to as the class 4 mutations, included splice mutations in cryptic splice sites and mutations which are either (i) currently classified as Variants of Unknown Significance (VUS, i.e. conflict of interpretations of pathogenicity, uncertain significance, or clinical significance not provided by the ClinVar database) or (ii) novel variants (described neither in the ClinVar, nor in the COSMIC) that were suggested to be pathogenic by our in silico prediction pipeline, as is described above.
All variants classified as benign in the mutation databases (ClinVar and/or COSMIC) or novel and VUS variants suggested to be benign by our in silico prediction pipeline (described above) were considered as “benign”. These benign variants were excluded from further statistical analyses.
Commonly mutated pathways
A pathway was considered as altered if at least one gene in the pathway contained pathogenic or likely pathogenic somatic mutation. The association of proteins with functional pathways was judged according to the information obtained from the largely used database Gene Cards (www.genecards.org), including information from e.g.: Kyoto Encyclopedia of Genes and Genomes (KEGG), NCBI databases, CIViC, GeneCards, UniProtKB/Swiss-Prot, Tocris, Reactome, STRING interaction database v11.050, and/or literature.
Functional assay
The ionizing radiation-induced expression of p21 (which is an established TP53 target), defects in acetylation of K382 and phosphorylation of S15 and S46 (which are posttranslational modifications needed for p53 to function51) were analyzed by western blot and flow cytometry. Suppression of cell proliferation in response to treatment with an mdm2 inhibitor nutlin-352 was also studied by a proliferation assay.
Cell lines
Human hTERT-RPE-1 cells (referred to as RPE cells; ATCC) were grown at 37 °C and 5% CO2 in DMEM supplemented with 6% FBS (Gibco), glutamine (2 mM), penicillin (100 U/ml), and streptomycin (100 g/ml). All cell lines were regularly checked for the absence of mycoplasma infection using MycoAlert Plus reagent (Lonza).
To generate RPE-TP53-KO cells, human hTERT-RPE-1 cells were transfected with a p53-CRISPR/Cas9 KO Plasmid (Santa Cruz, sc-416469) and p53-HDR Plasmid (1:1) and selected by puromycin (10 μg/ml) for 3 weeks53. The integration of the HDR cassette into the TP53 locus was confirmed by sequencing and loss of p53 expression by immunoblotting. Transiently expressed wild-type or mutant TP53 were cloned into pIRES2-EGFP-TP53 plasmid allowing gating for p53 expressing cells according to the EGFP signal. The pIRES2-EGFP-p53-WT plasmid (Addgene plasmid # 49242) and IRES2-EGFP reporter were gifted to us by Dylan Taatjes. The site-directed mutagenesis of individual variants was performed by Gibson assembly kit (NEB) of two PCR fragments into the NheI/BstxI sites of the pIRES2-EGFP-p53-WT plasmid. All plasmids were verified by Sanger sequencing. Plasmid DNA was transfected using Polyethylenimine HCl MAX 40 kDa (Palysciences) in ratio 6:1 to DNA and growth media were changed after 3 hours. Stable cell lines expressing pIRES2-EGFP-p53-WT or TP53 variants were selected with geneticin (Sigma) for 3 weeks.
Antibodies
used in the western blotting or flow cytometry experiments: p53 (sc-6243), TFIIH p89 (sc-293), and p21 (sc-397; Santa Cruz Biotechnology); Phospho-Ser15-p53 (#9284), Phospho-Ser45-53 (#2521), and Acetyl-Lys382-p53 (#2525; Cell Signaling Technology); HRP-conjugated secondary antibodies (Bio-Rad), and Alexa Fluor-labelled secondary antibodies (Life Technologies).
Flow cytometry
For the reconstitution of p53 expression in RPE-TP53-KO cells, the transiently transfected cells with pIRES2-EGFP-p53 WT or variant plasmid were seeded and after 48 hours fixated with 4% paraformaldehyde. Cells stained with antibodies against p53, p21, and DAPI were sorted by flow cytometer LSRII (BD Biosciences) and analyzed by the FlowJo software (FlowJo). The mean signal intensity of p53 or p21 per cell was determined in GFP positive cells. Signal intensity in GFP negative cells was subtracted as a background. At least 10000 cells were measured in three independent experiments.
Cell proliferation assays
The RPE-TP53-KO cells, stably reconstituted with pIRES2-EGFP-p53 WT or its variants, were seeded in 96-well plates (500 cells/well) and cultivated in the presence of nutlin-3 (0.5 μM, MedChem Express) for 7 days. Resazurin (30 μg/mL) was added to the growth media and the resulting fluorescence signal (Ex = 560 nm, Em = 590 nm) was measured after 1 h using the EnVision plate reader (PerkinElmer), as previously described elsewhere54. The level of cell growth suppression induced by nutlin was normalized to the wild-type p53. Samples were measured in hexaplicates in three independent experiments. The bars indicate standard deviation. Any statistical significance was determined by two-sided t-test using Prism software.
Immunohistochemical analysis
Immunohistochemical analysis was performed manually on 4 µm sections of FFPE tissue, using the avidin-biotin complex method with an antibody against the ARID1A (clone PSG3, dilution 1:1000; Santa Cruz Biotechnology, California, USA) and p53 (clone BP 53–12, dilution 1:200, Zytomed Systems, Berlin, Germany) antigens. Following the pretreatment in 0.01 M citrate buffer (pH 6.0) for ARID1A or 0.01 M citrate buffer (pH 9.0) for p53, the antigen retrieval was performed for 40 min in a water bath at 98°C. The expression of p53 and ARID1A was double-blindly evaluated by two pathologists. Discrepant cases were reviewed and a consensus score was assessed.
The ARID1A expression was scored as a percentage of tumor cells with nuclear staining of any intensity. Cases with less than 1% tumor cells with ARID1A expression were considered as negative.
The p53 protein expression was assessed as “aberrant” (defined as diffuse moderate to strong nuclear positivity of more than 50% of tumor cells or totally negative staining with a positive internal control in non-tumorous tissue) or “wild-type” (defined as low variable staining in less than 50% of the tumor nuclei)55.
Sanger sequencing and microsatellite instability analyses
Analyses were performed as previously described56. Variants detected by NGS with VAF >10% (range 11–90%) were selected for confirmation by direct Sanger sequencing. Where available, the corresponding non-tumor tissue of selected cases with detected class 4/5 mutation with VAF 45–55% was analyzed, to determine germline or somatic origin of the selected mutations. The specific designed primers are available upon reasonable request.
Statistical analyses
The software Statistica (StaSoft, Tulsa, OK) was used to perform the statistical analyses. Associations of mutations with clinico-pathological characteristics were evaluated using either the chi-squared test or logistic regression. The correlation between the number of mutations/mutated genes and the rates of C > T substitutions with clinico-pathological variables was evaluated using ANOVA. Time-to-event analysis was performed with three outcomes – disease-free survival (DFS: death from melanoma was considered as a failure), local recurrence-free survival (LFS: the period from primary diagnosis till the first local recurrence), and distant metastasis-free survival (MFS: the period from primary diagnosis till the first distant metastasis). These analyses included 105 patients without targeted therapy (including immunotherapy by check-point inhibitors). The date of the diagnosis was the date of the primary sample accession in the pathology database. The initial univariate analysis was performed using the log-rank test to determine the differences between cohorts and to construct the Kaplan-Meier estimated survival curves. Cox’s Proportional Hazard Ratio Models were used for univariate and multivariate analysis of risk factors for disease-free survival. The minimal adequate model was achieved through backward elimination of non-significant effects (p > 0.05). All of the performed tests were two-sided and a p-value <0.05 was considered as significant.
Data availability
The source data are included in this article and its Supplementary Information File or are available from the corresponding author upon reasonable request.
References
Duncan, L. M. The classification of cutaneous melanoma. Hematol Oncol Clin North Am 23, 501–513, ix, https://doi.org/10.1016/j.hoc.2009.03.013 (2009).
Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696, https://doi.org/10.1016/j.cell.2015.05.044 (2015).
Hayward, N. K. et al. Whole-genome landscapes of major melanoma subtypes. Nature 545, 175–180, https://doi.org/10.1038/nature22071 (2017).
Hodis, E. et al. A landscape of driver mutations in melanoma. Cell 150, 251–263, https://doi.org/10.1016/j.cell.2012.06.024 (2012).
Krauthammer, M. et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat Genet 47, 996–1002, https://doi.org/10.1038/ng.3361 (2015).
Mehnert, J. M. & Kluger, H. M. Driver mutations in melanoma: lessons learned from bench-to-bedside studies. Curr Oncol Rep 14, 449–457, https://doi.org/10.1007/s11912-012-0249-5 (2012).
Shain, A. H. et al. The Genetic Evolution of Melanoma from Precursor Lesions. N Engl J Med 373, 1926–1936, https://doi.org/10.1056/NEJMoa1502583 (2015).
Palmieri, G. et al. Molecular Pathways in Melanomagenesis: What We Learned from Next-Generation Sequencing Approaches. Curr Oncol Rep 20, 86, https://doi.org/10.1007/s11912-018-0733-7 (2018).
Zhang, T., Dutton-Regester, K., Brown, K. M. & Hayward, N. K. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res 29, 266–283, https://doi.org/10.1111/pcmr.12459 (2016).
Zhao, X. et al. The Prognostic Significance of Low-Frequency Somatic Mutations in Metastatic Cutaneous Melanoma. Front Oncol 8, 584, https://doi.org/10.3389/fonc.2018.00584 (2018).
Siroy, A. E. et al. Beyond BRAF(V600): clinical mutation panel testing by next-generation sequencing in advanced melanoma. J Invest Dermatol 135, 508–515, https://doi.org/10.1038/jid.2014.366 (2015).
Leichsenring, J. et al. Genetic profiling of melanoma in routine diagnostics: assay performance and molecular characteristics in a consecutive series of 274 cases. Pathology 50, 703–710, https://doi.org/10.1016/j.pathol.2018.08.004 (2018).
Krauthammer, M. et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 44, 1006–1014, https://doi.org/10.1038/ng.2359 (2012).
Nikolaev, S. I. et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet 44, 133–139, https://doi.org/10.1038/ng.1026 (2011).
Gumaste, P. V. et al. Skin cancer risk in BRCA1/2 mutation carriers. Br J Dermatol 172, 1498–1506, https://doi.org/10.1111/bjd.13626 (2015).
Linos, K. & Tafe, L. J. Isocitrate dehydrogenase 1 mutations in melanoma frequently co-occur with NRAS mutations. Histopathology 73, 963–968, https://doi.org/10.1111/his.13707 (2018).
Harbour, J. W. et al. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet 45, 133–135, https://doi.org/10.1038/ng.2523 (2013).
Emri, G. et al. Ultraviolet radiation-mediated development of cutaneous melanoma: An update. J Photochem Photobiol B 185, 169–175, https://doi.org/10.1016/j.jphotobiol.2018.06.005 (2018).
Bastian, B. C. The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol 9, 239–271, https://doi.org/10.1146/annurev-pathol-012513-104658 (2014).
Lo, J. A. & Fisher, D. E. The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science 346, 945–949, https://doi.org/10.1126/science.1253735 (2014).
Nik-Zainal, S. et al. The genome as a record of environmental exposure. Mutagenesis 30, 763–770, https://doi.org/10.1093/mutage/gev073 (2015).
Lattanzi, M. et al. Primary Melanoma Histologic Subtype: Impact on Survival and Response to Therapy. J Natl Cancer Inst, https://doi.org/10.1093/jnci/djy086 (2018).
Mar, V. J. et al. BRAF/NRAS wild-type melanomas have a high mutation load correlating with histologic and molecular signatures of UV damage. Clin Cancer Res 19, 4589–4598, https://doi.org/10.1158/1078-0432.CCR-13-0398 (2013).
Prokopcova, J., Kleibl, Z., Banwell, C. M. & Pohlreich, P. The role of ATM in breast cancer development. Breast Cancer Res Treat 104, 121–128, https://doi.org/10.1007/s10549-006-9406-6 (2007).
Goldstein, A. M. et al. Rare germline variants in known melanoma susceptibility genes in familial melanoma. Hum Mol Genet 26, 4886–4895, https://doi.org/10.1093/hmg/ddx368 (2017).
Korhonen, M. K., Vuorenmaa, E. & Nystrom, M. The first functional study of MLH3 mutations found in cancer patients. Genes Chromosomes Cancer 47, 803–809, https://doi.org/10.1002/gcc.20581 (2008).
Wu, C. E. et al. Targeting negative regulation of p53 by MDM2 and WIP1 as a therapeutic strategy in cutaneous melanoma. Br J Cancer 118, 495–508, https://doi.org/10.1038/bjc.2017.433 (2018).
Karachaliou, N. et al. Melanoma: oncogenic drivers and the immune system. Ann Transl Med 3, 265, https://doi.org/10.3978/j.issn.2305-5839.2015.08.06 (2015).
Caramel, J. et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 24, 466–480, https://doi.org/10.1016/j.ccr.2013.08.018 (2013).
Denecker, G. et al. Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ 21, 1250–1261, https://doi.org/10.1038/cdd.2014.44 (2014).
Maquat, L. E. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5, 89–99, https://doi.org/10.1038/nrm1310 (2004).
Bouaoun, L. et al. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum Mutat 37, 865–876, https://doi.org/10.1002/humu.23035 (2016).
Molven, A. et al. A large Norwegian family with inherited malignant melanoma, multiple atypical nevi, and CDK4 mutation. Genes Chromosomes Cancer 44, 10–18, https://doi.org/10.1002/gcc.20202 (2005).
Tavtigian, S. V. et al. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet 43, 295–305, https://doi.org/10.1136/jmg.2005.033878 (2006).
Curtin, J. A. et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 353, 2135–2147, https://doi.org/10.1056/NEJMoa050092 (2005).
Bauer, J. et al. BRAF mutations in cutaneous melanoma are independently associated with age, anatomic site of the primary tumor, and the degree of solar elastosis at the primary tumor site. Pigment Cell Melanoma Res 24, 345–351, https://doi.org/10.1111/j.1755-148X.2011.00837.x (2011).
Jakob, J. A. et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 118, 4014–4023, https://doi.org/10.1002/cncr.26724 (2012).
Long, G. V. et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol 29, 1239–1246, https://doi.org/10.1200/JCO.2010.32.4327 (2011).
Balch, C. M. et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 27, 6199–6206, https://doi.org/10.1200/JCO.2009.23.4799 (2009).
Brozyna, A. A. et al. Melanogenesis affects overall and disease-free survival in patients with stage III and IV melanoma. Hum Pathol 44, 2071–2074, https://doi.org/10.1016/j.humpath.2013.02.022 (2013).
Slominski, A. et al. The role of melanogenesis in regulation of melanoma behavior: melanogenesis leads to stimulation of HIF-1alpha expression and HIF-dependent attendant pathways. Arch Biochem Biophys 563, 79–93, https://doi.org/10.1016/j.abb.2014.06.030 (2014).
Silva, I. P. & Long, G. V. Systemic therapy in advanced melanoma: integrating targeted therapy and immunotherapy into clinical practice. Curr Opin Oncol 29, 484–492, https://doi.org/10.1097/CCO.0000000000000405 (2017).
da Silveira Nogueira Lima, J. P., Georgieva, M., Haaland, B. & de Lima Lopes, G. A systematic review and network meta-analysis of immunotherapy and targeted therapy for advanced melanoma. Cancer Med 6, 1143–1153, https://doi.org/10.1002/cam4.1001 (2017).
Wu, C. E. et al. ATM Dependent DUSP6 Modulation of p53 Involved in Synergistic Targeting of MAPK and p53 Pathways with Trametinib and MDM2 Inhibitors in Cutaneous Melanoma. Cancers (Basel) 11, https://doi.org/10.3390/cancers11010003 (2018).
Kim, H. S. et al. Oncogenic BRAF fusions in mucosal melanomas activate the MAPK pathway and are sensitive to MEK/PI3K inhibition or MEK/CDK4/6 inhibition. Oncogene 36, 3334–3345, https://doi.org/10.1038/onc.2016.486 (2017).
Hocker, T. & Tsao, H. Ultraviolet radiation and melanoma: a systematic review and analysis of reported sequence variants. Hum Mutat 28, 578–588, https://doi.org/10.1002/humu.20481 (2007).
Matsumoto, K., Umitsu, M., De Silva, D. M., Roy, A. & Bottaro, D. P. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci 108, 296–307, https://doi.org/10.1111/cas.13156 (2017).
Nemejcova, K. et al. Comparison of five different scoring methods in the evaluation of inflammatory infiltration (tumor-infiltrating lymphocytes) in superficial spreading and nodular melanoma. Pigment Cell Melanoma Res 32, 412–423, https://doi.org/10.1111/pcmr.12757 (2019).
McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biol 17, 122, https://doi.org/10.1186/s13059-016-0974-4 (2016).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47, D607–D613, https://doi.org/10.1093/nar/gky1131 (2019).
Gu, B. & Zhu, W. G. Surf the post-translational modification network of p53 regulation. Int J Biol Sci 8, 672–684, https://doi.org/10.7150/ijbs.4283 (2012).
Vassilev, L. T. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848, https://doi.org/10.1126/science.1092472 (2004).
Jaiswal, H. et al. ATM/Wip1 activities at chromatin control Plk1 re-activation to determine G2 checkpoint duration. EMBO J 36, 2161–2176, https://doi.org/10.15252/embj.201696082 (2017).
Pechackova, S. et al. Inhibition of WIP1 phosphatase sensitizes breast cancer cells to genotoxic stress and to MDM2 antagonist nutlin-3. Oncotarget 7, 14458–14475, https://doi.org/10.18632/oncotarget.7363 (2016).
Kobel, M. et al. The biological and clinical value of p53 expression in pelvic high-grade serous carcinomas. J Pathol 222, 191–198, https://doi.org/10.1002/path.2744 (2010).
Hajkova, N. et al. Germline mutation in the TP53 gene in uveal melanoma. Sci Rep 8, 7618, https://doi.org/10.1038/s41598-018-26040-0 (2018).
Acknowledgements
This work was supported by Ministry of Health, Czech Republic (Research project AZV 16-30954A and Conceptual development of research organization 64165, General University Hospital in Prague), by Charles University (Project Progress Q28/LF1 and SVV 260367), and by European Regional Development Fund (project EF16_013/0001674, BBMRI_CZ LM2015089, and OPPK - Research Laboratory of Tumor Diseases, CZ.2.16/3.1.00/24509). Flow cytometry analysis was performed in the Microscopy Centre - Light Microscopy Core Facility of the IMG supported by MEYS CR (Czech-Bioimaging, LM2015062). Special thanks belong to Zachary H.K. Kendall, B.A. for English editing, and to Lenka Matiaskova, B.A. and Andrea Berkyova (Institute of Pathology, First Faculty of Medicine, Charles University and General University Hospital in Prague) for their excellent technical assistance during DNA isolation.
Author information
Authors and Affiliations
Contributions
Study concept and design: P.D., Z.K., I.T., Contribution of the patient material with clinical data: M.D., O.K., M.B., K.N., Preparation and analysis of samples: N.H., J.H., E.K., Biostatistical analyses: J.H., N.H., R.M., Interpretation of data: N.H., J.H., I.T., functional assays and interpretation: L.M., A.S.V., M.K., Drafting of the manuscript: I.T., P.D., R.M., proofread of the manuscript: I.T., P.D., Z.K. All authors discussed the results, commented on the manuscript, and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ticha, I., Hojny, J., Michalkova, R. et al. A comprehensive evaluation of pathogenic mutations in primary cutaneous melanomas, including the identification of novel loss-of-function variants. Sci Rep 9, 17050 (2019). https://doi.org/10.1038/s41598-019-53636-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53636-x
This article is cited by
-
Discovery of a small molecule that inhibits Bcl-3-mediated cyclin D1 expression in melanoma cells
BMC Cancer (2024)
-
Refining mutanome-based individualised immunotherapy of melanoma using artificial intelligence
European Journal of Medical Research (2024)
-
Clear cell stromal tumor of the lung with multinucleated giant cells: a report of a case with YAP1-TFE3 fusion
Diagnostic Pathology (2023)
-
Uterine cellular leiomyomas are characterized by common HMGA2 aberrations, followed by chromosome 1p deletion and MED12 mutation: morphological, molecular, and immunohistochemical study of 52 cases
Virchows Archiv (2022)
-
Targeting wild-type TP53 using AMG 232 in combination with MAPK inhibition in Metastatic Melanoma; a phase 1 study
Investigational New Drugs (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
