
Abstract
Mycobacterium bovis infection has been described in many wildlife species across Africa. However, diagnostic tests are lacking for many of these, including warthogs (Phacochoerus africanus). Most literature on suids has focused on using serological tools, with few studies investigating the use of cell-mediated immune response (CMI) assays. A recent study showed that warthogs develop measurable CMI responses, which suggests that cytokine gene expression assays (GEAs) may be valuable for detecting M. bovis-infection, as shown in numerous African wildlife species. Therefore, the aim of the study was to develop GEAs capable of distinguishing between M. bovis-infected and uninfected warthogs. Whole blood was stimulated using the QuantiFERON-TB Gold (In-Tube) system, using ESAT-6 and CFP-10 peptides, before determining the relative gene expression of five reference (B2M, H3F3A, LDHA, PPIA and YWHAZ) and five target (CXCL9, CXCL10, CXCL11, IFNG and TNFA) genes through qPCR. The reference gene H3F3A was the most stably expressed, while all target genes were significantly upregulated in M. bovis-infected warthogs with the greatest upregulation observed for CXCL10. Consequently, the CXCL10 GEA shows promise as an ante-mortem diagnostic tool for the detection of M. bovis-infected warthogs.
Similar content being viewed by others


Introduction
There are few validated ante-mortem diagnostic tests available to determine the Mycobacterium bovis infection status of warthogs (Phacochoerus africanus). Warthogs may serve as potential maintenance hosts of bovine tuberculosis (bTB) in M. bovis-endemic areas and the lack of appropriate diagnostic tools is an obstacle to understanding the dynamics of this disease1,2.
Serological assays have been used to distinguish between M. bovis-infected and uninfected warthogs3. However, cell-mediated immune responses (CMI) have been shown to have greater sensitivity in many species4. The CMI is activated early after mycobacterial infection and leads to the production of Th1 cytokines4. These cytokines play an important role in the initial control of the infection reducing progression of disease and bacilli multiplication5. Many CMI assays are based on the in vitro stimulation of whole blood (WB) to detect antigen-specific production of Th1 cytokines, such as interferon gamma (IFN-γ) and tumour necrosis factor alpha (TNF-α)5,6. While these types of CMI assays have been used to detect M. bovis-infected cattle and African buffalo, very few have investigated their use in suids7.
The QuantiFERON TB Gold (In-Tube) system (QFT), which makes use of the antigens early secretory antigenic target 6 kDa (ESAT-6) and culture filtrate protein 10 kDa (CFP-10), has been used to stimulate M. bovis-specific responses in whole blood (WB) from a variety of species8,9,10,11,12. They are more specific than the commonly used bovine purified protein derivative, which is a crude extract of M. bovis. These antigens have been shown to be highly immunogenic in a wide range of species8,9,10,11,12,13.
Although assays to detect antigen-specific CMI responses are commonly used in humans and livestock7, their utility is limited in wildlife by the lack of host-specific reagents2. However, based on the homology of cytokine sequences between related domestic and wildlife species, cytokine gene expression assays (GEAs) may provide diagnostic techniques for wildlife9,10,14. Candidate biomarkers such as IFNG and those in the CXCL family, which are induced by IFN-γ, have been investigated using gene expression assays (GEAs) in wildlife9,10,14.
The aim of this study was to optimise GEAs for the detection of M. bovis infection in warthogs, targeting IFNG and TNFA, as well as genes that are induced by IFN-γ, including CXCL9, CXCL10 and CXCL11, which have been successfully used in other species to detect tuberculosis9,10,14. To achieve this, we developed relative quantitative real-time PCRs for transcripts of selected candidate reference and target genes of warthogs, identified the optimal reference gene (most stably expressed rgene), and calculated the relative target gene expression in response to ESAT-6 and CFP-10 in M. bovis-infected and uninfected warthogs.
Results
The median amount of RNA extracted from each blood sample was 1 560 ng (range: 324–11 772 ng). The 260/280 and 260/230 ratios had a median of 1.97 (range: 1.53–2.77) and 0.64 (range: 0.05–2.14), respectively.
All reference and target gene sequences derived from warthogs showed the greatest identity to the domestic pig sequence, as compared to that of the cow (Fig. S1). Additionally, the qPCR primer binding sites had a high identity with the pig sequences (Table 1). The qPCR products had characteristic melt curves (Table 1) and were confirmed to be specific by sequencing (data not shown). Furthermore, no products were formed in the no-RT control qPCRs, nor were there products in the no-template controls for each qPCR. For all qPCRs, intra-assay variability was low (<1%) and amplification efficiencies ranged from 90 to 115% (Table 1).
Of the five reference genes, H3F3A was the most stably expressed, and was therefore chosen as the reference gene for further analyses (Fig. S2). The efficiencies of all target gene qPCRs were similar to the selected reference gene (data not shown).
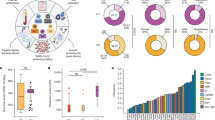
For M. bovis-infected warthogs, all target genes showed significant upregulation in response to antigen stimulation compared to uninfected animals (Fig. 1). Of these, CXCL9, CXCL10, and CXCL11 showed the greatest upregulation, with median fold increases of 32, 601 and 55, respectively (Fig. 1). Of the target genes, CXCL10 showed the greatest differential response between M. bovis-infected and uninfected warthogs (Fig. 1).

Antigen-specific expression of selected genes in response to ESAT-6/CFP-10 stimulation of blood from M. bovis uninfected (○) and M. bovis-infected (●) warthogs. Lines indicate gene specific diagnostic cut-off values. *p < 0.05, **p < 0.01, ***p < 0.001.
Cut-off values for all target genes were determined (Figs 1 and S3, Table 2). The sensitivity of CXCL10 was the highest (87%; 95% CI: 60–98%), while CXCL11 had the highest specificity (100%; 95% CI: 80–100%) (Table 2). Furthermore, CXCL10 had the highest Youden’s index value of 0.81, thus making it the most accurate marker of the five target genes (Table 2).
Discussion
This study, describes the successful development of novel cytokine gene expression assays to distinguish between M. bovis-infected and uninfected warthogs. Among the five reference genes evaluated, H3F3A was the most stably expressed. Although all target transcripts (CXCL9, CXCL10, CXCL11, IFNG and TNFA) showed significant upregulation, demonstrating their utility as potential diagnostic markers of M. bovis infection, CXCL10 was the most accurate and showed the greatest antigen-specific upregulation.
The mRNA coding regions and q-PCR primer binding sites of warthogs showed high identity with those of the domestic pig. This was expected since warthogs have common ancestry with domestic pigs and share many of their immune genes with other suids15,16. Consequently, the warthog GEA has potential to be used as another measure of CMI responses in other closely related suids.
The most stable reference gene was H3F3A, followed by YWHAZ. Previous studies for which H3F3A was reported to be a stable reference gene used other tissue types, such as PBMCs and alveolar macrophages, from M. bovis-infected cattle17,18. However, studies using WB identified YWHAZ as an appropriate reference gene in lion and hyena8,9. Nevertheless, H3F3A is a good reference candidate, as it is highly expressed in various tissue types19. Additionally, H3F3A and other genes in the H3.3 family are highly conserved across metazoans19, making this gene a candidate that might be used as a reference gene across a wide range of species.
For all five target genes, there was significantly greater antigen-induced expression in M. bovis-infected warthogs compared to uninfected animals. The CXCL genes showed greater antigen-induced expression levels in WB of M. bovis-infected warthogs than either IFNG or TNFA. The higher expression of the CXCL genes, compared to IFNG, have also been reported in lion and hyena9,10. This may be because CXCL genes are expressed by a greater number of cells (neutrophils, eosinophils, lymphocytes and monocytes) compared to IFNG (antigen stimulated Th1 cells) during antigen stimulation20,21,22,23,24,25,26. Studies have shown that chemokines and cytokines that are induced by IFN-γ can be used as sensitive and reliable biomarkers26,27. Therefore, the CXCL chemokines could be more suitable diagnostic candidates in GEAs.
Expression of CXCL10 was the greatest among the targets as well as the most accurate marker for M. bovis-infection in warthogs. Previous studies have shown that CXCL9 can be used as a diagnostic biomarker for M. bovis-infected lions, and that CXCL11 is significantly up-regulated in M. bovis exposed hyenas9,10. This indicates the necessity to identify the most appropriate biomarker for the species being studied. Nevertheless, CXCL10 may be a more suitable GEA biomarker for M. bovis infection in warthogs as a previous study identified IP-10, encoded by CXCL10, as a potential biomarker for M. bovis in warthogs8.
A limitation in this study was that the optimum incubation time for warthog WB was not determined prior to measuring the gene expression of each target gene28,29. In humans, peak mRNA levels for IFNG are detected after 4–6 h of incubation after which it returns to nearly background levels after 24 h29,30. This may be an additional explanation why IFNG had lower antigen-induced gene expression levels than CXCL10 in M. bovis-infected warthogs. Another limitation was that we were restricted to only include warthogs from M. bovis endemic regions, as samples were collected opportunistically.
Future studies should focus on determining if GEAs could be used in other suid species, to identify M. bovis-infected individuals. Furthermore, the optimum antigen incubation time of WB should be determined in this species for both gene expression and protein production.
In summary, all five target genes (CXCL9, CXCL10, CXCL11, IFNG and TNFA) were significantly upregulated in antigen-stimulated blood from M. bovis-infected warthogs, with CXCL10 showing the greatest upregulation. Therefore, GEAs targeting the CXCL genes, show great promise as potential diagnostic tools for detecting M. bovis infection in suids.
Materials and Methods
Animals, sampling and selection of study animals
Heparinized WB, serum and tissue samples were opportunistically obtained from warthogs from bTB endemic areas, as previously described8. Tissues were processed for mycobacterial culture and cultures were speciated using genetic region of difference analysis3,31. Sera were tested using the Indirect PPD ELISA as previously described1. Culture results were used to define warthogs as M. bovis-infected, whereas the culture and serology results were used to define the uninfected cohort. The Stellenbosch University Animal Care and Use committee provided ethical approval for this study (SU-ACUD15-00029). All methods were performed in accordance with the relevant guidelines and regulations set out in the ethics application.
Whole blood stimulation and RNA stabilization
The QFT system (Qiagen, Venlo, Netherlands) was used to stimulate warthog WB8. Briefly, 1 ml of WB was added to both a Nil tube (QFT-Nil, containing saline) and a TB antigen tube (QFT-TB, containing ESAT-6 and CFP-10 peptides). For optimal antigen exposure, the tubes were repeatedly inverted to ensure thorough mixing and contact of blood with the entire inner surface of the tube. Following incubation for 24 hours at 37 °C, blood was transferred to a 2 ml micro-centrifuge tube and centrifuged at 800 × g for 10 min. Plasma was harvested, and the cell pellet re-suspended in 1.3 ml of RNAlater (Ambion, Austin, TX, USA). Samples were subsequently stored at −80 °C until further analysis.
RNA extraction and cDNA preparation
The RNAlater-stabilised QFT-TB and QFT-Nil samples of 15 M. bovis-infected and 17 uninfected warthogs were centrifuged at 15 000 × g for 2 min and the supernatant discarded. The RNA was then extracted from the remaining blood cell pellet using the RiboPure Blood Kit (Ambion) according to the manufacturer’s guidelines. A single modification was made to the elution step; the volume of the elution solution was decreased to 60 µl. A NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA) was used to determine the RNA concentration and quality (260/280 and 260/230 ratio) of each sample. The QuantiTect Reverse Transcription (RT) kit (Qiagen), including a genomic DNA (gDNA) Wipeout step, was used to reverse transcribe 200 ng of RNA from each sample in a final volume of 20 µl, as per manufacturer’s guidelines.
Sequencing of warthog mRNA transcripts
To obtain warthog mRNA sequences of selected candidate reference (stably expressed in other species) and target genes (used to detect tuberculosis)9,10,14, degenerate primers based on the sequences of pig and cow, were designed to anneal to the transcripts of interest. To do this, pig and cow mRNA sequences of these genes were obtained from the Ensembl Genome Browser (http://www.ensembl.org/index.html) and aligned using the Clustal Omega online tool (http://www.ebi.ac.uk/Tools/msa/clustalo/). Using the Primer3Plus online software (http://primer3plus.com/cgi-bin/dev/primer3plus.cgi), primers were designed to anneal to sequences with the greatest identity within the untranslated regions (Table 3). Primers were then used to amplify the entire coding sequence from two randomly selected warthogs using a Veriti 96-Well Thermal Cycler (Applied Biosystems, Foster City, CA, USA). For each transcript, 1 µl of cDNA was added to 12.5 µl of OneTaq Hot Start 2x Master Mix with Standard Buffer (New England BioLabs Inc., Ipswich, MA, USA), 1 µl of each gene-specific forward and reverse primer (final concentration of 0.5 µM; Integrated DNA Technologies, Coralville, IA, USA) and 9.5 µl nuclease-free water. The reaction was initiated at 94 °C for 15 min, followed by 40 cycles of 94 °C for 30 s, a gene-specific annealing temperature for 30 s (Table 3), and 68 °C for 90 s, and concluded with a final extension at 68 °C for 5 min. The PCR products were sequenced at the Central Analytical Facility (Stellenbosch University, South Africa) using a 3130xl Genetic Analyzer (Applied Biosystems), according to the manufacturer’s guidelines, and analysed using MEGA 732. The warthog mRNA sequences were submitted to the GenBank genetic sequence database (http://www.ncbi.nlm.nih.gov/genbank/) (Table 3). The sequence identity of all warthog derived transcripts were determined by aligning these with those of the pig and cow using the web based multiple sequence alignment tool from EMBL-EB, MUSCLE (https://www.ebi.ac.uk/Tools/msa/muscle/). The result output was calculated as the percent identity.
qPCR design and optimization
Using Primer3Plus and warthog sequences, qPCR primers were designed to span putative exon-exon boundaries for each of the reference and target transcripts. All qPCRs were evaluated at annealing temperatures ranging from 58–62 °C and primer concentrations ranging from 0.25–0.75 µM. Hereafter, optimized reactions were done in triplicate using a CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories Inc., Hercules, CA, USA) and consisted of 5 µl iTaq Universal SYBR Green Supermix (Bio-Rad), 0.5 µl of each gene-specific forward and reverse qPCR primer (at a final primer concentration of 0.5 µM; Integrated DNA Technologies), 1 µl of cDNA and 3 µl of nuclease-free water. The reaction was initiated at 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and gene-specific temperatures for 30 s (Table 1), and concluded with a standard melt-curve analysis.
To confirm the specificity of all qPCRs, products from two warthogs were visualized in 1% agarose gel by electrophoresis and sequenced as described above (Table 1). The melt-curve for each qPCR product was characterized and used to confirm qPCR specificity of subsequent reactions. To confirm the absence of non-specific amplification, all qPCRs had a no-template control. In order to show the absence of amplifiable gDNA in RNA, qPCRs were performed with four randomly selected samples that had been treated with gDNA Wipeout buffer, but had not been reverse transcribed. For each qPCR, quantification cycles (Cq) were automatically determined by the CFX Manager Software (Bio-Rad). The efficiency of the qPCRs were determined by analysing a serial dilution (64 fold) of a pooled cDNA sample for each gene33. To validate the use of the relative quantification method, amplification efficiencies of the reference and target genes were compared as previously described34.
Data analysis
To determine the intra-assay variability of each qPCR, the coefficient of variance was calculated for triplicate reactions. The relative expression stability of the reference genes were compared by analysing qPCR Cq values for QFT-Nil and QFT-TB samples of four randomly selected M. bovis-infected warthogs, using the geNorm applet in Microsoft Excel35 and the NormFinder Excel Add-In36. The most stable reference gene was selected and used to further analyse the relative gene expression of the target genes.
The relative expression of each target gene was normalised by subtracting the Cq value of the selected reference gene from the Cq value of the target gene. This was done to calculate the relative abundance of the target gene mRNA for each sample (i.e. ΔCq). Thereafter, the ΔCq value derived from the QFT-Nil sample was subtracted from the ΔCq value derived from the QFT-TB sample for all animals (i.e. ΔΔCq). The relative fold change (2−ΔΔCq) was used to derive a QFT GEA result as a measure of the upregulation of the target transcript in response to antigen stimulation34.
The assay results for all target genes were then analysed in GraphPad Prism version 5 (GraphPad Software Inc., La Jolla, CA, USA). A Mann-Whitney U test was used to determine if there was a significant difference in the antigen-induced expression (2−ΔΔCq value) of each gene between M. bovis-infected and uninfected warthogs. Receiver operator characteristic (ROC) curve analysis was used to determine cut-off values for each target gene by using Youden’s index37. Results with a p-value < 0.05 were considered statistically significant.
References
Roos, E. O. et al. Seroprevalence of Mycobacterium bovis infection in warthogs (Phacochoerus africanus) in bovine tuberculosis-endemic regions of South Africa. Transbound. Emerg. Dis. 65, 1182–1189 (2018).
de Lisle, G. W., Bengis, R. G., Schmitt, S. M. & Brien, D. J. O. Tuberculosis in free-ranging wildlife: detection, diagnosis and management. Rev. Sci. Tech. 21, 317–334 (2002).
Roos, E. O. et al. Test performance of three serological assays for the detection of Mycobacterium bovis infection in common warthogs (Phacochoerus africanus). Vet. Immunol. Immunopathol. 182, 79–84 (2016).
Maas, M., Michel, A. L. & Rutten, V. P. M. G. Facts and dilemmas in diagnosis of tuberculosis in wildlife. Comp. Immunol. Microbiol. Infect. Dis. 36, 269–285 (2013).
Cavalcanti, Y. V. N., Brelaz, M. C. A., Lemoine Neves, J. K., de, A., Ferraz, J. C. & Pereira, V. R. A. Role of TNF-alpha, IFN-gamma, and IL-10 in the development of pulmonary tuberculosis. Pulm. Med. 2012, 1–10 (2012).
Waters, W. R., Palmer, M. V., Whipple, D. L., Carlson, M. P. & Nonnecke, B. J. Diagnostic implications of antigen-induced gamma interferon, nitric oxide, and tumor necrosis factor alpha production by peripheral blood mononuclear cells from Mycobacterium bovis-infected cattle. Clin. Diagn. Lab. Immunol. 10, 960–6 (2003).
de la Rua-Domenech, R. et al. Ante mortem diagnosis of tuberculosis in cattle: A review of the tuberculin tests, gamma-interferon assay and other ancillary diagnostic techniques. Res. Vet. Sci. 81, 190–210 (2006).
Roos, E. O. et al. IP-10: A potential biomarker for detection of Mycobacterium bovis infection in warthogs (Phacochoerus africanus). Vet. Immunol. Immunopathol. 201, 43–48 (2018).
Higgitt, R. L., Buss, P. E., van Helden, P. D., Miller, M. A. & Parsons, S. D. Development of gene expression assays measuring immune responses in the spotted hyena (Crocuta crocuta). African Zool. 52, 99–104 (2017).
Olivier, T. T. et al. Development of a gene expression assay for the diagnosis of Mycobacterium bovis infection in African lions (Panthera leo). Transbound. Emerg. Dis. 64, 774–781 (2015).
Parsons, S. D. C. et al. Modification of the QuantiFERON-TB Gold (In-Tube) assay for the diagnosis of Mycobacterium bovis infection in African buffaloes (Syncerus caffer). Vet. Immunol. Immunopathol. 142, 113–8 (2011).
Higgitt, R. L. et al. An interferon gamma release assay for the detection of immune sensitization to Mycobacterium bovis in African wild dogs (Lycaon pictus). J. Wildl. Dis. 55 (2018).
Pai, M., Riley, L. W. & Colford, J. M. Jr. Interferon-γ assays in the immunodiagnosis of tuberculosis: a systematic review. Lancet Infect. Dis. 4, 761–776 (2004).
Parsons, S. D. C. et al. Development of a diagnostic gene expression assay for tuberculosis and its use under field conditions in African buffaloes (Syncerus caffer). Vet. Immunol. Immunopathol. 148, 337–42 (2012).
Frantz, L. et al. The evolution of suidae. Annu. Rev. Anim. Biosci. 4, 61–85 (2016).
Groenen, M. A. M. et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491, 393–398 (2012).
Meade, K. G. et al. Innate gene repression associated with Mycobacterium bovis infection in cattle: Toward a gene signature of disease. BMC Genomics 8, 1–15 (2007).
Magee, D. A. et al. Innate cytokine profiling of bovine alveolar macrophages reveals commonalities and divergence in the response to Mycobacterium bovis and Mycobacterium tuberculosis infection. Tuberculosis (Edinb). 94, 441–50 (2014).
Jang, C. W., Shibata, Y., Starmer, J., Yee, D. & Magnuson, T. Histone H3.3 maintains genome integrity during mammalian development. Genes Dev. 29, 1377–1393 (2015).
Schoenborn, J. R. & Wilson, C. B. Regulation of Interferon-γ during innate and adaptive immune responses. Adv. Immunol. 96, 41–101 (2007).
Lande, R. et al. IFN-released by Mycobacterium tuberculosis-infected human dendritic cells induces the expression of CXCL10: Selective recruitment of NK and activated T Cells. J. Immunol. 170, 1174–1182 (2003).
Liu, M. et al. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 22, 121–130 (2011).
Ruhwald, M. et al. CXCL10/IP-10 release is induced by incubation of whole blood from tuberculosis patients with ESAT-6, CFP10 and TB7.7. Microbes Infect. 9, 806–812 (2007).
Ruhwald, M. et al. Evaluating the potential of IP-10 and MCP-2 as biomarkers for the diagnosis of tuberculosis. Eur. Respir. J. 32, 1607–1615 (2008).
Berry, M. P. R. et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466, 973–977 (2010).
Chegou, N. N., Heyckendorf, J., Walzl, G., Lange, C. & Ruhwald, M. Beyond the IFN-γ horizon: Biomarkers for immunodiagnosis of infection with Mycobacterium tuberculosis. Eur. Respir. J. 43, 1472–1486 (2014).
Hoff, S. T. et al. Human B cells produce chemokine CXCL10 in the presence of Mycobacterium tuberculosis specific T cells. Tuberculosis 95, 40–47 (2015).
Kasprowicz, V. O. et al. A molecular assay for sensitive detection of pathogen-specific T-Cells. PLoS One 6, e20606 (2011).
Kim, S. et al. Interferon gamma mRNA quantitative real-time polymerase chain reaction for the diagnosis of latent tuberculosis: a novel interferon gamma release assay. Diagn. Microbiol. Infect. Dis. 75, 68–72 (2013).
Kawabuchi, Y., Yamaguchi, Y., Ohshita, A., Minami, K. & Toge, T. Host-oriented peptide evaluation using whole blood assay for generating antigen-specific cytotoxic T lymphocytes. Anticancer Res. 24, 1193–200 (2004).
Warren, R. M. et al. Differentiation of Mycobacterium tuberculosis complex by PCR amplification of genomic regions of difference. Int. J. Tuberc. Lung Dis. 10, 818–822 (2006).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, 45e–45 (2001).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034 (2002).
Andersen, C. L., Ledet-Jensen, J. & Orntoft, T. Normalization of real-time quantitative RT-PCR data: a mode-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–5250 (2004).
Youden, W. J. Index for rating diagnostic tests. Cancer 3, 32–35 (1950).
Acknowledgements
The authors wish to acknowledge the contributions of the Veterinary Wildlife Services capture team from Kruger National Park, Guy Hausler, Dr. Jennie Hewlett, Leana Rossouw, Marius Kruger, Alicia McCall, Eduard Goosen and Dr David Cooper for their assistance during the sample collection phase of this study. We are grateful for the financial support of the NRF South African Research Initiative (SARChI, grant 86949), the American Association of Zoological Medicine Wild Animal Health Fund (#7-2016) and the South African Medical Research Council for this study. The content of this manuscript is the sole responsibility of the authors. Funding agencies had no part in the study design, implementation, analyses, or writing of this manuscript. Thus, the publication and its content does not necessarily represent the views of the funders.
Author information
Authors and Affiliations
Contributions
E.O.R., R.M.W., T.T.S., M.A.M. and S.D.C.P. were responsible for the conceptualizing the project. E.O.R., M.A.M., P.E.B. and L.-M.d.K.-L. were instrumental in the acquiring samples from the field. E.O.R., L.A.S., S.N. and T.T.S. were responsible for performing the lab work and generating data. F.O.-P., M.A.M. and S.D.C.P. supervised and encouraged E.O.R. to investigate the findings. E.O.R. was responsible for the data analysis. E.O.R., M.A.M. and S.D.C.P. drafted the manuscript. All authors critically revised before accepting and approving the final version (E.O.R., L.A.S., S.N., F.O.-P., P.E.B., L.-M.d.K.-L., R.M.W., P.D.v.H., T.T.S., M.A.M. and S.D.C.P.).
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Roos, E.O., Scott, L.A., Ndou, S. et al. Cytokine gene expression assay as a diagnostic tool for detection of Mycobacterium bovis infection in warthogs (Phacochoerus africanus). Sci Rep 9, 16525 (2019). https://doi.org/10.1038/s41598-019-53045-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53045-0
This article is cited by
-
Biological properties and diverse cytokine profiles followed by in vitro and in vivo infections with LSDV strain isolated in first outbreaks in Vietnam
Veterinary Research Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
