
Abstract
This study aimed to design and functionally characterize peptide mimetics of the nanobody (Nb) related to the β2-adrenergic receptor (β2-AR) (nanobody-derived peptide, NDP). We postulated that the computationally derived and optimized complementarity-determining region 3 (CDR3) of Nb is sufficient for its interaction with receptor. Sequence-related Nb-families preferring the agonist-bound active conformation of β2-AR were analysed using the informational spectrum method (ISM) and β2-AR:NDP complexes studied using protein-peptide docking and molecular dynamics (MD) simulations in conjunction with metadynamics calculations of free energy binding. The selected NDP of Nb71, designated P3, was 17 amino acids long and included CDR3. Metadynamics calculations yielded a binding free energy for the β2-AR:P3 complex of ΔG = (−7.23 ± 0.04) kcal/mol, or a Kd of (7.9 ± 0.5) μM, for T = 310 K. In vitro circular dichroism (CD) spectropolarimetry and microscale thermophoresis (MST) data provided additional evidence for P3 interaction with agonist-activated β2-AR, which displayed ~10-fold higher affinity for P3 than the unstimulated receptor (MST-derived EC50 of 3.57 µM vs. 58.22 µM), while its ability to inhibit the agonist-induced interaction of β2-AR with β-arrestin 2 was less evident. In summary, theoretical and experimental evidence indicated that P3 preferentially binds agonist-activated β2-AR.
Similar content being viewed by others


Introduction
Nanobodies (Nbs) are recombinant, antigen-specific, single-domain, variable fragments of camelid heavy-chain-only antibodies with a broad range of diagnostic, therapeutic and research applications1, including studies of G-protein-coupled receptors (GPCRs). GPCRs comprise the largest family of cell-surface receptors, as well as the most intensively studied drug target family, with implications in almost every major disease category2. In particular, GPCR crystallography has experienced impressive progress in recent years. Since the structures of rhodopsin and β2-adrenergic receptor (β2-AR) were resolved in 20003 and 20074,5, respectively, over 200 structures of more than 50 GPCRs have been solved (reviewed in6); yet data about the conformational changes associated with their activation is still sparse. As the biological activity induced by the binding of a ligand to orthosteric or allosteric sites on a GPCR is mediated by the stabilization of specific receptor conformations7, the emerging theme in GPCR activation/signalling is the role of the structural conformation of the receptor in G-protein/effector protein selection. The approaches used to obtain detailed descriptions of GPCR activation dynamics include molecular simulations and the use of Nbs that bind conformational epitopes, which occur only in native proteins8,9. Nbs were instrumental in solving (i) the first structure of activated β2-AR in complex with the Nb designated Nb80 and slowly dissociating agonist BI16710710, (ii) agonist-occupied β2-AR:Gs heterotrimer complex10 and (iii) β2-AR in complex with its low-affinity agonist adrenaline and the nanobody Nb6B9, which exhibited improved affinity and slower dissociation11. The same approach also facilitated the crystallization of agonist-bound active-state structures of other GPCRs (for reviews see8,12,13). Additionally, β2-AR-specific Nbs transiently expressed as “intrabodies” in HEK-293 cells retain their conformational specificity and have been used as a tool to study GPCR signalling via G-protein and β-arrestin recruitment14. Nbs that stabilize the active conformation, such as Nb80, bind to the intracellular domain of a GPCR that is otherwise occupied by Gα subunit or β-arrestin15, primarily through the third complementarity-determining region (CDR3), whereas CDR1 should stabilize only this interaction16. Therefore, we hypothesized that approximately only 25% of the length of original Nbs that is computationally derived and optimized is sufficient for its interaction with the receptor. The main objective of our study was, therefore, the computational design and functional characterization of peptide mimetics of the Nb CDR3 related to β2-AR, i.e., Nb-derived peptides (NDPs). We used the following computational approaches to test our hypothesis: (i) the informational spectrum method (ISM), a virtual spectroscopy method for investigations of protein-protein interactions and the structure/function relationship of proteins17,18 to design NDPs and to arrange a sequence of amino acids of NDPs that are functionally related to Nbs from the camelid family that are related to β2-AR, with reference to the informational properties of Nbs; (ii) explicit membrane molecular dynamics (MD) of derived NDPs docked into the intracellular space of the β2-AR active conformation to compute the protein-peptide binding free energy; and (iii) a novel computational approach in molecular dynamics stimulation that was introduced in 2002, known as metadynamics19,20 and implemented in the NAMD program21 with a CHARMM27 force field22,23, to examine the whole molecular conformational space and calculate the free energy during MD simulation. One selected computationally characterized NDP was then experimentally tested (i) by assessing its ability to bind β2-AR using techniques for studying protein-protein interactions, i.e., circular dichroism (CD) spectroscopy to detect changes in the conformation of interacting proteins24 and microscale thermophoresis (MST), a powerful analytical technique for characterizing biomolecular interactions based on the movement of molecules in microscopic temperature gradients (reviewed in25); and (ii) by interfering with its function using the previously developed bioluminescence resonance energy transfer (BRET)-based β-arrestin 2 recruitment assay26. Based on our results, molecular simulation-based theoretical calculations of binding free energy values are highly consistent with the experimental data, particularly the MST-derived half-maximal effective concentration (EC50) of the derived P3 for the agonist-activated β2-AR.
Results and Discussion
ISM analysis of Nbs and the interaction between β2-AR and Nbs
ISM was employed to identify common informational characteristics of Nbs in terms of their preference for agonist-occupied β2-AR and information about the characteristics of the interaction between β2-AR and the Nbs, as well as to identify the key domain of Nb involved in receptor targeting (Fig. 1). Based on CDR3 conservation, β2-AR Nbs were classified into four distinct families [A, B, C, and miscellaneous (MISC)]. Nbs that showed a clear preference for agonist (BI-167107)-occupied β2-AR belonged to family B14. Here, we analysed the extensively characterized Nb80 from family B that binds agonist-activated β2-AR10 and Nb84 and Nb71 from Nb families C and MISC, respectively, the latter two of which display a preference for binding active β2-AR conformations14. Common biological characteristics of the group of proteins that share common information are represented by peaks in their consensus informational spectrum (CIS)18. A cross-spectral analysis of Nbs stabilizing an active β2-AR conformation showed that Nbs shared common information corresponding to the informational spectrum (IS) frequency F(0.216) (Fig. 1a). An ISM analysis of individual spectra of Nbs that bound the β2-AR only in the presence of agonist, Nb80 and Nb71 (Fig. 1b), revealed the same dominant IS peak at the frequency F(0.216) in both individual spectra. Furthermore, we performed a cross-spectral analysis of β2-AR and Nb71 and identified that these two molecules shared the same common information corresponding to the IS frequency F(0.216), indicating their interaction (Fig. 1c). This finding prompted us to speculate that the common information for Nbs is also the most important for the interaction of Nb71 with the β2-AR. In a subsequent analysis, we wanted to determine the Nb domains that were most important for the identified information feature because they would be a key part of the Nb in its interaction with the receptor. We assumed that this isolated portion of the Nb would be sufficient for the interaction with the β2-AR and for stabilizing an active receptor conformation.

Bioinformatics analysis of Nbs and β2-AR and Nb71 using ISM. (a) Cross-spectral analysis (CIS) of Nb80, Nb84 and Nb71 stabilizing the active conformation of β2-AR, with the characteristic peak at F(0.216). (b) IS of Nb71 and (c) CS of β2-AR and Nb71. The CS of β2-AR and Nb71 showed a common peak corresponding to the IS frequency F(0.216).
Identification of the key protein domain responsible for the interaction between β2-AR and Nbs
Next, a computational peptide scanning analysis was performed to define linear protein regions of the β2-AR that exhibited the greatest contributions to the amplitude and signal-to-noise ratio at the characteristic frequency and therefore were responsible for the interaction(s) described by the particular spectral characteristic. By performing computational peptide scanning of Nb71 at F(0.216), we identified the region encompassing amino acid residues (aar) 88–104 (Fig. 2a) as essential for the information represented by this frequency. The identified domain of Nb, denoted by P3, was the key domain responsible for the interaction with the receptor. It was a peptide of 17 aa, EDTAVYYCNANWDLLSD. As shown in Fig. 2b, P3 had an amino acid sequence that reflected common informational properties shared with Nb71. The identified NDP was proposed to be a mimetic of Nb71 and assumed sufficient for the interaction with the agonist-bound β2-AR and for stabilizing an active receptor conformation. P3 is located inside CDR3, consistent with some previous findings. First, highly diverse CDR3 regions in all antigen receptors are suggested to be the key determinants of specificity in antigen recognition27. Second, an antibody uses only a single loop, its CDR3, to interact directly with the antigen28. Because CDR3 within Nb80 is responsible for most of the binding interactions16 and because the peptidomimetics of the CDR3 loop were likely sufficient for binding to the receptor and inhibiting the interaction of β2-AR with its intracellular GPCR-interacting proteins, such as Gαs and β-arrestins, the peptidomimetics of CDR3 structurally mimicked the CDR3 loop of an Nb29.

Mapping of the putative interaction sites of β2-AR and Nb71. (a) Position of the domain in the primary structure of Nb71 (residues 88–104). (b) The IS of P3, which is 17 aa long.
Molecular docking of peptides
CABS-dock docking results were the output of the 1,000 top conformations of β2-AR-peptide complexes, along with the 10 best and individual trajectories for each final conformation. We chose a complex where the peptide was docked in the intracellular loop using the β2-AR-Nb80 binding pattern as a reference. The list of aar and interactions observed in the β2-AR-Nb80 complex (PDB 3P0G crystal structure)16 are presented in Table 1. These data guided us in selecting properly docked conformations of our peptides. Finally, the best conformations of our four peptides were isolated. Since only one (P3) out of four peptides had reported experimental activity, only P3 data are presented. However, the sole use of flexible ligand docking is not the most reliable method to estimate complex stability and ligand binding affinity; therefore, the obtained complexes were subjected to MD simulations and free energy calculations.
MD simulations
A prepared complex of β2-AR:P3 was subjected to MD simulations to determine its stability. Its structure is shown in Fig. 3. During the 100 ns of the production phase, the β2-AR:P3 complex remained stable and intermolecular interactions persisted, with eventual breaking and re-arranging on peptide terminals due to its flexibility. The interactions observed in the β2-AR:P3 complex are presented in Fig. 4a and Table 2. The total energy plot and the RMSD plots of both the receptor and peptide showed convergence of the system (Supplementary Figs S1–3). MD alone is neither sufficient to identify the proper conformation of the ligand nor to estimate its affinity for the receptor, and it should be combined with one of the methods used to calculate free energy. The basic idea behind the free energy calculations is to calculate the probability density along the postulated reaction coordinate, and typically, biased sampling is required. Therefore, the next logical step was to select a method that systematically explored the conformational space of the peptide and receptor to calculate their binding free energy values. For that purpose, we chose metadynamics19,30, which has also been successfully applied in bimolecular simulations dedicated to the study of protein-protein interactions (reviewed in20).

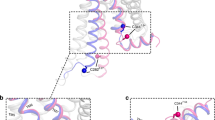
Structure of the β2-AR receptor in complex with the cocrystallized agonist P0G (8-[(1R)-2–1-hydroxyethyl]-5-hydroxy-2H-1,4-benzoxazin-3(4H)-one) and the docked peptide P3, prepared for the MD simulation.

Conformation of (a) the docked peptide in β2-AR after the first 100 ns production phase and (b) Nb80 in β2-AR. Marked interactions are the same as observed in the β2-AR-Nb80 crystal structure. Orange: electrostatic interactions; purple/grey: hydrophobic interactions. Amino acid residues shown in black belong to the β2-AR, while green residues belong to P3.
Metadynamics simulations
We performed well-tempered metadynamics simulations of the β2-AR:P3 complex after the MD production phase to estimate the binding free energy between β2-AR and the peptide. For the centroid of β2-AR aar, we chose the backbone carbon atoms Arg63, Asn69, Arg131, Ile135, Tyr141, Thr274, Ser329 and Pro330. We chose all residues of the peptide. The initial distance between the two centroids in the docked conformation was 4,890 Å, and we altered this distance by up to 60 Å. The choice of protocol was not trivial, and the main reason was the flexibility of the peptide ligand. The peptide itself possessed high flexibility, giving rise to slow equilibration and a noisy potential of mean force (PMF). After numerous preliminary metadynamics simulation experiments, we concluded that extraction of the peptide to an intracellular water layer along the z-axis of the system, parallel to the axis of the receptor and perpendicular to the cell membrane was an appropriate protocol. A good starting peptide conformation was identified by assessing its conformation after 100 ns of production where the P3 preserved its orientation and a similar binding pattern that between β2-AR and Nb80. (Fig. 4a,b, Tables 1 and 2). For further confirmation of stability after the production phase, we plotted the RMSD values of receptor and peptide (Supplementary Figs S2 and 3). Regarding the stability of β2-AR during the production and metadynamics phases, we plotted the distance between the backbone carbon atoms of Trp68 (TM3) and Ala271 (TM6) during production and the metadynamics phase (Supplementary Figs S4 and 5), the RMSDs of the receptor and the total energy profile (Supplementary Figs S6 and 7). The changes in distance during both phases showed the stability of the active conformation, and the values were within the limits of the reference values: 10.766 Å for the inactive conformation and 18.084 Å for the active conformation of β2-AR (values from PDB structures 2RH1 and 3P0G, respectively).
The distance evolution, i.e., its projection on the z-axis between receptor and peptide, is presented in Fig. 5. Regarding the PMF energy profile, the PMF energy initially increased to ~15 kcal/mol until the distance reached approximately 17 Å in the initial bound conformation as shown in Fig. 6. This energy change originated from the breaking of non-bonded interactions between aar of the receptor and peptide and peptide stretching due to the applied force on the peptide and its flexibility. During metadynamics, receptor-peptide interactions are gradually broken and re-formed. Peptide conformational changes increases the conformational energy, resulting in a very noisy PMF output. After the distance exceeds 17 Å, the PMF slowly decreases because of complete transfer of the peptide to the unbound form and convergence of the internal conformational energy of the peptide, forming a stable PMF area between 20 and 30 Å. After pushing the peptide to the upper boundary, the PMF energy continues to rise as a result of continuous peptide stretching. Afterwards, the peptide is shifted to the upper cell boundary and remains there (Fig. 5). The PMF profile is presented in Fig. 6. The binding free energy value is the negative difference between the initial and final states of PMF evolution. We averaged the stable PMF area between 20 and 30 Å to calculate the binding free energy while avoiding metadynamics artefacts and eliminating noise in the energy. The corresponding PMF difference, according to descriptive statistical analysis, was 7.23 kcal/mol; i.e., we estimated the binding free energy of the β2-AR:P3 complex as ΔG = −7.23 kcal/mol, or Ki = 7.9 μM at 310 K. The standard error of the calculation was 0.04 kcal/mol, so the final result was written as ΔG = (−7.23 ± 0.04) kcal/mol, or Kd = (7.9 ± 0.5) μM.

β2-AR:P3 distance evolution during metadynamics simulation.

Evolution of potential of mean force during the metadynamics simulation of the initially docked peptide conformation.
The peptide conformational energy minima were meant to be achieved during the 100 ns simulation time. However, due to high conformational freedom of the peptide producing a noisy PMF profile, we had no choice but to select an area within approximately 20 to 30 Å to average the PMF, as it represents the unbound peptide form. The complete metadynamics simulation movie is in Supplementary Material S8, and the non-peptide-bound form is observed at approximately 00:00:07. We are aware that convergence of PMF cannot be reached using only one starting peptide conformation and only one binding/unbinding event, but the good agreement with experimental results (MST and BRET) and limited access to GPU resources prompted us to keep this approach and consider the obtained result as acceptable. In this respect, we considered the calculated potential of mean force as semiquantitative. We feel that for fully converged potential of mean force one would require a simulation length of several tens of microseconds, which exceeds available computer power.
In subsequent steps, we attempted to provide experimental evidence for the computationally characterized interaction between the β2-AR and P3.
Circular dichroism (CD)
Based on the CD spectra for P3 recorded in the far-UV range, this peptide does not have a distinctive secondary structure and is rather unstructured (Fig. 7, dark blue line). Experimental evidence from CD spectropolarimetry for the computationally characterized interaction between β2-AR and P3 is presented in Fig. 7. Cell lysates from untransfected (control) HEK-293 cells and HEK-293 cells transfected with β2-AR/Rluc8 were titrated with P3 in the presence or absence of the β2-AR agonist isoproterenol. Comparison of the graphs in Fig. 7 reveals that P3 strongly interacted with cell lysates prepared from β2-AR/Rluc8-transfected HEK-293 cells that were preincubated with the β2-AR agonist isoproterenol (Fig. 7a) relative to its interaction with lysates from either unstimulated β2-AR transfected HEK-293 cells (Fig. 7b) or isoproterenol-stimulated, untransfected HEK cells (Fig. 7c). This claim is based on the hypothesis that all P3 is bound to β2-AR and then the complex contributes to the entire CD signal. In the case of HEK-293 cells, a high contribution of free P3 to the CD signal was shown at P3 concentrations ≥20 µM (Fig. 7c), particularly at wavelengths below 214 nm. We also concluded that the induced CD signal was higher when β2-AR-transfected HEK-293 cells were pretreated with agonist than in unstimulated cells (cf. panels a and b in Fig. 7). The only difference between the untransfected and β2-AR-transfected HEK-293 cells was that the level of β2-AR was significantly higher in transfected HEK-293 cells (confirmed by total luminescence measurements) than in untransfected HEK-293 cells, in which low endogenous expression of the β2-AR was indicated by the very sensitive cAMP ALPHAscreenTM assay31 and the low expression of the β2-AR mRNA32.

CD spectra of cell lysates prepared from β2-AR-transfected/untransfected HEK-293 cells incubated with different concentrations of P3 in the presence or absence of the agonist isoproterenol. Lysates of HEK-293 cells transiently transfected with β2-AR/Rluc8 (a and b) and mock-transfected (control) HEK-293 cells (c) were titrated with P3 in the presence (a,c) or absence of the β2-AR agonist isoproterenol (b). In the presence of agonist, the cell lysate was preincubated with 1 µM isoproterenol, and then P3 was added at a final concentration of 5, 10, 20 or 30 µM. The CD spectrum of P3 (P3, dark blue line) in buffer is shown for comparison. All experiments were performed in PBS, pH 7.2, containing 0.1% DMSO at 25 °C.
Microscale thermophoresis (MST) data
Next, we used the recently developed MST method to quantify β2-AR:P3 interactions. Measurements were performed directly in cell lysates prepared from HEK-293 cells transiently transfected with β2-AR tagged with the green fluorescent protein 2 (β2-AR/GFP2), without the need for a prior protein purification/labelling step. MST-derived dose-response curves for the interactions of isoproterenol and P3 with β2-AR are shown in Fig. 8. The EC50 of the well-validated β-adrenoceptor agonist isoproterenol for β2-AR was in the submicromolar range, i.e., 0.275 ± 0.065 µM, which corresponded well to the relatively low affinity of β2-AR agonists10. On the other hand, P3 displayed EC50 values of 58.22 ± 13.59 µM and 3.57 ± 0.81 µM for non-activated and agonist-activated β2-AR, respectively. Isoproterenol-activated β2-AR thus displayed an approximately 10-fold higher affinity for P3 than unstimulated β2-AR (Fig. 8b), corroborating the calculated Ki (7.9 μM) obtained from the metadynamics simulation. This finding is also consistent with the CD spectropolarimetry observations and provides additional experimental evidence for the preference of P3 for agonist-activated β2-AR. Similarly, Nb80 efficiently binds to only agonist-occupied β2-AR10.

Detection of the interaction of P3 with the β2-AR using MST. In the MST experiment, the concentration of β2-AR/GFP2 was maintained at a constant value by adding equal amounts of cell lysate while increasing the concentration of the known β2-AR agonist isoproterenol (control, panel a) or tested NDP, i.e., P3, in the absence (panel b, solid line) or presence of 10 µM isoproterenol (panel b, dotted line). After a short incubation, the samples were centrifuged and loaded into MST NT.115 premium capillaries, and the MST analysis was performed using Monolith NT.115pico, as described in the Materials and Methods. The change in the normalized fluorescence (ΔFnorm) value of each point was divided by the amplitude of the fitted curve to calculate the bound fraction, resulting in values ranging from 0 to 1. Data (mean ± S.E.) were obtained from 3–5 independent measurements.
BRET2-based β-arrestin 2 recruitment assay
Because the protein-peptide binding energies of the β2-AR:P3 complex and MST predicted a micromolar-range interaction of P3 with the agonist-activated β2-AR, we next evaluated the ability of P3 to interfere with the agonist-induced interaction of β2-AR with β-arrestin 2, as P3 and β-arrestin 2 should compete for the same binding site, and Nb71, which was used as the model to derive P3, is a powerful inhibitor of β-arrestin recruitment to β2-AR14. Due to the hydrophilic nature of P3 and its presumptive binding to the intracellular receptor site, we modified the previously developed BRET-based β-arrestin 2 recruitment assay26 to enable P3 to bind to β2-AR. Therefore, we used either homogenized or Triton-X100-permeabilized cells (Fig. 9) instead of whole cells. First, previously characterized β2-AR ligands were tested to validate our modified BRET2 assay. The maximal agonist-induced BRET signal (BRETmax) was reduced by approximately 50% when the experiment was performed with homogenized or permeabilized cells relative to the maximal signal when the experiment was performed with whole cells (BRETmax of ≈40 vs. 80 mBU), whereas the potency of isoproterenol and pindolol remained comparable among the various conditions (see panels a and b in Fig. 9). The obtained BRET2 EC50 values for the isoproterenol-induced interaction of the β-arrestin 2 Arg393Glu, Arg395Glu mutant with β2-AR (13.65 ± 1.98, 28.89 ± 1.98 and 13.74 ± 1.37 nM in whole, homogenized and permeabilized cells, respectively) were also highly consistent with previously reported data26. Figure 9b shows the BRET2 antagonist dose-response curves generated in the presence of increasing concentrations of the adrenergic receptor antagonist pindolol. The potency of pindolol (IC50 of 1.49 ± 0.07, 3.76 ± 0.08 and 2.24 ± 0.22 nM in whole, homogenized and permeabilized cells, respectively) at inhibiting the isoproterenol-induced BRET2 signal was consistent with a previously reported range26. The hydrophilic NDP P3 showed no effect on the isoproterenol-induced BRET2 signal in whole cells, but when either homogenized or permeabilized cells were used, the peptide caused a small dose-dependent reduction (up to 10%) in the isoproterenol-induced BRET2 signal (Fig. 9c), with an estimated EC50 in a low-nanomolar range, i.e., ~1 nM. We presumed that the inability of P3 to effectively compete with β-arrestin 2 for the same binding site was due to its reduced size (less than 25% of the length of the original Nb). Researchers have also postulated that Nb71 inhibits agonist-mediated β-arrestin recruitment to the β2-AR by blocking receptor phosphorylation14. Therefore, we also hypothesized that the computationally derived NDP of the Nb71 CDR3 is not able to block receptor phosphorylation. The β-arrestin 2 Arg393Glu, Arg395Glu mutant used in our study should bind to the receptor regardless of its phosphorylation status due to the reversed charge of an amino acid in the polar core (Arg393Glu)33,34, and this mutation prolongs the lifetime of the receptor:β-arr2 complex due to disruption of β-arrestin 2:AP-2 binding (Arg395Glu34). Similarly, peptidomimetics of the Nb80 CDR3 loop were recently shown to only moderately inhibit isoproterenol-induced cAMP production29, suggesting that additional residues outside the CDR3 loop are important for effective interference with the Gα/β-arrestin interactions. Alternatively, the relative inefficiency of P3 in the BRET recruitment assay might have been due to the lower affinity of P3 (micromolar range) than purified β-arrestin for phosphorylated β2-AR, which is in the nanomolar range (a reported Kd of 1.8 nM)35.

Comparison of ligand-induced BRET2 signals. HEK-293 cells were transiently transfected with β2-AR/RLuc8 together with the GFP2/β-arr2 Arg393Glu, Arg395Glu at a 1:10 cDNA ratio, and the BRET2 assay was performed with whole, homogenized or Triton X-100-permeabilized cells. Increasing concentrations of (a) the agonist isoproterenol (10−12 to 10−5 M final concentration), (b) the antagonist pindolol (10−12 to 10−6 M final concentration) and isoproterenol (10 µM final concentration) or (c) P3 (10−12 to 10−5 M final concentration) and isoproterenol (10 µM final concentration) were added to cells, and BRET2 was measured as described in Materials and Methods. Data are presented as the means ± S.E. of triplicate measurements.
In summary, this study presents evidence obtained from a combination of computer-based methodological approaches supported by in vitro experimental data used to design and characterize NDPs. It is a small step towards obtaining a better understanding of GPCR dynamics at the molecular level in the context of GPCR interactions with their protein partners. The combined use of experimental and computational techniques represents a powerful framework for achieving progress in this direction and could lead to further modification and optimization of NDPs as efficient modulators of GPCRs and other applications, including drug discovery and therapy.
Methods
Materials
Molecular biology and cell culture reagents for were from Sigma-Aldrich (St. Louis, MO, USA) and Gibco Invitrogen Corporation (Breda, The Netherlands). Pindolol and isoproterenol were from Sigma-Aldrich. Coelenterazine 400a from Biotrend Chemikalien GmbH (Köln, Germany). Selected NDPs (P1-P4) were custom-synthesized at Biomatik Corporation, Cambridge, Ontario, Canada.
Informational spectrum method (ISM)
The principle of the ISM has been thoroughly explained18,36 and has been successfully applied to the structure-function analysis of different proteins18, the prediction of new protein interactors37 and the identification of protein domains responsible for long-range interactions38.
Computational peptide scanning
Computational peptide scanning was utilized to define linear protein regions responsible for the interaction(s) described by the particular spectral characteristic. The sequence of Nb71 was scanned by the ISM algorithm with overlapping windows of different lengths to identify regions with the highest amplitudes at the predefined Fourier frequency.
Datasets
The sequence of human β2-AR used for the bioinformatics analysis was retrieved from the UniProt database with accession number P07550. The sequences of Nbs are presented in US patent US20130137856 and the PDB entry 3P0G FASTA sequence.
Receptor preparation
The active state crystal structure of the β2-AR was obtained from the RSCB protein databank (PDB entry code 3P0G). All lipids, water molecules, and ions and Nb80 were removed. Only the P0G ligand was retained.
Molecular docking of peptides
Peptide-protein docking was conducted using the online CABS-dock server39. Using the given protein receptor 3D structure, binding site and peptide sequence, a docking search for the binding site is performed that allows for full flexibility of the peptide and small fluctuations in the receptor backbone. The output of the simulation is the three-dimensional coordinates of the protein in complex with the ligand accompanied by full docking process trajectories and CABS force field docking scores, including energies of the receptor, ligand and their interaction. The binding site is indirectly defined by unlikely-to-bind regions of β2-AR, which exclude all aar regions except intracellular loops. The number of simulation cycles was set to 50. The best solutions, including the peptide in the intracellular space and lowest CABS-dock energy, were selected for further MD simulations.
Ligand parameterization
Ligand was assigned CGenFF force field atomic charges (ParamChem)40 and van der Waals parameters, whereas force constants were obtained from the Hessian equation calculated after geometric optimization of the HF/6–31 G(d) level of theory in Gaussian 03 W41. All parameters were generated in the VMD Parameterize extension42.
MD simulations
The β2-AR-agonist-NDP complex, with peptide coordinates obtained from the docking output, was inserted into a 70 × 70 Å 2-oleoyl-1-palmitoyl-sn-glyecro-3-phosphocholine (POPC) lipid bilayer. A 10 Å water layer was added from the positive side of the z-axis and 60 Å from the negative side. Bad-contact water molecules were removed from the lipid membrane bilayer using the appropriate tcl script. Additionally, the system was neutralized with 0.15 M NaCl, resulting in a 61,183 (~60,000) atom ensemble. The system was subject to a 10,000 step energy minimization, 250 NVE ps equilibration, and 100 ns NPT MD production. Pressure and temperature were set to 1 bar and 310 K, respectively, using a Berendsen thermostat, and the applied integration step was 1 fs. In all simulations, periodic boundary conditions with particle-mesh Ewald calculations were implemented. The cut-off was set to 12 Å. A CHARMM2222,23 force field was used for protein and lipids, and CGenFF43,44 was used for ligands.
Metadynamics simulation
Metadynamics is a powerful method for calculating free energy. It was initially developed by Laio and Parrinello19 and later improved to well-tempered metadynamics20. Before metadynamics simulation, one must determine the collective variables that will be varied during simulation and in regard to which the PMF will be calculated. We chose one variable, the distance between centroids of protein amino acid (aa) and peptide residues. Their atoms belonged to backbone C of the binding aar in the intracellular loops of the receptor and all residues in the peptide. The lower boundary (minimal distance value) was set to the initial distance between centroids of receptor-ligand atom groups, which was obtained from the coordinates of the docked structure optimized by MD production. The upper boundary (maximal value) was set to the distance at which the peptide was located sufficiently far away from the receptor in the water layer close to the side of the PBC cell. In this way, we ensured that no intermolecular interactions between the protein and peptide were present along all three coordinate axes. During our metadynamics simulation, the peptide pushed towards the intracellular water layer, parallel to the z-axis of the cell and perpendicular to the cell membrane. The resulting change in free energy between initial and equilibrated states of the peptide in the water layer was designated the binding free energy of the complex. Collective variable trajectory frequency (frequency of generating free energy files) was set to 10,000 ps. The lower wall constant (lowest value of applied force, units in kcal/(mol*A)) was set to 120.0, the upper wall constant (highest value of applied force) was set to 180.0, and width (the force resolution) was set to 0.1. For the main atoms, we selected backbone carbon atoms from the receptor, and for the reference, we selected backbone carbon atoms from the peptide. The hill weight (amount of PMF energy that is gradually added to a system during simulation) was set to 0.1 kcal/mol, the hill width was set to 1.0 Å, and the new hill frequency was set to 100 ps. Bias temperature was set to 1550 K. All free energy files generated during simulation were collected. The total metadynamics simulation time was 40 ns, and the integration step was 1 fs. Descriptive statistical analysis of PMF and the corresponding Figures was performed using Origin 8 (OriginLab Corporation, Northampton, MA, USA).
Fusion constructs
Human HA-tagged β2-AR (HAβ2-AR) cDNA and human β-arrestin 2 N-terminally tagged with the green fluorescent protein variant 2 GFP2 (GFP2/β-arr2) were from cDNA Resource Center (www.cdna.org) and from BioSignal Packard Inc., Montreal, Canada, respectively. C-terminal Renilla luciferase 8 (RLuc8) tagged HAβ2-AR (β2-AR/RLuc8), C-terminal GFP2-tagged β2-AR (β2-AR/GFP2) and mutant GFP2/β-arr2 (GFP2/β-arr2 Arg393Glu, Arg395Glu) were described in previous studies26,45,46.
Cell culture and transfection
HEK-293 cells (European Collection of Animal Cell Cultures, Salisbury, UK) were routinely maintained and passaged as described previously45,47. Transient transfections were performed when cells reached ~90% confluence using the Lipofectamine®-Plus™ Reagent. The expression levels of RLuc8- and GFP2-tagged constructs were monitored by recording total luminescence and fluorescence as previously described48.
Circular dichroism (CD)
All CD spectra were recorded on a Jasco J-1500 spectropolarimeter at 25 °C using a 1 mm quartz cuvette in the UV range of 250–200 nm. The CD spectra were measured every 0.5 nm with a scanning rate of 10 nm/min. Lysed cells (A280 = 1.5) (HEK-293, β2-AR/Rluc8 transfected HEK-293 cells) were dialysed against PBS buffer, pH 7.2, containing 1% glucose and 0.1% DMSO. P3 was solubilized in 0.1% DMSO in DPBS supplemented with Ca2+/Mg2+, 1 g/L glucose, and 36 mg/L sodium pyruvate buffer. The concentration of the P3 stock solution was 1 mM. CD spectra of a 10-fold dilution of the initial cell lysate in the presence of P3 at final concentrations of 0, 5, 10, 20, and 30 µM were recorded. The density of HEK-293 and β2-AR/Rluc8-transfected HEK-293 cells were equal, as determined spectrophotometrically. In the case of agonist addition (10 µM), the cell lysate was incubated with agonist for 5 minutes at 25 °C before the CD spectra were recorded. Each CD spectrum was subtracted from the CD spectrum of the buffer.
Microscale thermophoresis (MST)
HEK-293 cells plated in a 75 cm2 flask were transiently co-transfected with constructs encoding β2-AR/GFP2 (5.0 μg), and cell lysates were prepared after 48 h as described below. The 75 cm2 flasks were transferred to ice, and cells were washed once with ice-cold DPBS. Then, 750 µL of NP40 lysis buffer was added to each 75 cm2 flask and incubated on ice for 5 minutes. Cells were scraped from the 75 cm2 tissue culture flasks using cell scrapers (Falcon), and detached cells were incubated on ice for an additional 20 minutes. Cell lysates were transferred to cold 1.5 mL tubes and clarified by centrifugation (15,300 rpm for 30 minutes at 4 °C). Next, 700 µL of cleared lysate was transferred to a new cold 1.5 mL tube, and the level of β2-AR/GFP2 was verified by measuring the total fluorescence. Ten microlitres of cell lysate were used as the target, while the non-fluorescent ligands isoproterenol and P3 were titrated in a series of 1:1 dilutions in MST buffer containing 0.05% Tween 20. Ten microlitres of the serial dilution of the ligand were mixed with 10 μL of the cell lysate. After a short incubation at room temperature, samples were centrifuged and loaded into Monolith™ NT.115 MST Premium Capillaries (NanoTemper Technologies GmbH, Munich, Germany) and measured using a Monolith NT.115pico and MO. The control software was set to room temperature (25 °C) (LED/excitation power setting 95%, MST power setting low). Data were analysed using MO.Affinity Analysis software (version v2.2.4, NanoTemper Technologies) at the standard MST-on time of 5 s and presented as a bound fraction. The change in the normalized fluorescence (ΔFnorm) value of each point was divided by the amplitude of the fitted curve to calculate the bound fraction, resulting in values ranging from 0 to 1. This approach is independent of both the initial Fnorm value and the amplitude of the binding curve and thus enabled us to compare the EC50 values of interactions with different amplitudes.
BRET-based β-arrestin 2 recruitment assay
We used a previously described BRET-based βarr2 recruitment assay26,45,48, with some modifications. Because P3 binds to intracellular receptor domains, BRET assays were performed using either homogenized cells or permeabilized cells. HEK-293 cells cultured in a 75 cm2 flask were transiently co-transfected with constructs encoding β2-AR/RLuc8 (0.1 μg) alone or together with the double GFP2/β-arr2 Arg393Glu, Arg395Glu mutant (4.9 μg). Homogenized/permeabilized cells were prepared 48 h after transfection. Homogenized cells were washed twice with DPBS, scraped from 75 cm2 tissue culture flasks using cell scrapers (Falcon) and pelleted by centrifugation at 1,000 rpm for 5 minutes. The cell pellets were resuspended in 1 mL of DPBS supplemented with Ca2+/Mg2+, 1 g/L glucose, and 36 mg/L sodium pyruvate, incubated on ice for 10 minutes, and homogenized using a glass homogenizer (BDH).
Cells were permeabilized by adding 0.01% Triton™ X-100 in DPBS and incubating for 15 minutes at 37 °C. Homogenized/permeabilized cells were then diluted in supplemented DPBS such that the luminescence signal in 180 μL of DPBS containing homogenized/permeabilized cells distributed in 96-well microplates (white Optiplate; Packard BioScience, Meriden, CT, USA) was approximately 30,000 arbitrary units. After the coelenterazine 400a was injected (final concentration of 5 μM), the luminescence signals were recorded (TriStar LB 942 microplate reader, Berthold Technologies, Bad Wildbad, Germany). Signals at 410 nm and 515 nm were recorded sequentially, and the 515/410 ratio (BRET2 signal) was calculated. The results were reported in milliBRET units (mBU); BRET2 ratio × 1000. The expression levels of RLuc8- and GFP2-tagged constructs in each experiment were assessed by measuring the total luminescence and fluorescence as described above. Determinations were performed in triplicate. The obtained data were transferred to GraphPad Prism 7.0 (GraphPad software, San Diego, CA, USA) and BRET EC50/IC50 values (nM ± SEM) generated using a sigmoidal dose-response curve fit.
Data availability
All data generated or analysed during this study are included in this published article and its Supplementary Information Files.
References
Hassanzadeh-Ghassabeh, G., Devoogdt, N., De Pauw, P., Vincke, C. & Muyldermans, S. Nanobodies and their potential applications. Nanomedicine (Lond) 8, 1013–1026 (2013).
Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schioth, H. B. & Gloriam, D. E. Trends in GPCR drug discovery: new agents, targets and indications. Natl. Rev. Drug Discov. 16, 829–842 (2017).
Palczewski, K. et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289, 739–745 (2000).
Cherezov, V. et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 (2007).
Rasmussen, S. G. et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 450, 383–387 (2007).
Thal, D. M., Glukhova, A., Sexton, P. M. & Christopoulos, A. Structural insights into G-protein-coupled receptor allostery. Nature 559, 45–53 (2018).
Katritch, V., Cherezov, V. & Stevens, R. C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 (2013).
Manglik, A., Kobilka, B. K. & Steyaert, J. Nanobodies to study g protein-coupled receptor structure and function. Annu. Rev. Pharmacol. Toxicol. 57, 19–37 (2017).
Steyaert, J. & Kobilka, B. K. Nanobody stabilization of G protein-coupled receptor conformational states. Curr. Opin. Struct. Biol. 21, 567–572 (2011).
Rasmussen, S. G. et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011).
Ring, A. M. et al. Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579 (2013).
Granier, S. & Kobilka, B. A new era of GPCR structural and chemical biology. Natl. Chem. Biol. 8, 670–673 (2012).
Stevens, R. C. et al. The GPCR network: a large-scale collaboration to determine human GPCR structure and function. Natl. Rev. Drug Discov. 12, 25–34 (2013).
Staus, D. P. et al. Regulation of beta2-adrenergic receptor function by conformationally selective single-domain intrabodies. Mol. Pharmacol. 85, 472–481 (2014).
Kang, Y. et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567 (2015).
Rasmussen, S. G. et al. Structure of a nanobody-stabilized active state of the beta2 adrenoceptor. Nature 469, 175–180 (2011).
Schmier, S. et al. In silico prediction and experimental confirmation of HA residues conferring enhanced human receptor specificity of H5N1 influenza a viruses. Sci. Rep. 5, 11434 (2015).
Veljkovic, N. et al. Discovery of new therapeutic targets by the informational spectrum method. Curr. Protein Pept. Sci. 9, 493–506 (2008).
Laio, A. & Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 99, 12562–12566 (2002).
Barducci, A., Bussi, G. & Parrinello, M. Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys. Rev. Lett. 100, 020603 (2008).
Phillips, J. C. et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 (2005).
MacKerell, A. D. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998).
Mackerell, A. D. Jr., Feig, M. & Brooks, C. L. III Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 (2004).
Greenfield, N. J. Circular dichroism (CD) analyses of protein-protein interactions. Methods Mol. Biol. 1278, 239–265 (2015).
Asmari, M., Ratih, R., Alhazmi, H. A. & El Deeb, S. Thermophoresis for characterizing biomolecular interaction. Methods 146, 107–119 (2018).
Vrecl, M., Jorgensen, R., Pogačnik, A. & Heding, A. Development of a BRET2 screening assay using beta-arrestin 2 mutants. J. Biomol. Screen. 9, 322–333 (2004).
Xu, J. L. & Davis, M. M. Diversity in the CDR3 region of VH is sufficient for most antibody specificities. Immunity 13, 37–45 (2000).
Desmyter, A., Decanniere, K., Muyldermans, S. & Wyns, L. Antigen specificity and high affinity binding provided by one single loop of a camel single-domain antibody. J. Biol. Chem. 276, 26285–26290 (2001).
Martin, C. et al. Rational design of nanobody80 loop peptidomimetics: towards biased beta2 adrenergic receptor ligands. Chemistry 23, 9632–9640 (2017).
Barducci, A., Bonomi, M. & Parrinello, M. Metadynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 1, 826–843 (2011).
Elster, L., Elling, C. & Heding, A. Bioluminescence resonance energy transfer as a screening assay: focus on partial and inverse agonism. J. Biomol. Screen. 12, 41–49 (2007).
Atwood, B. K., Lopez, J., Wager-Miller, J., Mackie, K. & Straiker, A. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genom. 12, 14 (2011).
Celver, J., Vishnivetskiy, S. A., Chavkin, C. & Gurevich, V. V. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J. Biol. Chem. 277, 9043–9048 (2002).
Milano, S. K., Pace, H. C., Kim, Y. M., Brenner, C. & Benovic, J. L. Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry 41, 3321–3328 (2002).
Sohlemann, P., Hekman, M., Puzicha, M., Buchen, C. & Lohse, M. J. Binding of purified recombinant beta-arrestin to guanine-nucleotide-binding-protein-coupled receptors. Eur. J. Biochem. 232, 464–472 (1995).
Veljkovic, N., Glisic, S., Perovic, V. & Veljkovic, V. The role of long-range intermolecular interactions in discovery of new drugs. Expert Opin. Drug. Discov. 6, 1263–1270 (2011).
Doliana, R. et al. EMILINs interact with anthrax protective antigen and inhibit toxin action in vitro. Matrix Biol. 27, 96–106 (2008).
Veljkovic, V. et al. Identification of hemagglutinin structural domain and polymorphisms which may modulate swine H1N1 interactions with human receptor. BMC Struct. Biol. 9, 62 (2009).
Kurcinski, M., Jamroz, M., Blaszczyk, M., Kolinski, A. & Kmiecik, S. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res. 43, W419–W424 (2015).
Ghosh, J. et al. Molecular parameter optimization gateway (ParamChem): workflow management through TeraGrid ASTA. in Proceedings of the 2011 TeraGrid conference: extreme digital discovery 1–8 (ACM, 2011).
Frisch, M. J. et al. Gaussian 16 Rev. B.01 (Gaussian Inc., 2016).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14(33–38), 27–38 (1996).
Vanommeslaeghe, K. et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690 (2010).
Yu, W., He, X., Vanommeslaeghe, K. & MacKerell, A. D. Jr. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 33, 2451–2468 (2012).
Mandic, M. et al. Demonstration of a direct interaction between beta2-adrenergic receptor and insulin receptor by BRET and bioinformatics. PLoS One 9, e112664 (2014).
Vrecl, M., Drinovec, L., Elling, C. & Heding, A. Opsin oligomerization in a heterologous cell system. J. Recept. Signal Transduct. Res. 26, 505–526 (2006).
Kubale, V., Blagotinsek, K., Nohr, J., Eidne, K. A. & Vrecl, M. The conserved arginine cluster in the insert of the third cytoplasmic loop of the long form of the D2 dopamine receptor (D2L-R) acts as an intracellular retention signal. Int. J. Mol. Sci. 17, E1152 (2016).
Vrecl, M. et al. Beta-arrestin-based BRET2 screening assay for the “non”-beta-arrestin binding CB1 receptor. J. Biomol. Screen. 14, 371–380 (2009).
Acknowledgements
We acknowledge funding from the Slovenian Research Agency programmes P4-0053 to M. Vrecl, P4-0121 to M. Šnajder and N. Poklar Ulrih, and P1-0012 to J. Mavri, as well as a grant No. 173001 from the Ministry of Education, Science and Technological Development of the Republic of Serbia to M. Sencanski, S. Glisic and N. Veljkovic. M. Vrecl and M. Sencanski participate in the European COST Action CA 18133 (ERNEST). Special thanks to application specialist Dr. Jakub Nowak (NanoTemper Technologies) for his consultation on MST experimental design/data analysis and to NanoTemper Technologies for providing access to the Monolith NT.115pico device. The authors would also like to thank American Journal Experts for language editing and proofreading.
Author information
Authors and Affiliations
Contributions
Milan Sencanski, Janez Mavri, Sanja Glisic, Nataša Poklar Ulrih and Milka Vrecl conceived the study and designed the experiments. Sanja Glisic and Nevena Veljkovic performed the ISM analysis. Milan Sencanski performed MD and metadynamics analyses. Marko Šnajder performed CD spectropolarimetry. Milka Vrecl performed MST and BRET experiments. Nataša Poklurar Ulrih and Janez Mavri provided equipment and analysis tools. All authors were involved in data analysis/interpretation and manuscript preparation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sencanski, M., Glisic, S., Šnajder, M. et al. Computational design and characterization of nanobody-derived peptides that stabilize the active conformation of the β2-adrenergic receptor (β2-AR). Sci Rep 9, 16555 (2019). https://doi.org/10.1038/s41598-019-52934-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52934-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
