
Abstract
Human cytomegalovirus (CMV) is a major cause of morbidity in fetuses following intrauterine infection. The glycoprotein (g) envelope trimeric gH/gL/gO and pentameric gH/gL/pUL128/pUL130/pUL131A complexes are required for CMV entry into fibroblasts and endothelial/epithelial cells, respectively, and both are targets for neutralizing antibodies. The role of sequence variability among viral strains in the outcome of congenital CMV infection is controversial. Variation in the CMV UL75 gene encoding glycoprotein H (gH), the UL115 (gL), the UL74 (gO), and the UL128 locus (UL128L) encoding three structural proteins (pUL128, pUL130, and pUL131A) was determined in 82 newborns with congenital CMV infection and 113 infants with postnatal or unproven congenital CMV infection. Genotyping was performed by sequencing analysis of PCR‐amplified fragments and the PCR-restriction fragment length polymorphism (RFLP) method, and the viral load was measured by quantitative real‐time PCR. The obtained results demonstrated that (1) different CMV variants and mixed CMV infections can be detected in newborns infected congenitally; (2) the gH1 genotype, UL130 variant 6, and UL131A variant 1 were associated with some signs/symptoms within cohort of pediatric patients, mainly consisting of infants with symptomatic CMV infection. The results revealed that pUL130, pUL131A, and gH polymorphisms seemed to be associated with the outcome of CMV infection in infants.
Similar content being viewed by others
Introduction
Human cytomegalovirus (CMV) is an opportunistic ß-herpesvirus that infects 40% to nearly 100% of the adult population worldwide1,2. It is the most common cause of congenital infection worldwide and an important cause of morbidity and mortality in immunocompromised individuals. CMV infection is generally asymptomatic or mild in healthy individuals, whereas the virus undergoes latency in myeloid cells of the bone marrow and may reactivate at any time.
CMV is the leading viral cause of neonatal developmental disabilities, including sensorineural hearing loss (SNHL) and central nervous system (CNS) damage3. In the developed world, congenital CMV (cCMV) infections occur in 0.5–2.0% of pregnancies4,5. CMV infection can result in intrauterine growth restriction (IUGR), preterm birth, fetal or neonatal abnormalities and death. Approximately 5–10% of CMV-infected newborns are symptomatic, with findings including unilateral or bilateral SNHL, chorioretinitis, microcephaly, hepatomegaly, splenomegaly, thrombocytopenia, petechiae, jaundice, and seizures4,6,7. Moreover, approximately 15% of newborns with asymptomatic cCMV infection at the birth develop long-term neurological sequelae in the next years of life8,9. The highest risk of severe symptoms in the fetus and newborn exists in cases of primary maternal infection in the first-trimester10. However, natural immunity is not protective against CMV infection, and the majority of infected newborns are born to mothers who are already seropositive7,11. Primary or nonprimary maternal infection, including reactivation of the latent virus or reinfection with a different strain, can lead to crossing the placenta and infecting the fetus. Approximately 20–30% of nonimmune women who are infected during pregnancy transmit the virus to their offspring, whereas the rate of transmission following nonprimary infection is between 0.6–1%7.
CMV replicates in different cells, including epithelial cells, endothelial cells, fibroblasts and smooth muscle cells, which are the predominant targets12. This broad cell tropism facilitates both systemic spread within the host with efficient proliferation of the virus and interhost transmission. A number of factors can affect the interaction between the host immune system and the virus, and the major determinants of CMV disease appear to be host factors. The polymorphisms in the CMV genes encoding envelope glycoproteins, important targets of the host immune response, could potentially be associated with variability in the control of virus infection. Sequence variability among the viral strains may affect viral dissemination, disease severity, and immune response13. However, placental transmission appears to be independent of a specific virus strain, and different CMV genotypes are detected in infected neonates14,15,16,17,18,19. An association between CMV variants in surface proteins and clinical outcome is still controversial. The glycoprotein H (gH) is encoded by the UL75 gene and has two genotype (gH1 and gH2) based on variability in the 37 amino acid N-terminal domain13. Both gH genomic variants do not correlate with symptomatic cCMV infection20; however, an association between the gH genotype and hearing loss in infants in another study was found17. The UL74 gene encodes viral glycoprotein O (gO) and at least eight genetic variants of gO, including five major genotypes (gO1, gO2, gO3, gO4 and gO5) with sub-genotypes (gO1a, gO1b, gO1c, gO2a, gO2b), have been identified21,22. Genetic linkage between gO and glycoprotein N (gN), encoded by the UL73 gene, has been reported in CMV-infected infants22, while strong genetic linkage between the gO1 and the gH1 genotypes has been found in immunosuppressed adult patients23. Nucleotide variations is high in gN and gO genes (40–50%), lower differences exist in glycoprotein B (gB) and gH genes (5–10%), while the glycoprotein L (gL) gene is highly conserved among clinical strains.
Herpesviruses use envelope glycoproteins to enter host cells, including the viral gB that is necessary for entry into all cell types24. This viral fusogen is highly immunogenic and is the target of neutralizing antibodies25. CMV gH is another dominant target of specific antibodies that can be strain-specific26. CMV gH is crosslinked through disulfide bonds with gL. CMV requires two membrane glycoproteins, gB and gH/gL, to enter host cells, but gH/gL binds cellular receptors before triggering gB27. It was also reported that gB and gH/gL form stable gB-gH/gL complexes in cell-free virions independent of receptor binding28. The gH/gL dimer exists on the CMV surface as part of a trimeric complex with gO (gH/gL/gO), known as the gCIII complex, or a pentameric complex with the UL128 protein (pUL128), pUL130 and pUL131A (gH/gL/pUL128-131A)29. gO and pUL128-131A bind to the same site on gH/gL through a disulfide bond with gL-Cys144. Introduction of a double mutation at the disulfide bond level of the pentamer impaired syncytium formation and reduced interference with CMV entry into epithelial cells30. The gH/gL/gO complex is made of three disulfide-bonded proteins, gH, gL, and gO and is sufficient for attachment to and infection of fibroblasts31. The platelet-derived growth factor receptor alpha (PDGFR-α) has been identified as a receptor for entry into cells32,33,34. It is suggested that the trimer complex may be required for entry into all cell types33,35,36,37. The gH/gL/gO trimer binds with high affinity through the gO subunit to PDGFR-α, which is expressed by fibroblasts but not by epithelial and endothelial cells32,38. It was recently shown that the N terminus of gO contributes to efficient spread in fibroblasts by promoting the interaction of virions with cellular PDGFR-α39. The gH/gL/pUL128-131A complex consists of five proteins, namely, gH, gL, pUL128, pUL130 and pUL131A; it is required for the infection of endothelial, epithelial, and myeloid cells but is dispensable for the infection of fibroblasts40,41,42,43,44,45. Pentamer-dependent entry into epithelial and endothelial cells by endocytosis followed by low-pH-dependent fusion, while CMV strains enter fibroblasts by pH-independent fusion with the plasma membrane45. The UL128-131A gene locus (UL128L) of CMV is indispensable for both productive infection of endothelial cells and viral transfer to leukocytes31.
Recent studies have revealed findings concerning pentamer structure, location of epitopes for neutralizing antibodies and potential binding sites for cell surface receptors37. These data suggest that receptor binding triggers a conformational change in the pentamer, allowing it to interact with gB and initiate the membrane fusion process. The dimer gH/gL is thought to act as an intermediary, transmitting the fusion trigger to gB46,47. It is suggested that complexes containing gH/gL play a key role in host cell tropism32,33,34,48. Moreover, high expression of the pentamer on the epithelial cell surface leads to the interference of virus entry into cells, possibly through sequestration of cell surface receptors, providing strong evidence for a cell-specific receptor49. High levels of pentamer expression have been associated with an increase in cell-association of the virus and with cell-to-cell transmission50. Pentamer proteins are the dominant target of the most potent neutralizing antibodies, highlighting their critical role in CMV infection51,52. While antibodies that target gB and gH/gL prevent infection of all cell types, antibodies specific to the pentamer are a thousand-fold more potent than antibodies against gB or gH/gL complex specifically in neutralizing CMV infection in epithelial and endothelial cells32,52. Moreover, antibodies against pentamer are capable of protecting cytotrophoblasts against CMV infection53. A response against pentamer in seronegative pregnant women with primary infection is associated with a lower risk of transmission of the virus to the fetus54. These findings make the gH/gL/pUL128-131A complex a promising vaccine candidate.
In this study, we determined the distribution of CMV genes that code proteins forming trimer and pentamer complexes in infants with congenital and postnatal infection. We show that some genomic variants are detected more frequently in congenital than in postnatal patients and are associated with an increased risk of specific disease outcomes. Because mainly symptomatic patients were enrolled in the study, our findings may not pertain to all CMV-infected infants. These studies confirm the findings that various CMV variants can be vertically transmitted. The results support the hypothesis that variability in pentamer genes is an important factor that affects clinical sequelae following CMV infection.
Results
Study population and clinical outcome
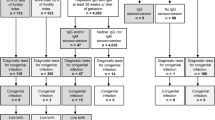
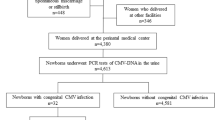
One hundred and ninety-five pediatric patients with CMV DNAemia were enrolled in the study. The patients were selected based on clinical diagnosis and diagnostic markers. Congenital CMV infection was confirmed in 82 newborns (positive CMV DNA in urine ≤ 21 day of life), and the remaining 113 children were classified as having postnatal or unproven congenital CMV infection (pCMV). Among infants in pCMV group, negative CMV DNA in urine ≤ 21 day of life was found in 83% cases. The average age at which the newborns with cCMV infection were examined was 9.6 days (median age 8.0 days; range 1–21 days), while in the group of infants with pCMV infection, it was 3.4 months (median age 3.0 months; range 1–11.5 months). The demographic and clinical characteristics of congenitally or postnatally infected children are summarized in Table 1. Almost all examined patients demonstrated cytomegaly symptoms, and this high frequency of symptomatic infection was due to selection bias. The most prevalent symptoms in CMV-infected children from both groups were neurological dysfunction and hematological disorders (especially anemia and thrombocytopenia). In addition, newborns with cCMV infection usually demonstrated CNS damage (73%), including microcephaly and abnormal brain ultrasound findings (cystic lesions, intracranial calcification, and ventriculomegaly). In contrast to postnatal infection, symptoms such as CNS damage (P < 0.0001), neurological dysfunction (P = 0.003), intrauterine growth restriction (P < 0.0001), unilateral or bilateral hearing loss (P < 0.0001), ocular defects (P = 0.015), thrombocytopenia (P = 0.0001), and petechiae (P < 0.0001) occurred more commonly after cCMV infection. Some complications, such as pneumonia and hepatitis were more common in infants in pCMV group (P = 0.005 and P = 0.022, respectively).
Viral load
The CMV DNA was detected in 94 samples obtained from 82 newborns with cCMV infection and in 125 samples obtained from 113 infants with postnatal or unproven congenital CMV infection. The viruria was found in all newborns with cCMV infection and in 83/113 (73.5%) of infants from pCMV group after 21 days of life. The CMV DNA concentration in blood samples obtained from congenitally infected newborns ranged from 2.1 × 102 to 3.2 × 105 copies/mL (median 4.2 × 103 copies/mL; mean 3.8 × 104 ± 8.6 × 104 copies/mL), while in urine samples, it ranged from 3.1 × 102 to 8.7 × 108 copies/mL (median 1.2 × 106 copies/mL; mean 3.4 × 107 ± 1.2 × 108 copies/mL). In infants from pCMV group, the viremia levels ranged from undetected to 8.2 × 105 copies/mL (median 3.6 × 102 copies/mL; mean 2.7 × 104 ± 1.4 × 105 copies/mL), whereas the viruria levels ranged from undetected to 2.2 × 108 copies/mL (median 1.6 × 104 copies/mL; mean 3.5 × 106 ± 2.4 × 107 copies/mL). In both patient groups, the CMV DNA concentration in urine was significantly higher than in blood samples (P < 0.0001, Wilcoxon test). The concentration of CMV DNA in urine samples was higher among the children with cCMV infection compared to the children who had pCMV infection (P < 0.0001; Mann–Whitney U-test). Similarly, the median blood virus load was significantly higher in children with cCMV infection than in those from pCMV group (P = 0.0002; Mann–Whitney U-test).
Prevalence of CMV variants
The trimer gH/gL/gO gene products were amplified for all examined pediatric patients (Table 2). Genotyping of gH was performed by analysis of nested PCR (nPCR)-amplified fragments and the two genomic variants were identified by different amplicon size lengths. The applied PCR-restriction fragment length polymorphism (PCR-RFLP) method allowed the detection of major gL or gO genotypes (Fig. 1). Genotypes determined by RFLP were sequenced, and no discrepancies were found. In contrast, the UL128 locus gene products were amplified and sequenced successfully for 79/82 (96.3%) newborns with cCMV infection and 101/113 (89.4%) infants in pCMV group (Table 3).

Visualization of PCR-RFLP products for gL and gO genotyping. Gel image: 1–6, gL variants; 7–12, gO variants. 1. gL1 (bands 40 bp, 90 bp, 100 bp, 108 bp, 133 bp, 150 bp), 2. gL2 (40 bp, 100 bp, 133 bp, 150 bp), 3. gL3 (100 bp, 133 bp, 150 bp, 165 bp, 225 bp), 4. gL4 (90 bp, 100 bp, 133 bp, 165 bp), 5. gL1 and gL3 (40 bp, 90 bp, 100 bp, 108 bp, 133 bp, 150 bp, 165 bp, 225 bp), 6. gL2 and gL3 (40 bp, 100 bp, 133 bp, 150 bp, 165 bp, 225 bp); 7. gO1 (390 bp), 8. gO2 (134 bp, 203 bp), 9. gO3 (134 bp, 271 bp), 10. gO1 and gO2 (134 bp, 203 bp, 390 bp), 11. gO1 and gO3 (134 bp, 271 bp, 390 bp), 12. gO1 and gO4 (110 bp, 203 bp, 390 bp). Alignments markers (15 bp, 1 kbp).
The distribution of the gH genotypes were similar in both patient groups (P > 0.05). To explore the data, one hundred and five infants with CMV infection, including 28 cases of cCMV and 77 cases of pCMV, were examined for the presence of UL75 genotypes as described in our previous study17. No significant advantages of the gH1 genotype in congenital cases and gH2 in postnatal cases were observed. The gH1 genotype was detected in 54/82 (65.9%), while the gH2 in 46/82 (56.1%) newborns with cCMV infection (Table 2). Among children with pCMV infection, the gH1 genotype was found in 63/113 (55.8%), while the gH2 genotype was found in 74/113 (65.5%) cases. The nucleotide sequence analysis confirmed the gH genomic variants previously identified by a nPCR analysis. Mixed infections with both gH1 and gH2 genotypes were detected in 18/82 (22.0%) patients with cCMV infection and in 24/113 (21.2%) cases with pCMV infection.
The UL115 gene encoding gL had a low proportion of nucleotide and amino acid variability in clinical isolates (5–9% and 1.4–2.5%, respectively), while low sequence conservation for UL74 (gO) was observed (sequence variability: 20–50% and 19.4–26.4%, respectively). All obtained amino acid sequences are illustrated in Fig. 2A–C. The gL3 genotype was prevalent in congenital infections, whereas this genomic variant was less common in postnatal infection (P < 0.0001). The most prevalent gO genotype in both groups, newborns with cCMV infection and infants with pCMV infection, was gO1 (approximately 91%). The gO4 genotype was detected more frequently in congenital compared to postnatal infection (P = 0.018). Congenital infection with mixed gL and gO genotypes was detected in 56.1% and 31.7% cases, respectively. Mixed pCMV infections were detected in 30.1% (gL) and 11.5% (gO) of infants. The children exhibited mixed infection with two gO genotypes, especially gO1 and gO4 or gO3, and with two or three distinct CMV gL genotypes. Among examined infants, mixed infections were identified mostly with viral gL1-gL3 (29 cases), gL2-gL3 (22 cases), gL3-gL4 (14 cases), and gL1-gL3-gL4 (5 cases) genomic variants. Analysis of the gH/gL/gO genotypes revealed that multiple CMV strains were detected in congenitally infected patients. Unexpectedly, considering all trimer gH/gL/gO genotypes, mixed infections were more commonly detected in congenital than in postnatal infections (75.6% vs. 48.7%; P = 0.0002).

Amino acid alignments of gH (A), gL (B), and gO (C) in clinical isolates from children with congenital and postnatal CMV infection.
The obtained UL128, UL130, and UL131A gene sequences were clustered into thirteen (named from 1 to 13), nineteen (1–19), and seven (1–7) variants, respectively. An amino acid sequence alignment of the UL128, UL130, and UL131A products is shown in Fig. 3A–C. The UL128L showed a low degree of variability. The ratios of sequence variability of UL128 ranged from 3 to 9% at the DNA level and from 0.6 to 2.3% at the amino acid level. The overall variability of the UL130 ranged from 2 to 10% at the DNA level and from 0.5 to 2.8% at the amino acid level, whereas the variability of the UL131A was 2–4% at the DNA level and 0.8% at the amino acid level.

The phylogenetic analysis of UL128 (A), UL130 (B), and UL131A (C) from clinical isolates of newborns with congenital CMV infection and infants with postnatal or unproven congenital CMV infection. The conserved cysteine residues are in box.
Several viral variants were identified in newborns with cCMV infection, indicating that all variants of the virus could be passed from mother to child. The UL128 variant 3 was identified most commonly in both pediatric patient groups (43.0% and 44.6%), and no statistically significant association with the UL128 variant distribution among children with congenital or postnatal CMV infection was observed (Table 3). The UL130 variant 6 was prevalent in newborns with cCMV and infants in pCMV group (46.8% and 43.6%, respectively). The UL130 variant 8 was identified more commonly in cCMV than pCMV patients (P = 0.027). Analysis of the UL131A region in clinical isolates revealed that the variant 1 was prevalent in both patient groups, and it occurred more frequently in congenital than postnatal infections (79.7% vs. 58.4%; P = 0.002). In contrast, the UL131A variant 4 was found in 20.3% newborns with cCMV and in 34.7% infants in pCMV group (P = 0.045). Some mixing of pUL128, pUL130, and pUL131A gene variants was observed, but the frequency was ≤ 10% among isolates.
Linkages among components of the gH/gL/gO complex
Potential linkage disequilibrium between the gH, gO, and gL genotypes was investigated in isolates obtained from all pediatric patients. The analysis included only those isolates for which the genotypes were determined (Table 4). One hundred and thirteen of 122 isolates with the gH1 genotype had also a gO1 genotype (92.6%), 2 had gO2 (1.6%), 1 had gO3 (0.8%), and 6 had gO4 (4.9%). Among 123 isolates with gH2 genotype, 112 had also a gO1 genotype (91.1%). Mixed CMV infections with various gO genotypes were detected in 25/122 (20.5%) isolates with gH1 genotype and in 18/123 (14.6%) isolates with gH2 genotype. The prevalent gO1 genotype was not associated with gH genomic variant (P > 0.05). Analysis of the Spearman’s rank correlation coefficients (ρ) and associated P-values showed no significant correlation between the gH, gL, and gO genotypes, including gH-gO (ρ = 0.02), gH-gL, and gL-gO (ρ = 0.12) paired groups (Spearman’s correlation test P > 0.05). In addition, there was no significant association between the gH, gO, and gL genotypes (Cohen’s kappa coefficient ĸ = −0.07; P > 0.05). We observed a weak negative correlation between pUL130 and pUL131A variants (ρ = −0.18; P < 0.05). No significant association was found between other gene pairs encoding the gH/gL/pUL128-131A complex (ρ-values from −0.05 to 0.11; P > 0.05). In the examined patients we also observed no conformity between the gH, gL, pUL128, pUL130, and pUL131A variants (ĸ = −0.05; P > 0.05).
CMV gene variability is associated with an increased risk of specific disease outcomes
In the patients carrying the gH1 genotype, hearing loss was diagnosed with higher incidence compared to those with the gH2 genotype (P = 0.023; Table 5). As was previously found in a smaller group of cases17, infants carrying the gH2 genotype were diagnosed with deafness at a lower incidence compared to those with the gH1 genotype (P = 0.004). In addition, gH2-infected children exhibited an increased risk of developing purpuric and petechial rashes. The gH2 genotype was associated with a diminished risk of hearing loss and psychomotor retardation in infants in adjusted and unadjusted models (Table 5).
Infection with gH1 genotype was also associated with a three-fold increased risk of neurological dysfunction and microcephaly (P < 0.05). Infection with the gH2 genotype was associated with a decreased risk of hearing loss and ventriculomegaly (P = 0.007 and P = 0.024, respectively), though this genotype was observed only in a small number of the symptomatic infants (data not shown).
The UL130 variant 6 was detected in 62.5% of newborns with IUGR (P = 0.013) and was associated with at least a three-fold increased risk of this symptom (Table 5). The risk of ocular defects was more than three-fold increased following congenital infection with this variant (P = 0.024). Infection with the UL131A variant 1 was associated with a four-fold increased risk of IUGR, neurological dysfunction, and hepatitis (P = 0.001, P = 0.025, and P = 0.021, respectively) and with at least a two-fold increased risk of CNS damage (P = 0.005). In contrast, gL2-infected patients exhibited a decreased risk of developing hematological disorders (P = 0.025 for neonates with cCMV infection; P = 0.018 for all pediatric patients).
CMV glycoprotein polymorphisms and viral load
The viral load in the blood and urine samples correlate with the infection of some CMV genotypes. The gL2 and gO3 genotypes were associated with a low shedding of CMV in urine (P = 0.005 and P = 0.008, respectively; Wilcoxon test). The viral load in the blood and urine samples did not correlate with the presence of the other CMV variants.
Discussion
Several studies have analyzed sequences from CMV clinical isolates and revealed that genomic variability caused additional infectivity or immunomodulation functions55,56,57,58,59,60. The increasing evidence suggests a correlation of clinical importance of CMV genetic diversity with pathogenesis15,16,17,18,20,61. To assess whether the variability in the CMV genes encoding trimeric and pentameric complexes are associated with congenital transmission, we compared the frequencies of variants in children with and without confirmed congenital infection. Our study focused on the variation in the all CMV genes encoding envelope protein complexes, gH/gL/gO and gH/gL/pUL128-131A, and the distribution of genotypes in pediatric patients. We have determined the sequence variation in the UL128-UL131A genes, named these specific viral variants and identified variants transmitted during congenital and postnatal infection. Our study provides the first demonstration that the CMV variants of the pentamer complex seems to be assigned to a clinical outcome in children, including, gH1, UL130 variant 6, and UL131A variant 1. In addition, the gL3 and gO4 genotypes of CMV might be an important virological marker of congenital infection in children.
Detailed sequence analysis showed that UL128, UL130, and UL131A genes were highly conserved in clinical isolates, suggesting an important role of these genes in viral pathogenesis55,61,62,63. Most amino acid substitutions were limited to the N-terminal region of the UL128 and UL131A genes, although they can also be found in other parts of the UL130 gene. The UL128 and UL130 proteins share conserved cysteine residues that are characteristic of CC- and CXC-chemokines, respectively. Our findings showed 96–99%, 84–97%, and 97–99% sequence identity at the aa level in the UL128, UL130, and UL131A proteins, respectively, in comparison to the protein sequences obtained from the publishing database NCBI. The overall identity of these CMV genes was similar and ranged between 92% and 96% in isolates from Italian patients55. Sun et al. analyzed the UL128L in 23 congenitally infected Chinese infants and found that the nucleotide and amino acid identity among strains scored 96–97%63. Baldanti et al. suggested that a high conservation of the UL128L is essential for CMV growth in endothelial cells and virus transfer to leukocytes55. According to the results of Vogel et al. sequencing with subsequent phylogenetic analysis showed that all UL128L genes of all clinical isolates from AIDS patients were highly conserved61. The conserved cysteine residues in exons 2 and 3 of UL128 were detected in all variants61. These conserved cysteines in CMV UL128 protein are especially important for CC chemokine motifs. Vogel et al. found no evidence for a connection between UL144/UL128/UL130/UL131A genotypes and the incidence of CMV retinitis in AIDS patients61. We have observed that none of the detected UL128 variants were significantly linked with symptomatic CMV infection. Our results revealed that the UL130 variant 6 and the UL131A variant 1 were associated with an increased risk of specific symptoms in children. No significant difference was detected between the CMV variants determined on the basis of the UL128L and the signs of infection in Chinese infants63. The UL130 variant 6 showed a few amino acid substitutions such as P40L, L60I, and S78L that were undetected in common CMV variants. It was found that a 2-bp insertion (TT) led to a frameshift mutation in the UL130 (Towne strain) and a 1-bp insertion (A) in UL131A (AD-169 strain) genes leading to a frameshift mutation have impaired the immunogenicity of viral strains58,59. The mutations include nucleotide substitutions that introduce in-frame translational termination codons or splicing are predicted to ablate gene functions, insertions, and deletions that affect one or more genes. These mutations impact the growth properties and tropism of the virus. Moreover, these changes have likely impaired the immunogenicity, particularly with respect to induction of epithelial cell and endothelial cell neutralizing antibodies57,60.
The findings of Maidji et al. support a potential role of endothelial cells in CMV transmission from the uterus to endovascular cytotrophoblasts64. The syncytiotrophoblast constitutes a barrier to vertical transmission, and first-trimester chorionic villi are largely resistant to CMV infection, whereas cytotrophoblasts and other villous cells are susceptible65. It is suggested that virions transmitted to cytotrophoblasts could then spread the infection to the placenta and to fetal blood vessels in the villus core. Considering the structure of the human placenta as an active barrier against infection, the pentamer gH/gL/pUL128-131A complex is of utmost importance in vertical CMV transmission53. The pUL128/pUL130/pUL131A, when assembled with the gH/gL heterodimer to form the pentameric complex, is necessary for entry into endothelial and epithelial cells, as well as for CMV transmission to leukocytes. The pentamer complex mediates virus entry into epithelial and endothelial cells by endocytosis and fusion. The entry process into fibroblasts is independent of the proteins of the pentamer complex, and the UL128L genes may be lost during long-term cultivation in human fibroblasts. Thus, this genetic locus is supposed to play an important role in cell tropism. Recent studies of the virus tropism concentrated on the UL128L of wild-type CMV strains. The pUL128/pUL130/pUL131A complex is also the target of the most potent neutralizing antibodies following natural infection31,41,52. The neutralization potency of CMV infection in epithelial cells by monoclonal antibodies against the UL128L complex and the gH/gL complex was 1000-fold and 10-fold higher, respectively, than that by CMV-specific hyperimmune globulin66. The pentamer gH/gL/pUL128-131A complex is of interest in the CMV vaccine field.
gH/gL dimer may be complexed either with pUL128L forming the pentamer complex, or gO, forming the gH/gL/gO complex. Greater amounts of pentamer gH/gL/pUL128/pUL130/pUL131A complex may result in there being less gH/gL available for the formation of trimer gH/gL/gO complex. Two viral factors, UL148 and US16, were identified to impact the composition of gH/gL complexes in CMV strains67,68. Moreover, the expression levels of UL128-131A and gO seem to influence the abundance of gH/gL/pUL128-131A and gH/gL/gO complexes and virion infectivity69. There are at least eight genotypes of gO that differ by 10 to 30% of amino acids. Used set of primers was adequate for amplifying a region of the UL74 gene that was digested for identification of the five major gO genotypes, with two of them divided in five sub-genotypes. However, no relationship was observed between gO genotype and the outcome of CMV infection in infants. The distribution of the gO genotypes in clinical isolates from different disease settings confirmed that no correlation exists between gO type and CMV disease23. The present results performed on larger groups of pediatric patients confirmed our earlier findings that infection with the gH2 genotype diminishes the risk of hearing loss, whereas detection of the gH1 genotype is associated with hearing loss17. In addition, an association between the gH1 genotype and the incidence of neurological dysfunction was found. The heterogeneity between gH1 and gH2 genotypes is characterized by the deletion of a proline at position 36 and the substitution of lysine for histidine at position 37 in the AD-169 strain.
Most of congenital infections occur during nonprimary maternal infection, and it has been estimated that approximately three-quarters of congenital CMV infections occur in the setting of recurrent maternal infection during pregnancy5. A previously acquired maternal CMV infection does not provide complete protection against infection of the fetus, but it reduces this risk. It was found that antibodies targeting the CMV pentameric complex are able to neutralize genetically diverse clinical isolates of CMV66. Moreover, a pentameric complex of proteins elicited far more neutralizing antibodies than gB41. Antibodies to anti-UL128L complex are increased in the groups of non-transmitting mothers54. The increase in antibodies after CMV infection in pregnant women is associated with a decreased risk of congenital infection, suggesting that anti-UL128L complex antibodies play a critical role in protection against transmission to the fetus54,70. However, it is unknown if antibodies specific for pentamer complex can block infection of trophoblasts and restrict cCMV transmission. It was observed that these antibodies inhibit infection of term cytotrophoblasts, while no inhibition in the first-trimester trophoblast progenitor cells was found53,71.
This study has a number of strengths, but also some limitations. This is the first study to focus on the genes encoding the gH/gL/pUL128-131A complex of CMV in infants infected congenitally or postnatally. The main strength of the present study is the clinical evaluation of children with a CMV infection and important clinical implications. We have found significant associations of CMV variants with specific clinical symptoms. It should be noted that our results are not representative of the entire infant population and that the detected CMV variant distribution is only valid for symptomatic patients. Because the sample population of patients with specific symptoms was small, the significance of these results must be handled with caution. Further studies using larger patient groups from different geographical regions are needed to confirm our findings. Moreover, investigations of the role of intra-host recombination and functions of variants of key viral genes are necessary to elucidate the role of CMV diversity in pathogenesis. It should be underlined that we have no universal CMV screening for newborns in our country. As pCMV infection was diagnosed in children above three weeks of age, it is possible that, apart from the postnatally infected majority, there are also congenitally infected infants (with no clinical findings at birth) in this group.
In summary, this report demonstrates that a relationship between the CMV variant in trimeric and pentameric complexes, including gH, pUL130 and pUL131A, and clinical outcome exists. This study suggests that the CMV variant may be one of the virological markers of congenital CMV disease.
Methods
Patients
A total of 195 children with CMV infection were enrolled from February 2008 to April 2011 and again from January 2015 to August 2018 at the Children’s Memorial Health Institute, Warsaw, and the Polish Mother’s Memorial Hospital Research Institute in Lodz, Poland. The geographical origins of the patients were in half the Central Poland (Warsaw, Lodz and the surrounding areas) and all Poland. Patients were divided into two groups: cCMV group, which represent infants who were CMV-positive in urine samples collected ≤ 21st day of life, and pCMV group, which represented infants with the postnatal acquisition of CMV (CMV-negative in urine samples ≤ 21st day of life, seroconversion > 21 day of life) or probable postnatal CMV infection/unproven cCMV infection (unavailable result of CMV DNA in the urine ≤ 21st day of life). Blood and/or urine samples were obtained from 82 newborns with cCMV infection and from 113 infants in pCMV group. One hundred and five children with CMV infection, including 28 cases of cCMV and 77 cases of pCMV, were earlier examined for the presence of UL75, as described in our previous study17. CMV infection was confirmed by the presence of CMV-specific antibodies and/or CMV DNA detection in whole blood and/or urine samples. Congenital infection was confirmed within the first 2–3 weeks of life by CMV DNA detection in the urine samples. When the samples were collected after 3 weeks of life, infants were classified as having postnatal or unproven congenital CMV infection. The children were classified as having symptomatic infection with any clinical manifestations (jaundice, petechiae, IUGR, hepatosplenomegaly, hepatitis, cholestasis, hearing loss, microcephaly, neurological dysfunction, CNS damage in neuroimaging [ultrasound and/or magnetic resonance imaging], chorioretinitis, pneumonia) or laboratory findings, including thrombocytopenia, granulocytopenia, and anemia. Due to selection bias, 96.3% of newborns and 94.7% of infants with CMV infection in this study were symptomatic. Serum samples were assessed for anti-CMV IgG and IgM antibodies with the use of ELISA AxSYM CMV IgM and IgG (Abbott Laboratories, Abbott Park, IL, USA) or CLIA LIASON CMV IgM and IgG assays (DiaSorin, Saluggia, Italy). The demographic and clinical characteristics of pediatric patients with CMV infection were summarized in Table 1. The study protocols were approved by the Bioethics Committee of the Medical University of Lodz (RNN/278-279/16/KE, RNN/120/09/KE) and the Ethics Committee of the Polish Mother’s Memorial Hospital Research Institute (90/KBE/207). Parents or guardians provided written informed consent to participate in this study on behalf of the children. All experiments were performed in accordance with relevant guidelines and regulations.
DNA extraction
Total genomic DNA was extracted from peripheral blood and urine samples using a QIAamp DNA Blood Mini Kit or QIAamp DNA Mini Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s instructions. The concentration and purity of DNA were assessed using a 2000c UV-VIS Spectrophotometer (Thermo Scientific, Wilmington, DE, USA).
Quantification of CMV DNA
The CMV DNA copy numbers in DNA isolates were determined using a 7900HT Fast Real-Time PCR system (Applied Biosystems, Foster City, CA, USA) as previously described72,73. DNA was amplified using primers specific for the CMV gB (UL55) gene. The sensitivity of the assay was determined to be approximately 2 × 102 copies/mL. A negative control without template DNA was included in every amplification run.
Genotyping of three genes encoding the gH/gL/gO complex of CMV
The UL75 gene (gH) was amplified by nPCR by using two sets of primers, as described elsewhere17. The genomic variants gH1 and gH2 were identified by different amplicon size lengths (240 bp and 237 bp, respectively). The identification of UL74 (gL) and UL115 (gO) was performed by nPCR-RFLP with the primers 5′-TAACGGGCGCTTGTTTACGT-3′ and 5′-CAGCAAAACGACCAGAATCAG-3′ (UL74) and 5′-GACGCACGGCGCGGTTGGTACG-3′ and 5′-CGTGCCGCAGACTTGATGTGCCG-3′ (UL115) in the first round and primer sets designed in our Laboratory or described previously by Stanton et al.74. The sequences of all primers used in PCR assays are listed in Table 6. Each run consisted of an initial denaturation at 95 °C for 1 min following by 45 consecutive cycles of denaturation at 95 °C for 3 min and different annealing temperatures (68 °C for UL74 and 55 °C for UL115) for 30 s, extension at 72 °C for 1 min, and the final extension at 72 °C for 10 min. The PCR was performed in a Veriti® 96 Well Thermal Cycler (Applied Biosystems). Each PCR was performed in a volume of 50 μl as follows: 0.5 μg (blood) or 0.2 μg (urine) template DNA (5 μl), 5 μl 10 × DreamTaq™ Buffer (20 mM Tris-HCl, pH 8.0; 1 mM DTT, 0.1 mM EDTA, 100 mM KCl, 0.5% Nonidet P40, 0.5% Tween 20, 50% glycerol), 4 μl 2.5 mM dNTP, 0.5 μl gene-specific primers (100 pmol/μl of each), 0.25 μl DreamTaq™ polymerase (5 U/μl, Fermentas, Glen Burnie, MD, USA), and 34.75 μl nuclease-free water. DNA isolates from MRC-5 cells (ATCC CCL-171, American Type Culture Collection, Rockville, MD, USA) infected with the CMV strains AD-169 (ATCC VR-538), Towne (ATCC VR-977) or Davis (ATCC VR-807) was used as positive controls in each of PCR runs, and nuclease-free water was used as a negative control. The nPCR products were digested with the restriction enzymes HpaII (UL74), Eco24I [BanII], and CseI [HgaI] (UL115) (Fermentas, Hanover, MD, USA) according to the manufacturer’s recommendations. The restriction enzymes and the CMV genotypes identified by different fragment lengths are described in Table 7. Briefly, a digestion with HpaII was used to characterize the five major gO genotypes, while double digestion with Eco24I and CseI was used to identify the four gL genotypes. The nPCR products and the digested DNA were analyzed using a QIAxcel DNA Screening Kit and a QIAxcel system (Qiagen) and the AL420 method. The QX Alignment Marker 15 bp/1 kbp and QX DNA Size Marker 50–800 bp were included in the analysis. Randomly selected samples (50–60 samples per each gene) were sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit and the 96-capillary 3730xl DNA Analyzer (Applied Biosystems).
CMV UL128 locus genotyping
To successfully amplify the genes encoding the CMV UL128L, nPCR (UL130 and UL131A) and hemi-nested PCR (UL128) assays were used. The sequences of all primers used in UL128L genotyping are listed in Table 6. The reaction mixture for UL128 and UL131A gene amplifications consisted of 0.5 μg (blood) or 0.2 μg (urine) template DNA (5 μl) and 45 μl reaction solution, as described above, while for the UL130 gene, it was as follows: 5 μl template DNA, 10 μl 5 × KAPA HiFi Fidelity Buffer, 1.5 μl 10 mM KAPA dNTP, 0.15 μl gene-specific primers, 1 μl KAPA HiFi polymerase (1 U/μl, KAPA Biosystems, Boston, MA, USA), and 32.2 μl nuclease-free water. The UL128 and UL131A reactions were performed at 95 °C for 5 min, followed by 30 cycles of 95 °C for 1 min, annealing at 55 °C for UL128 and 57 °C for UL131A for 1 min, and 72 °C for 1 min, followed by a final extension at 72 °C for 10 min. The PCR was performed in a Veriti® 96 Well Thermal Cycler (Applied Biosystems). The PCR parameters for UL130 were as follows: 95 °C for 3 min and 35 cycles each at 98 °C for 20 s, at 59 °C for 15 s, and at 72 °C for 15 s with a final extension at 72 °C for 5 min. Positive and negative controls were included in the PCR assays, as described above. The amplicons were verified by the QIAxcel capillary electrophoresis system (Qiagen) and were sequenced using an Applied Biosystems 3730xl sequencer (Applied Biosystems).
CMV sequences were aligned to the reference sequences deposited in the NCBI using BLAST.
Statistical analysis
The gene variant distributions and the association between them and the outcome of CMV infection were analyzed using Fisher’s exact test or the chi-square test. A nonparametric Mann–Whitney U-test with correction for continuity and Wilcoxon rank-sum test were performed to calculate the P-values for the relationship between the viral load and infection outcome. Pairwise correlations between gene variants were evaluated with Spearman’s rank correlation coefficient (ρ). A logistic regression model was used to evaluate the association between the specific CMV variants and clinical characteristics of patients, adjusting for potential cofounders, e.g., DNAemia level. The data were analyzed using descriptive statistics, including the median, range, and 95% confidence intervals (CIs). Analyses were performed using the SPSS statistical software package for Windows 24.0 (SPSS, Chicago, IL). A P-value ≤0.05 was considered as statistically significant.
References
Griffiths, P., Baraniak, I. & Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 235, 288–297 (2015).
Cannon, M. J., Schmid, D. S. & Hyde, T. B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 20, 202–213 (2010).
Boppana, S. B., Ross, S. A. & Fowler, K. B. Congenital cytomegalovirus infection: clinical outcome. Clin. Infect. Dis. 57, S178–181 (2013).
Kenneson, A. & Cannon, M. J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 179, 253–276 (2007).
Wang, C., Zhang, X., Bialek, S. & Cannon, M. J. Attribution of congenital cytomegalovirus infection to primary versus non-primary maternal infection. Clin. Infect. Dis. 52, e11–13 (2011).
Boppana, S. B., Pass, R. F., Britt, W. J., Stagno, S. & Alford, C. A. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr. Infect. Dis. J. 11, 93–99 (1992).
Britt, W. J. Maternal immunity and the natural history of congenital human cytomegalovirus infection. Viruses. 10, E405 (2018).
Dollard, S. C., Grosse, S. D. & Ross, D. S. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev. Med. Virol. 17, 355–363 (2007).
Dahl, H. H. et al. Etiology and audiological outcomes at 3 years for 364 children in Australia. PLoS One. 8, e59624 (2013).
Enders, G., Daiminger, A., Bäder, U., Exler, S. & Enders, M. Intrauterine transmission and clinical outcome of 248 pregnancies with primary cytomegalovirus infection in relation to gestational age. J. Clin. Virol. 52, 244–246 (2011).
de Vries, J. J. et al. The apparent paradox of maternal seropositivity as a risk factor for congenital cytomegalovirus infection: a population-based prediction model. Rev. Med. Virol. 23, 241–249 (2013).
Sinzger, C., Digel, M. & Jahn, G. Cytomegalovirus Cell Tropism in Human Cytomegalovirus. Current Topics in Microbiology and Immunology (eds Shenk, T. E. & Stinski, M. F.) 63–83 (Springer, 2008).
Chou, S. Molecular epidemiology of envelope glycoprotein H of human cytomegalovirus. J. Infect. Dis. 166, 604–607 (1992).
Pignatelli, S. et al. Intrauterine cytomegalovirus infection and glycoprotein N (gN) genotypes. J. Clin. Virol. 28, 38–43 (2003).
Pignatelli, S. et al. Cytomegalovirus gN genotypes distribution among congenitally infected newborns and their relationship with symptoms at birth and sequelae. Clin. Infect. Dis. 51, 33–41 (2010).
Paradowska, E. et al. Distribution of cytomegalovirus gN variants and associated clinical sequelae in infants. J. Clin. Virol. 58, 271–275 (2013).
Paradowska, E. et al. Cytomegalovirus glycoprotein H genotype distribution and the relationship with hearing loss in children. J. Med. Virol. 86, 1421–1427 (2014).
Arav-Boger, R. Strain variation and disease severity in congenital CMV infection – in search of a viral marker. Infect. Dis. Clin. North. Am. 29, 401–414 (2015).
Paradowska, E. et al. Human cytomegalovirus UL55, UL144, and US28 genotype distribution in infants infected congenitally or postnatally. J. Med. Virol. 87, 1737–1748 (2015).
Pati, S. K. et al. Genotypic diversity and mixed infection in newborn disease and hearing loss in congenital cytomegalovirus infection. Pediatr. Infect. Dis. J. 32, 1050–1054 (2013).
Zhou, M., Yu, Q., Wechsler, A. & Ryckman, B. J. Comparative analysis of gO isoforms reveals that strains of human cytomegalovirus differ in the ratio of gH/gL/gO and gH/gL/UL128-131 in the virion envelope. J. Virol. 87, 9680–9690 (2013).
Yan, H. et al. Genetic linkage among human cytomegalovirus glycoprotein N (gN) and gO genes, with evidence for recombination from congenitally and post-natally infected Japanese infants. J. Gen. Virol. 89, 2275–2279 (2008).
Rasmussen, L., Geissler, A., Cowan, C., Chase, A. & Winters, M. The genes encoding the gCIII complex of human cytomegalovirus exist in highly diverse combinations in clinical isolates. J. Virol. 76, 10841–10848 (2002).
Isaacson, M. K. & Compton, T. Human cytomegalovirus glycoprotein B is required for virus entry and cell-to-cell spread but not for virion attachment, assembly, or egress. J. Virol. 83, 3891–3903 (2009).
Vanarsdall, A. L., Ryckman, B. J., Chase, M. C. & Johnson, D. C. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J. Virol. 82, 11837–11850 (2008).
Boppana, S. B., Rivera, L. B., Fowler, K. B., Mach, M. & Britt, W. J. Intrauterine transmission of cytomegalovirus to infants of women with preconceptional immunity. N. Engl. J. Med. 344, 1366–1371 (2001).
Wille, P. T., Wisner, T. W., Ryckman, B. & Johnson, D. C. Human cytomegalovirus (HCMV) glycoprotein gB promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. MBio. 4, e00332–13 (2013).
Vanarsdall, A. L., Howard, P. W., Wisner, T. W. & Johnson, D. C. Human cytomegalovirus gH/gL forms a stable complex with the fusion protein gB in virions. PLoS Pathog. 12, e1005564 (2016).
Ryckman, B. J. et al. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J. Virol. 82, 60–70 (2008).
Ciferri, C. et al. Structural and biochemical studies of HCMV gH/gL/gO and Pentamer reveal mutually exclusive cell entry complexes. Proc. Natl. Acad. Sci. USA 112, 1767–1772 (2015).
Hahn, G. et al. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 78, 10023–10033 (2004).
Kabanova, A. et al. Platelet-derived growth factor-alpha receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat. Microbiol. 1, 16082 (2016).
Stegmann, B. et al. A derivative of platelet-derived growth factor receptor alpha binds to the trimer of human cytomegalovirus and inhibits entry into fibroblasts and endothelial cells. PLoS Pathog. 13, e1006273 (2017).
Wu, Y. et al. Human cytomegalovirus glycoprotein complex gH/gL/gO uses PDGFR-alpha as a key for entry. PLoS Pathog. 13, e1006281 (2017).
Gerna, G., Percivalle, E., Perez, L., Lanzavecchia, A. & Lilleri, D. Monoclonal Antibodies to Different Components of the Human Cytomegalovirus (HCMV) Pentamer gH/gL/pUL128L and Trimer gH/gL/gO as well as Antibodies Elicited during Primary HCMV Infection Prevent Epithelial Cell Syncytium Formation. J. Virol. 90, 6216–6223 (2016).
Zhou, M., Lanchy, J. M. & Ryckman, B. J. Human cytomegalovirus gH/gL/gO promotes the fusion step of entry into all cell types, whereas gH/gL/UL128-131 broadens virus tropism through a distinct mechanism. J. Virol. 89, 8999–9009 (2015).
Malito, E., Chandramouli, S. & Carfi, A. From recognition to execution-the HCMV pentamer from receptor binding to fusion triggering. Curr. Opin. Virol. 31, 43–51 (2018).
Vanarsdall, A. L., Wisner, T. W., Lei, H., Kazlauskas, A. & Johnson, D. C. PDGF receptor-alpha does not promote HCMV entry into epithelial and endothelial cells but increased quantities stimulate entry by an abnormal pathway. PLoS Pathog. 8, e1002905 (2012).
Stegmann, C., Rothemund, F., Laib Sampaio, K., Adler, B. & Sinzger, C. The N terminus of human cytomegalovirus glycoprotein O is important for binding to the cellular receptor PDGFRα. J. Virol. 93, e00138–19 (2019).
Wang, D. & Shenk, T. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J. Virol. 79, 10330–10338 (2005).
Wang, D. & Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 102, 18153–18158 (2005).
Adler, B. et al. Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J. Gen. Virol. 87, 2451–2460 (2006).
Gerna, G. et al. Dendritic-cell infection by human cytomegalovirus is restricted to strains carrying functional UL131-128 genes and mediates efficient viral antigen presentation to CD8+ T cells. J. Gen. Virol. 86, 275–284 (2005).
Nogalski, M. T., Chan, G. C., Stevenson, E. V., Collins-McMillen, D. K. & Yurochko, A. D. The HCMV gH/gL/UL128-131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS Pathog. 9, e1003463 (2013).
Ryckman, B. J., Jarvis, M. A., Drummond, D. D., Nelson, J. A. & Johnson, D. C. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 80, 710–722 (2006).
Sathiyamoorthy, K., Chen, J., Longnecker, R. & Jardetzky, T. S. The complexity in herpesvirus entry. Curr. Opin. Virol. 24, 97–104 (2017).
Stampfer, S. D. & Heldwein, E. E. Stuck in the middle: structural insights into the role of the gH/gL heterodimer in herpesvirus entry. Curr. Opin. Virol. 3, 13–19 (2013).
Chandramouli, S. et al. Structural basis for potent antibody-mediated neutralization of human cytomegalovirus. Sci. Immunol. 2, 12 (2017).
Ryckman, B. J., Chase, M. C. & Johnson, D. C. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc. Natl. Acad. Sci. USA 105, 14118–14123 (2008).
Murrell, I. et al. The pentameric complex drives immunologically covert cell–cell transmission of wild-type human cytomegalovirus. Proc. Natl. Acad. Sci. USA 114, 6104–6109 (2017).
Fouts, A. E., Chan, P., Stephan, J. P., Vandlen, R. & Feierbach, B. Antibodies against the gH/gL/UL128/UL130/UL131 complex comprise the majority of the anti-cytomegalovirus (anti-CMV) neutralizing antibody response in CMV hyperimmune globulin. J. Virol. 86, 7444–7447 (2012).
Macagno, A. et al. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J. Virol. 84, 1005–1013 (2010).
Chiuppesi, F. et al. Vaccine-derived neutralizing antibodies to the human cytomegalovirus gH/gL pentamer potently block primary cytotrophoblast infection. J. Virol. 89, 11884–11898 (2015).
Lilleri, D. et al. Fetal human cytomegalovirus transmission correlates with delayed maternal antibodies to gH/gL/pUL128-130-131 complex during primary infection. PLoS One. 8, e59863 (2013).
Baldanti, F. et al. Human cytomegalovirus UL131A, UL130 and UL128 genes are highly conserved among field isolates. Arch. Virol. 151, 1225–1233 (2006).
Dargan, D. J. et al. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J. Gen. Virol. 91, 1535–1546 (2010).
Wilkinson, G. W. et al. Human cytomegalovirus: taking the strain. Med. Microbiol. Immunol. 204, 273–284 (2015).
Murphy, E. et al. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. USA 100, 14976–14981 (2003).
Murphy, E. & Shenk, T. Human cytomegalovirus genome. Curr. Top. Microbiol. Immunol. 325, 1–19 (2008).
Lilleri, D., Kabanova, A., Lanzavecchia, A. & Gerna, G. Antibodies against neutralization epitopes of human cytomegalovirus gH/gL/pUL128-130-131 complex and virus spreading may correlate with virus control in vivo. J. Clin. Immunol. 32, 1324–1331 (2012).
Vogel, J. U. et al. Role of human cytomegalovirus genotype polymorphisms in AIDS patients with cytomegalovirus retinitis. Med. Microbiol. Immunol. 202, 37–47 (2013).
Sijmons, S. et al. High-throughput analysis of human cytomegalovirus genome diversity highlights the widespread occurrence of gene-disrupting mutations and pervasive recombination. J. Virol. 89, 7673–7695 (2015).
Sun, Z. R. et al. Structure characterization of human cytomegalovirus UL131A, UL130 and UL128 genes in clinical strains in China. Genet. Mol. Res. 8, 1191–1201 (2009).
Maidji, E., Percivalle, E., Gerna, G., Fisher, S. & Pereira, L. Transmission of human cytomegalovirus from infected uterine microvascular endothelial cells to differentiating/invasive placental cytotrophoblasts. Virol. 304, 53–69 (2002).
Fisher, S., Genbacev, O., Maidji, E. & Pereira, L. Human cytomegalovirus infection of placental cytotrophoblasts in vitro and in utero: implications for transmission and pathogenesis. J. Virol. 74, 6808–6820 (2000).
Ha, S. et al. Neutralization of diverse human cytomegalovirus strains conferred by antibodies targeting viral gH/gL/pUL128-131 pentameric complex. J. Virol. 91, e02033–16 (2017).
Li, G., Nguyen, C. C., Ryckman, B. J., Britt, W. J. & Kamil, J. P. A viral regulator of glycoprotein complexes contributes to human cytomegalovirus cell tropism. Proc. Natl. Acad. Sci. USA 112, 4471–4476 (2015).
Luganini, A., Cavaletto, N., Raimondo, S., Geuna, S. & Gribaudo, G. Loss of the human cytomegalovirus US16 protein abrogates virus entry into endothelial and epithelial cells by reducing the virion content of the pentamer. J. Virol. 91, e00205–17 (2017).
Zhang, L., Zhou, M., Stanton, R., Kamil, J. & Ryckman, B. J. Expression levels of glycoprotein O (gO) vary between strains of human cytomegalovirus, influencing the assembly of gH/gL complexes and virion infectivity. J. Virol. 92, e00606–18 (2018).
Lilleri, D. & Gerna, G. Maternal immune correlates of protection from human cytomegalovirus transmission to the fetus after primary infection in pregnancy. Rev. Med. Virol. 27, 2 (2017).
Zydek, M. et al. HCMV infection of human trophoblast progenitor cells of the placenta is neutralized by a human monoclonal antibody to glycoprotein B and not by antibodies to the pentamer complex. Viruses. 6, 1346–1364 (2014).
Paradowska, E. et al. Detection of cytomegalovirus in human placental cells by polymerase chain reaction. APMIS. 114, 764–771 (2006).
Paradowska, E. et al. TLR9 -1486T/C and 2848C/T SNPs Are Associated with Human Cytomegalovirus Infection in Infants. PLoS One. 11, e0154100 (2016).
Stanton, R., Westmoreland, D., Fox, J. D., Davison, A. J. & Wilkinson, G. W. Stability of human cytomegalovirus genotypes in persistently infected renal transplant recipients. J. Med. Virol. 75, 42–46 (2005).
Grosjean, J. et al. Direct genotyping of cytomegalovirus envelope glycoproteins from toddler’s saliva samples. J. Clin. Virol. 46, S43–48 (2009).
Murrell, I. et al. Impact of sequence variation in the UL128 locus on production of human cytomegalovirus in fibroblast and epithelial cells. J. Virol. 87, 10489–10500 (2013).
Acknowledgements
This work was supported in part by the National Science Centre of Poland (No. 2014/13/B/NZ7/02317) (http://www.ncn.gov.pl/); contributions from the Statutory Fund of IMB PAS are also gratefully acknowledged. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. There was no additional external funding received for this study.
Author information
Authors and Affiliations
Contributions
E.P. conceptualized the study and supervised the study, analyzed the data, and drafted the manuscript. A.J. and M.S. performed experiments and analyzed the data. B.K. performed the ELISA experiments. Material from patients was organized by J.C.K., B.K., K.D.F., M.W.L. and T.W.G. Clinical information were prepared by J.C.K. and M.W.L. Critical manuscript revisions were done by A.J., J.C.K., K.D.F., B.K., M.S., M.W.L. and T.W.G.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Paradowska, E., Jabłońska, A., Studzińska, M. et al. Distribution of the CMV glycoprotein gH/gL/gO and gH/gL/pUL128/pUL130/pUL131A complex variants and associated clinical manifestations in infants infected congenitally or postnatally. Sci Rep 9, 16352 (2019). https://doi.org/10.1038/s41598-019-52906-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52906-y
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
