
Abstract
Hyperglycemia during myocardial infarction (MI) has a strong and direct association with mortality. In stable patients and experimental models, statins favor the elevation of glycaemia. The present study investigated whether short-course treatment with statins during MI can influence glucose homeostasis and thus the clinical outcome. In this prospective study, euglycemic hyperinsulinemic clamp (EHC) was performed at second (D2) and sixth (D6) day after MI in patients randomized to simvastatin (S)10 or 80 mg/day during hospitalization (n = 27). In addition, patients (n = 550) were treated without (WS) or with simvastatin (S) at 20, 40 or 80 mg/day had HOMA2S on admission (D1) and fifth (D5) day after MI. According to EHC, insulin sensitivity increased by 20 ± 60% in S10 and decreased by −6 ± 28% in S80 (p = 0.025). Consistently, the changes in HOMA2S between D1 and D5 were 40 ± 145% (WS), 22 ± 117% (S20), 16 ± 61% (S40) and −2% ± 88% (S80) (p = 0.001). In conclusion, statin during the acute phase of MI reduces insulin sensitivity in a dose-dependent manner.
Similar content being viewed by others

Introduction
Both hyperglycemia and hypoglycemia during the acute phase of myocardial infarction (MI), particularly in those with ST-elevation MI (STEMI)1,2, has evoked clinical concern ever since it was established as a robust predictor of worse short- and long-term prognosis3,4. From a mechanistic standpoint, hyperglycaemia can reduce thrombolysis, increase platelet aggregation, induce coronary vasoconstriction, decrease oxygen transport, and prolong endothelial inflammation after MI4,5,6. Consistently, observational data indicate that glucose normalization is associated with improved survival in hyperglycaemic patients hospitalized with MI7. In fact, in the only randomized controlled trial with MI patients where blood glucose was successfully reduced, glucose-lowering therapy improved clinical outcome8.
Regardless of acute phase glycaemia, the residual risk of death in patients with MI remains high. Mitigation of this residual risk has been attempted through the inclusion of high dose of potent statins in the early treatment of acute coronary syndromes (ACS), a rationale that was largely based on a broad range of potentially beneficial mechanisms9, even though evidence from clinical trials does not fully support this practice10,11. Indeed, we recently reported the lack of benefit with high doses of statin in 4,191 patients in the first 30 days after ACS12.
A factor that may possibly contribute for the lack of benefit has been raised in studies demonstrating increased insulin resistance (IR) after chronic statin therapy11,13. In line with this, in cellular and animal models, a decrease in both insulin sensitivity and secretion can occur immediately after statin administration14,15. Consequently, it can be hypothesized that statin treatment initiated in the acute phase of MI may favor hyperglycaemia during the acute phase. If this is so, such negative effect may indeed restrict or even surpass a spectrum of beneficial mechanisms related to the use of these drugs during ACS9. To date, this hypothesis has not been investigated. Hence, this study aimed to identify the influence of statins on glucose homeostasis during MI.
In the present study, we selected non-diabetic patients to obtain a sample of individuals whose metabolic state reflects a direct and self-adjusted relationship between sensitivity and insulin secretion. We also chose individuals admitted with STEMI because they traditionally have large myocardial lesions and are therefore more susceptible to changes in glucose homeostasis due to the potential neurohumoral and inflammatory stimuli1. We used two methods for measuring insulin sensitivity whose consistency was validated by our group in STEMI patients16. Euglycemic hyperinsulinemic clamps (EHC) were used for being the gold standard method in a small group of patients and the Homeostasis Model Assessment (HOMA) was used for being a fast and accessible index feasible to be applied in a larger group of STEMI patients allowing us to confirm the findings and investigate interactions.
Materials and Methods
Study designs
We prospectively enrolled STEMI patients referred to Hospital de Base do Distrito Federal, Brasilia, Brazil, who survived hospital course and were able to undergo glucose homeostasis assessments as described below. Inclusion criteria for both substudies were as follows: (i) less than 24 hours after the onset of MI symptoms, (ii) ST-segment elevation of a least 1 mm (frontal plane) or 2 mm (horizontal plane) in two contiguous leads, and (iii) myocardial necrosis, as evidenced by increase to at least one value above the 99th percentile above the reference limit of CK-MB (25 U/L) and troponin I (0.04 ng/mL) followed by a decline of both. Exclusion criteria were glycosylated hemoglobin (HbA1c) ≥ 6.5%, prior diagnosis of diabetes or use of antidiabetic therapies. The Institutional Ethics Committee, Comitê de Ética em Pesquisa da Secretaria de Estado de Saúde do Distrito Federal (CEP/SES-DF), approved protocols of both substudies. All methods were performed in accordance with the relevant guidelines and regulations.
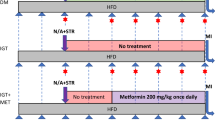
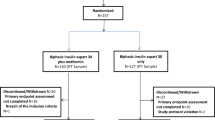
In the substudy 1 (interventional), consecutive STEMI patients (n = 37) aged between 20 and 60 years were prospectively enrolled into this substudy to investigate the effect of statin therapy on insulin sensitivity (ClinicalTrials.gov: NCT01205347). Exclusion criterium was the use of statins in the last 6 months. After enrollment, 10 patients were excluded for not completing the second assessment. The reasons for exclusion were withdrawal of consent (n = 4) and significant deterioration of clinical status (n = 6), i.e. death, cardiogenic shock, or recurrent coronary event. Patients were randomized at the second day of hospitalization (D2) for treatment with simvastatin (Zocor®, Merck, Sharp & Dohme, São Paulo, Brazil) 10 mg/day (n = 13) or 80 mg/day (n = 14). Although there is concern about the risk of myopathy with simvastatin 80 mg/day, the decision to use this dose was based on the wide availability of this statin in the public health system where enrolled patients were followed up over the period in which this substudy was carried out.
The EHC were performed after a 12-h fast at D2 and at the sixth day (D6) of STEMI onset. EHCs were not done at the first day of hospitalization (D1) in order to avoid delaying of medical assistance in the first 24 hours of STEMI and to ensure a 12-h overnight fasting period. As EHC is a laborious and time-consuming procedure, a limited number of patients was enrolled in substudy 1. Accuracy validation of the substitution indices for the insulin sensitivity (IS) obtained by the EHC in MI patients was performed by our group and published elsewhere16. MI mass was measured by cardiac magnetic resonance imaging (CMRi) 30 days after STEMI to verify whether there was imbalance of infarcted mass between groups. Blood samples were collected at admission (D1) and at the fifth day after STEMI onset (D5) for biochemical analyses.
Substudy 2 (observational) was designed to evaluate, in a multivariate approach, the interaction between statin use and IR induction. Hence, 375 consecutive participants of the prospective observational cohort Brasilia Heart Study (ClinicalTrials.gov: NCT02062554, retrospectively registered) were enrolled. A complete medical evaluation was performed upon hospital admission (D1) followed by an initial blood sample collection with a mean fasting time of 8 ± 3 hours. The second sample was collected after a 12-h overnight fast at fifth day of hospitalization (D5). Attending physicians exclusively decided on medical treatments without any participation of the investigators and were blinded to all results from the methods used in this study. At the end of data collection patients were grouped according to the use of statin during hospitalization: without statin (WS), simvastatin 20 mg/day (S20), simvastatin 40 mg/day (S40) and simvastatin 80 mg/day (S80).
Both substudies were developed in compliance with STROBE and CONSORT guidelines. Diagram Flow of substudies can be found in the Fig. 1.

Diagram flow of substudies 1 and 2.
Laboratory analyses
The following plasma measurements were performed: glucose (Glucose GOD-PAP, Roche Diagnostics, Mannheim, Germany, coefficient of variation 1.7%, range 241 mg/dL), total cholesterol (CHOD-PAP, Roche Diagnostics, Mannheim, Germany, coefficient of variation 4.0%, range 318 mg/dL), triglycerides (GPO-PAP, Roche Diagnostics, Mannheim, Germany, coefficient of variation 6.4%, range 117 mg/dL), high-density lipoprotein cholesterol (HDL-C) (HDL-C without sample pre-treatment, Roche Diagnostics, Mannheim, Germany, coefficient of variation 4.1%, range 74 mg/dL), HbA1c (Variant II, Bio-Rad Laboratories, Hercules, CA, USA), high-sensitivity C-reactive protein (CRP) (Cardiophase, Dade Behring, Marburg, Germany, coefficient of variation 6.7%, 13.8 g/L). Low-density lipoprotein cholesterol (LDL-C, range 280 mg/dL) was calculated by the Friedewald formula. Technicians, who were blinded to the statin treatment, performed all laboratorial analyses.
Glucose homeostasis model assessment
We employed the HOMA Calculator version 2.2.2 in order to assess insulin sensitivity (HOMA2S), based on plasma insulin, and β cell function (HOMA2B), based on plasma C-peptide, in all enrolled patientes17. Insulin (Roche Diagnostics, Mannheim, USA, coefficient of variation 4.2%, range 117 µI/mL) and C-peptide (Immulite 2000, Diagnostic Products Corporation, Los Angeles, CA, USA, coefficient of variation 7.1%, range 22.6 ng/dL) were measured on first day (D1) and fifth day of admission (D5). The Disposition Index was calculated as HOMA2B*HOMA2S/100.
Euglycemic hyperinsulinemic clamps
EHCs were performed at 07:00-h on second day (D2) and sixth day (D6). In order to arterialize and collect blood samples, we heated the arm to 50 °C, catheterized the antecubital vein and maintained its patency with slow saline drip. In the contralateral arm, we performed insulin and glucose infusion and 30 minutes after the beginning of the infusion, we started collecting glucose and insulin plasma concentrations. During 180 minutes, we infused insulin (Novolin R®; Novo-Nordisk, Bagsvaerd, Denmark) at a rate of 7 pmol.kg–1.min–1 and maintained euglycemia (~100 mg/dL) by simultaneous 50% glucose intravenous administration whose infusion rate was corrected every 10 minutes. The insulin sensitivity index (ISi) was calculated in two steps by using the equation ISi = M/(G × ΔI) corrected for body weight. M is the steady state glucose infusion rate (milligrams per min), G is the steady-state blood glucose concentration (mg/dL), and ΔI is the difference between basal and steady state plasma insulin concentrations (micro units/mL). ISi was estimated by increasing fractional glucose disappearance for each unit increase in plasma insulin18,19.
Cardiac magnetic resonance imaging (CMRI)
CMRI studies were carried out in the intervention group using a MRI scanner with a 1.5-T (Signa CV/i, GE Medical Systems, Waukesha, WI), equipped with a gradient of high performance (gradient strength 40mT/m; maximum slew rate 150 mT/m/s) and a four-elements phased array cardiac coil. MI mass was quantified using gadolinium-based delayed enhancement myocardial images thirty days after STEMI.
Statistical methods
Data were presented as mean ± SD for normally distributed data or median and interquartile range (IQR) for skewed data. Comparisons between S10 and S80 groups in the interventional substudy were made by Wilcoxon-Mann-Whitney test or a two-tailed t test. Baseline patient characteristics were stratified by the dose of simvastatin given using ANOVA 1-way test or Fisher exact test when appropriate. Categorical variables were compared by using the chi-square test. Analysis of covariance (ANCOVA) was used to assess the association between the treatment arm and the change in glycaemia, insulin, C-peptide, HOMA2S, and HOMA2B from D1 to D5, all of which were calculated as a delta, i.e. D5-D1. Adjustments for baseline levels, age, and gender were performed in all comparisons between simvastatin groups. Assumptions of the ANCOVA models (linearity, normality of distribution and equal variance) were checked using histograms, normal probability plots, and residual scatter plots.
Principal components factor analysis (PCA) was performed retaining factors with eigenvalues of at least 1.0. Missing variables were handled by listwise deletion, i.e. including only those sets of data where all the variables are present. PCA is a method of identifying patterns in data and only is appropriate for use with highly correlated variables. This statistical approach groups linear combinations into principal components of correlated variables, but the components themselves are derived so that they are uncorrelated with each other. Variables with an absolute value of the coefficient ≥0.5 were considered to be influential within each factor. Variables included in the analysis were Delta HOMA2S, Delta HOMA2B, and Delta Disposition Index. A two-sided p-value of 0.05 was considered statistically significant. All analyses were performed with SPSS 20 and Stata 13 for Mac. The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
Results
Interventional substudy
Baseline characteristics
There were no significant differences between simvastatin groups regarding sex, gender, clinical characteristics upon admission, or anthropometric characteristics. Infarction mass, reperfusion therapy, and the use of beta-blockers were also similar (Table 1). Admission levels of glycaemia, LDL-C, HDL-C, triglycerides, and CRP were not different.
Euglycemic hyperinsulinemic clamps
At D2, ISi was not different between patients treated with simvastatin 10 or 80 mg (Supplementary Table 1). Between D2 and D5, the group treated with 10 mg increased ISi from 2.8 ± 1.8 to 3.1 ± 1.9 10−4.kg−1.min−1/(μU/mL), and patients who took 80 mg decrease from 2.8 ± 1.3 to 2.5 ± 0.9 10−4.kg−1.min−1/(μU/mL) (p < 0.05 for both). The differences in Delta ISi between simvastatin groups were confirmed by both Student’s t-test (p = 0.04) and ANCOVA adjusted for sex, age, and ISi at D2 (p = 0.025).
Observational substudy
Baseline characteristics
As commented above, patients were grouped according to the dose of simvastatin during hospitalization. There was no significant difference across groups (both in the whole cohort and in non-diabetics) regarding clinical characteristics as well as infarction mass estimated by MRi, reperfusion therapy, and in-hospital use of beta-blockers, clopidogrel, and AAS or tirofiban (Table 2). Of note, time to reperfusion therapy after the onset of chest pain and medications after discharge were also similar across groups. No statin-associated muscle symptoms were observed in simvastatin users, even in those at 80 mg/day.
Glucose-insulin homeostasis
At D1, there were no differences between groups in terms of plasma glycaemia, insulin, or C-peptide levels of non-diabetic patients. HOMA2S and HOMA2B were also not different (Table 3). However, between D1 and D5, the controls and the S80 groups presented opposing behaviors on glucose metabolism, followed by an intermediate profile in the S20 and S40 groups. While in the control group glycaemia decreased by 15 ± 46 mg/dL, HOMA2S increased by 40 ± 145% and HOMA2B decreased by 38 ± 100%; in the S80 group glycaemia increased by 1 ± 33% (p = 0.01), HOMA2S decreased by 2 ± 89% (p = 0.001) and HOMA2B increased by 12 ± 100% (p = 0.001).
Subsequently, we evaluated whether simvastatin dose could independently predict a reduction in HOMA2S between D1 and D5 in non-diabetics (Supplementary Table 1). Indeed, HOMA2B at D1 (p = 0.0001), the dose of simvastatin (p = 0.009) and the number of components of metabolic syndrome (p = 0.009) were the only independent markers for a fall in HOMA2S between D1 and D5. Particularly, each 20 mg of simvastatin led to an 18% increase in chance of reducing insulin sensitivity from D1 to D5.
It is well known that insulin secretion and sensitivity are connected via a negative feedback loop, where pancreatic β-cells compensate for changes in whole body insulin sensitivity by a proportional and reciprocal change in insulin secretion in a rectangular hyperbolic function (y = constant/x)20. Based on this assumption, the product of HOMA2B and HOMA2S, i.e. the disposition index (DI), remains approximately constant if only one of these parameters is changed. Therefore, in Fig. 2 the expected slope should follow a −45° line. As shown in Fig. 1, we found the following linear regression equations for each group −0.574x + 0.11 (WS), −0.620x + 0.28 (S20), −0.534x + 0.26 (S40) and −0.436x + 0.30 (S80), indicating a dose-dependent change in the coupling between HOMA2S and HOMA2B (p < 0.05). Accordingly, as seen in Table 3, the S80 group showed a significantly impaired change in the DI between D1 and D5 (p = 0.007) when compared to other groups. In addition, by performing PCA, we found two components that summarize roughly 75% of variation of Delta HOMA2S, Delta HOMA2B, and Delta DI (Kaiser-Meyer-Olkin measure of sampling adequacy of 0.51) (Supplementary Fig. 1). While component 1 mostly represent positive values for Delta HOMA2S and Delta DI, component 2 mostly summarizes the positive variance of Delta HOMA2B and Delta DI (Supplementary Table 2). ANCOVA analysis showed that the S80 group presented a significantly lower mean ± SD on component 1 (−0.38 ± 0.2, p = 0.0229) compared to controls (+0.12 ± 0.3), S20 (+0.13 ± 0.3) and S40 (+0.01 ± 0.2). On component 2, however, the groups were equilibrated. Together, these findings suggest that, compared to the controls, S20 and S40 groups, the major source of variance of S80 group is related to HOMA2S impairment.

Compensatory changes between HOMA2S and HOMA2B during the first 5 days after MI. A significantly lower slope was found in patients taking simvastatin at 80 mg/day (S80) (β of −33.6 ± 6, p = 0.018) as compared with their counterparts (β of −23.3 ± 10, −18.6 ± 4, and −27.0 ± 6, respectively, in WS, S20 and S40 groups).
Discussion
Although it is increasingly more evident that statins may favor the incidence of diabetes after prolonged treatment11, it was still unclear whether statins could derange glucose homeostasis after short-term initiation of therapy. In a small trial with atorvastatin, after two months of treatment, there was reduced insulin sensitivity and C-peptide as well as increased blood glucose and HbA1c in a dose-dependent manner13. In another study, 156 patients with MI received atorvastatin 20 or 40 mg/day for one year. Patients with the higher statin dose had a greater increase in plasma glucose, insulin, C-peptide levels and progression of insulin resistance as estimated by the quantitative insulin sensitivity check index (QUICKI) score21.
As commented above, cellular model data indicate that statins can readily reduce the phosphorylation of insulin receptor tyrosine, the activation of MAP kinase in fibroblasts and inhibit glucose-induced calcium [Ca2+] signaling14,15. Through this set of mechanisms, statins would potentially prevent the post-translational prenylation of small G-proteins which is required for membrane targeting and activation of both insulin secretion in pancreatic β-cells and insulin signal transduction in adipocytes22,23. To the best of our knowledge, the present study is the first to demonstrate in STEMI patients that a rapid deterioration of insulin signaling and secretion can really be observed soon after statin treatment.
In the very early phase of MI, IS declines because of a spectrum of mechanisms that include the neurohumoral activation and acute phase of inflammatory response3,24,25. Although statins can quickly reduce inflammatory activity26 and sympathetic activation27, its above-commented ability to inhibit prenylation seems to predominate in the modulation of IS during acute stress according to our EHC data28. Indeed, in in vitro model with 3T3-L1 adipocytes, statins promote downregulation of insulin-sensitive GLUT-4 and up-regulation of GLUT-1; these effects are reversed by mevalonate, demonstrating that inhibition of isoprenoid biosynthesis causes insulin resistance in adipocytes29. In our observational substudy, IS estimated by HOMA2S decreased and insulin secretion estimated by HOMA2B increased in a directly proportional manner; this pairing resulted in an unchanging DI. At the higher simvastatin dose (80 mg/day), however, this balance was lost probably reflecting the statin-mediated inhibition of insulin secretion.
EHC remains as the gold standard for evaluating IS even in stress conditions due to the suppression of hepatic glucose secretion by constant infusion of insulin. Yet, EHC is a laborious and time-consuming procedure, unsuitable to be executed in large sample sizes, particularly in STEMI patients. With this in mind, we recently tested the accuracy of surrogate homeostasis models in STEMI patients and found that the HOMA2S can be used with accuracy, thus representing a viable tool for larger clinical studies16. Following this strategy, we were able to verify the dose-dependent effect on insulin sensitivity and secretion and to detect in multivariate models that besides statin dose, the presence of metabolic syndrome components and HOMA2B obtained at admission are independent predictors of IS decline after MI.
This study was designed to test the existence of an inverse and dose-dependent association between statin therapy initiated after STEMI and insulin sensitivity. For this, we used two distinct methods for assessing insulin sensitivity and we confirmed the hypothesis. Nevertheless, important limitations need to be recognized and considered when interpreting our findings. First, the study exclusively enrolled STEMI patients to homogenize the sample with individuals with a greater extent of myocardial injury. This strategy prevents our findings from being extrapolated to other acute coronary syndromes. Second, all patients used exclusively simvastatin and there may be heterogeneity between statins in the effect on glucose homeostasis. Third, because of the limited sample size, we cannot verify if a clinical impact exists after the acutely statin-induced IR. This hypothesis, needs to be properly tested and our study is not fit for it. Finally, although no difference was apparent between patients grouped according to statin therapy in the observational substudy, we cannot exclude selection bias. However, since the findings are equivalent in the randomized controlled substudy and in the observational substudy, altogether this set of data adds consistency for the statin and insulin sensitivity interaction during STEMI.
Conclusion
In conclusion, statin therapy initiated upon admission of STEMI quickly reduces insulin sensitivity in a dose-dependent manner as estimated by both EHC and HOMA2S% assessments.
Ethics approval and consent to participate
Institutional Ethics Committee approved protocols of both substudies and all patients signed an informed consent term. The study was registered at ClinicalTrials.gov by the numbers NCT02062554 and NCT01205347.
Consent for publication
All authors have read and approved the manuscript submission as it is.
Registration numbers
At ClinicalTrials.gov: NCT01205347, at September 20, 2010 and NCT02062554 retrospectively registered at February 13, 2014.
Data availability
The datasets generated and/or analyzed during the current study are not publicly available due to ongoing proprietary work but are available from the corresponding author on reasonable request.
References
Sinnaeve, P. R. et al. Association of elevated fasting glucose with increased short-term and 6-month mortality in ST-segment elevation and non-ST-segment elevation acute coronary syndromes: the Global Registry of Acute Coronary Events. Arch Intern Med 169, 402–409, https://doi.org/10.1001/archinternmed.2008.572 (2009).
Karetnikova, V., Gruzdeva, O., Uchasova, E., Osokina, A. & Barbarash, O. Glucose levels as a prognostic marker in patients with ST-segment elevation myocardial infarction: a case-control study. BMC Endocr Disord 16, 31, https://doi.org/10.1186/s12902-016-0108-8 (2016).
Ceriello, A. Acute hyperglycaemia: a ‘new’ risk factor during myocardial infarction. Eur Heart J 26, 328–331 (2005).
Worthley, M. I. et al. The deleterious effects of hyperglycemia on platelet function in diabetic patients with acute coronary syndromes mediation by superoxide production, resolution with intensive insulin administration. J Am Coll Cardiol 49, 304–310 (2007).
Timmer, J. R. et al. Hyperglycemia is an important predictor of impaired coronary flow before reperfusion therapy in ST-segment elevation myocardial infarction. J Am Coll Cardiol 45, 999–1002 (2005).
Kim, J. A., Montagnani, M., Koh, K. K. & Quon, M. J. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation 113, 1888–1904 (2006).
Kosiborod, M. et al. Glucose normalization and outcomes in patients with acute myocardial infarction. Arch Intern Med 169, 438–446 (2009).
Malmberg, K. Prospective randomised study of intensive insulin treatment on long term survival after acute myocardial infarction in patients with diabetes mellitus. DIGAMI (Diabetes Mellitus, Insulin Glucose Infusion in Acute Myocardial Infarction) Study Group. Bmj 314, 1512–1515 (1997).
Sposito, A. C. & Chapman, M. J. Statin therapy in acute coronary syndromes: mechanistic insight into clinical benefit. Arterioscler Thromb Vasc Biol 22, 1524–1534 (2002).
Morrissey, R. P., Diamond, G. A. & Kaul, S. Statins in acute coronary syndromes: do the guideline recommendations match the evidence? J Am Coll Cardiol 54, 1425–1433 (2009).
Sattar, N. et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 375, 735–742 (2010).
Berwanger, O. et al. Effect of Loading Dose of Atorvastatin Prior to Planned Percutaneous Coronary Intervention on Major Adverse Cardiovascular Events in Acute Coronary Syndrome: The SECURE-PCI Randomized Clinical Trial. Jama 319, 1331–1340, https://doi.org/10.1001/jama.2018.2444 (2018).
Koh, K. K. et al. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J Am Coll Cardiol 55, 1209–1216 (2010).
Xu, X. Q., McGuire, T. F., Blaskovich, M. A., Sebti, S. M. & Romero, G. Lovastatin inhibits the stimulation of mitogen-activated protein kinase by insulin in HIRcB fibroblasts. Arch Biochem Biophys 326, 233–237 (1996).
Yada, T., Nakata, M., Shiraishi, T. & Kakei, M. Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet beta-cells. Br J Pharmacol 126, 1205–1213 (1999).
Moura, F. A. et al. Validation of surrogate indexes of insulin sensitivity in acute phase of myocardial infarction based on euglycemic-hyperinsulinemic clamp. American journal of physiology. Endocrinology and metabolism 306, E399–403, https://doi.org/10.1152/ajpendo.00566.2013 (2014).
Caumo, A., Perseghin, G., Brunani, A. & Luzi, L. New insights on the simultaneous assessment of insulin sensitivity and beta-cell function with the HOMA2 method. Diabetes Care 29, 2733–2734 (2006).
DeFronzo, R. A., Tobin, J. D. & Andres, R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 237, E214–223 (1979).
Katz, A. et al. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab 85, 2402–2410 (2000).
Kahn, S. E. et al. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 42, 1663–1672 (1993).
Gruzdeva, O. et al. Effect of different doses of statins on the development of type 2 diabetes mellitus in patients with myocardial infarction. Diabetes Metab Syndr Obes 10, 481–489, https://doi.org/10.2147/DMSO.S149463 (2017).
Kowluru, A. Small G proteins in islet beta-cell function. Endocrine reviews 31, 52–78, https://doi.org/10.1210/er.2009-0022 (2010).
Takaguri, A., Satoh, K., Itagaki, M., Tokumitsu, Y. & Ichihara, K. Effects of atorvastatin and pravastatin on signal transduction related to glucose uptake in 3T3L1 adipocytes. Journal of pharmacological sciences 107, 80–89 (2008).
Falciglia, M. Causes and consequences of hyperglycemia in critical illness. Curr Opin Clin Nutr Metab Care 10, 498–503 (2007).
Goyal, A. et al. Glucose levels compared with diabetes history in the risk assessment of patients with acute myocardial infarction. Am Heart J 157, 763–770, https://doi.org/10.1016/j.ahj.2008.12.007.
Sposito, A. C. et al. Timing and dose of statin therapy define its impact on inflammatory and endothelial responses during myocardial infarction. Arterioscler Thromb Vasc Biol 31, 1240–1246, https://doi.org/10.1161/ATVBAHA.110.218685 (2011).
Lewandowski, J., Symonides, B., Gaciong, Z. & Sinski, M. The effect of statins on sympathetic activity: a meta-analysis. Clinical autonomic research: official journal of the Clinical Autonomic Research Society 25, 125–131, https://doi.org/10.1007/s10286-015-0274-1 (2015).
Sasaki, J., Iwashita, M. & Kono, S. Statins: beneficial or adverse for glucose metabolism. J Atheroscler Thromb 13, 123–129, JST.JSTAGE/jat/13.123 (2006).
Chamberlain, L. H. Inhibition of isoprenoid biosynthesis causes insulin resistance in 3T3-L1 adipocytes. FEBS Lett 507, 357–361 (2001).
Acknowledgements
The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written. This work was supported by a grant from the Brazilian National Research Council (CNPq) grant number 301465/2017-7. Prof. Sposito is recipient of a Research Career Awards from the CNPq.
Author information
Authors and Affiliations
Contributions
L.S.F.C. and A.C.S. collected outpatient data, conducted data analyses and prepared the manuscript. L.S.F.C., F.A.M., O.R.A. and R.M.R.C. performed the euglycemic clamps and systematized the mechanistic substudy (prepared patients, randomized treatment, etc). J.Q.S., A.M.C.S., W.N.J. and A.C.S. reviewed the manuscript. A.C.S. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sposito, A.C., Carvalho, L.S.F., Moura, F.A. et al. Statin Short-term Inhibition of Insulin Sensitivity and Secretion During Acute Phase of ST-Elevation Myocardial Infarction. Sci Rep 9, 16401 (2019). https://doi.org/10.1038/s41598-019-52111-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52111-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
