
Abstract
A number of bifidobacterial species are found at a particularly high prevalence and abundance in faecal samples of healthy breastfed infants, a phenomenon that is believed to be, at least partially, due to the ability of bifidobacteria to metabolize Human Milk Oligosaccharides (HMOs). In the current study, we isolated a novel strain of Bifidobacterium kashiwanohense, named APCKJ1, from the faeces of a four-week old breastfed infant, based on the ability of the strain to utilise the HMO component fucosyllactose. We then determined the full genome sequence of this strain, and employed the generated data to analyze fucosyllactose metabolism in B. kashiwanohense APCKJ1. Transcriptomic and growth analyses, combined with metabolite analysis, in vitro hydrolysis assays and heterologous expression, allowed us to elucidate the pathway for fucosyllactose metabolism in B. kashiwanohense APCKJ1. Homologs of the key genes for this metabolic pathway were identified in particular in infant-derived members of the Bifdobacterium genus, revealing the apparent niche-specific nature of this pathway, and allowing a broad perspective on bifidobacterial fucosyllactose and L-fucose metabolism.
Similar content being viewed by others

Introduction
Breast-feeding influences neonatal intestinal microbiota development and composition1,2,3, with significant differences reported between the gut microbiota of breast-fed infants and their formula-fed counterparts4. It currently is believed that Human Milk Oligosaccharides (HMOs) represent some of the most influential components of human breastmilk and colostrum in terms of their impact on microbiota composition by acting as a selective growth substrate2,3,5, with recent work correlating differences in the neonatal microbiota with variations in HMO composition of corresponding mothers milk6. After lactose, HMOs are the most abundant carbohydrate component of breastmilk2,7, and are composed of a heterogeneous mix of at least 200 distinct glycan structures8. HMOs consist of a lactose moiety linked, through a variety of different glycosidic connections, to L-fucose, sialic acid, lacto-N-biose (LNB) or N-acetyllactosamine (LacNAc); the latter two cases creating the tetrasaccharides lacto-N-tetraose (LNT) or lacto-N-neotetraose (LNnT), respectively. LN(n)T may be elongated by additional LNB or LacNAc residues at the reducing end, and/or may be fucosylated or sialylated at different positions2,8,9. HMOs can be viewed as a model prebiotic, since it complies with the definition of the latter as a ‘non-digestible food ingredient that beneficially affects the host by selectively stimulating growth and/or activity of one or a limited number of bacteria in the colon, thereby improving host health10. Recent studies have furthermore shown the beneficial properties of HMOs extending to protection against infant-associated gastrointestinal conditions, such as Campylobacter and Group B Streptococcal infections, and necrotising enterocolitis (NEC)11.
Bifidobacteria are Gram-positive, anaerobes belonging to the Actinobacteria phylum, and are commonly observed as commensal members of the mammalian, avian and insect intestinal tract12. Enrichment of bifidobacteria has been observed in the faecal microbiota of breastfed infants13, to whom they are believed to provide an array of health benefits5,14,15,16. The dominance of bifidobacteria in this niche is attributed to the presence of a small number of key species, which are capable of selectively utilising (particular) HMOs as their sole carbohydrate source5,8,17,18. These infant-associated species include Bifidobacterium bifidum, Bifidobacterium longum subsp. infantis and Bifidobacterium breve. B. bifidum carries out extracellular hydrolysis of complex HMOs, including LN(n)T, and fucosylated and sialylated structures, prior to the import and metabolism of (many of) the resultant mono- and di-saccharides19,20,21,22,23,24,25. B. breve and B. longum subsp. infantis internalise (certain) intact HMO moieties, such as LNT, LNnT and LNB, and sequentially degrade them for subsequent metabolism of the generated monosaccharides8,19,26,27,28,29,30. Furthermore, B. infantis internalizes and then metabolizes various fucosylated and sialylated HMOs31,32,33.
Bifidobacterium kashiwanohense has been previously isolated from the faeces of neonates34,35, though it is not considered a common component of the neonatal gut microbiota. There is little knowledge of this species and its metabolic capabilities. The ability of two B. kashiwanohense strains to grow on the HMO components 2′-fucosyllactose (2′-FL) and 3-fucosyllactose (3-FL) was recently shown, although these strains did not appear to metabolize the (fucosyllactose component) L-fucose33. While the presence of fucosylated HMOs in breastmilk has been shown to vary greatly, depending on genetic and geographical factors36, fucosylated HMOs are generally thought to represent ~50–80% of all HMOs found in human breastmilk37. Furthermore, free 2′-FL and 3-FL represent between 12–45% and 0.5–3% of the total HMO content, respectively36. 2′-FL (Fucα1-2[Galβ1-4]Glc) consists of L-fucose linked by an α1-2 bond to the galactose residue in lactose, while its isomer 3-FL (Fucα1-3Galβ1-4Glc) consists of L-fucose linked by an α1-3 bond to the glucose moiety in lactose37. The ability to metabolize 2′-FL and/or 3-FL is therefore considered to be important for infant gut colonisation38. Of note, FL-utilising properties of certain members of bifidobacterial species have a crucial impact on metabolite production and early life gut microbiota composition38. Metabolite profiles for B. kashiwanohense following growth on 2′-FL or 3-FL suggest that the inability of B. kashiwanohense to grow on L-fucose is due to the lack of two enzymes, L-fucose mutarotase and fucose permease33. The latter study, and another recent study39, however, demonstrated L-fucose metabolism resulting in the formation of 1,2-propanediol (1,2-PD), during growth on fucosyllactose by B. breve, B. longum subsp. infantis and B. longum subsp. suis, which suggests the presence of a common pathway for L-fucose metabolism. Notably, trophic interactions between the butyrate-producing gut commensal Eubacterium hallii and B. breve and/or B. longum subsp. infantis driven by FL and L-fucose utilisation have recently been shown to occur in the gut of infants39. Importantly, E. hallii represents one of the first producers of butyrate in the infant gut, while it is also able to convert 1,2-PD to propionate40.
In the current study, we sequenced the genome of B. kashiwanohense strain APCKJ1, isolated from faeces of a breastfed infant using 2′-FL as a selective carbohydrate. Employing various approaches we dissected fucosyllactose metabolism in APCKJ1, and also identified key genes involved in L-fucose metabolism in B. breve. Subsequent bioinformatic analysis revealed the distribution of fucose/fucosyllactose utilisation genes among members of the genus Bifidobacterium. This work not only elucidates details of fucosyllactose metabolism in B. kashiwanohense, but also allows general insights into bifidobacterial L-fucose metabolism.
Results
Isolation of a novel B. kashiwanohense isolate
A single stool sample from an exclusively breast-fed neonate, aged 4 weeks, was employed to isolate 2′-FL-utilising Bifidobacterium isolates. Several Bifidobacterium isolates were obtained from this faecal sample, based on their resistance to mupirocin (which selects for bifidobacteria41,42) and ability to grow on 2′-FL as the sole carbohydrate source. All assessed isolates were found to belong to the species B. kashiwanohense and appeared to be clonal. As this is an infrequently encountered species (from infant faecal samples), yet clearly exhibiting an ability to grow on 2′-FL, one of these isolates, designated APCKJ1, was selected for further characterisation.
Carbohydrate utilisation by B. kashiwanohense APCKJ1
In order to further investigate the carbohydrate utilisation capabilities of B. kashiwanohense APCKJ1, growth assays were carried out for this strain inoculated in modified de Man Rogosa and Sharpe (mMRS) medium supplemented with 1% (wt/vol) of one of 16 different sugars, including nine HMOs or HMO-components (Fig. 1). Growth was assessed by measuring the OD600nm following 24 hours of anaerobic growth at 37 °C. APCKJ1 was shown to reach its highest level of cell density following growth on lactose (OD600nm > 3.0), while also reaching high optical densities following growth on the monosaccharides glucose, ribose, sorbitol, as well as the disaccharide melibiose and the trisaccharide raffinose. Good growth (final OD600nm > 1.5) was observed for just two of the nine HMO components tested: the isomers 2′-FL and 3-FL. This confirmed the observation that APCKJ1 is capable of metabolising 2′-FL as its sole carbohydrate source. L-fucose, however, did not support growth of APCKJ1 to any substantial degree, suggesting the strain is incapable of metabolizing this sugar when present in the growth medium in its free form.

Carbohydrate utilization profiles of B. kashiwanohense APCKJ1. Final OD600nm values after 24 hours of growth of B. kashiwanohense APCKJ1 in modified MRS containing a range of carbohydrates at 1% (wt/vol) as the sole carbon source. The results are mean values obtained from two separate experiments. Error bars represent the standard deviation. Figure adapted from thesis Figure 5.1; James, 201897.
The cell-free supernatants of APCKJ1 following growth on 2′-FL or 3-FL (or the control carbohydrate lactose) were analysed by High Performance Liquid Chromatography (HPLC) for the presence of specific metabolic end-products (Fig. 2). As additional controls we also analysed uninoculated mMRS containing lactose, 2′-FL, 3-FL or no carbohydrate source. This analysis revealed that growth of B. kashiwanohense APCKJ1 on either 2′-FL or 3-FL causes the accumulation of lactate and acetate in the growth medium (Fig. 2), which are known metabolic end products of lactose and fucosyllactose metabolism in bifidobacteria33,39,43. Furthermore, a peak was observed corresponding to 1,2-PD, which has been put forward as an end product of L-fucose metabolism33,39. These findings indicate that B. kashiwanohense APCKJ1 is not only capable of degrading 2′-FL/3-FL and metabolizing its lactose component, but that it also utilises the L-fucose constituent of these HMOs.

Fermentation of lactose, 2′-FL and 3-FL by B. kashiwanohense APCKJ1. HPLC chromatogram profiles of fermentations of mMRS containing 1% lactose, 1% 2′-FL or 1% 3-FL inoculated with B. kashiwanohense APCKJ1, following 24 hours growth anaerobically at 37 °C, as well as a blank mMRS control. Figure adapted from thesis Figure 5.2; James, 201897.
General genome features of B. kashiwanohense APCKJ1, and transcriptome analysis during growth on 2′-FL or 3-FL
To identify the genes involved in 2′-FL/3-FL metabolism we first sequenced the genome of B. kashiwanohense isolate APCKJ1, revealing a 2,445,409-base pair (bp) circular molecule, containing 53 tRNA genes and 5 rRNA operons. The G + C content of the genome is 56.19%, with a total of 2,110 predicted protein encoding sequences (CDS). Scores for the sequencing reads and quality are given in Table S1. We then assessed global gene expression by microarray analysis of B. kashiwanohense APCKJ1 when grown in mMRS supplemented with 2′-FL, and compared with the transcriptome obtained during growth in mMRS supplemented with sorbitol or with lactose. Genes that were shown to be significantly up- or down-regulated in transcription above the designated cut-off (fold-change > 5.0, P < 0.001) are shown in Table 1. A fold-change of 5.0 was chosen as the threshold for differential expression, as this allowed a clear resolution between genes that exhibit a high level of increased transcription and (a large number of) genes eliciting a lower level of increased transcription, which was presumed to be due to background variation. When grown on 2′-FL (and compared to the transcriptome of the strain when grown on sorbitol or lactose), the transcriptionally upregulated genes of strain APCKJ1 included all genes (with the exception of a predicted regulator, fumR) of a gene cluster corresponding to locus tags BKKJ1_2069-2079 (and designated here as the fum locus [for fucose metabolism]) (Table 1 and Fig. S1). This gene cluster was found to contain two predicted and adjacent α-fucosidase-encoding genes, BKKJ1_2069 and BKKJ1_2070 (designated fumA1 and fumA2, respectively). Microarray analysis also revealed that transcription of lacA, encoding a predicted β-galactosidase, and the adjacent lacS, encoding a predicted lactose permease, was increased when cells were grown in a medium containing 2′-FL and compared to transcription patterns when grown on sorbitol or lactose, respectively. The lacA and lacS gene cluster correspond to locus tags BKKJ1_2066 and 2067, respectively, and are located adjacent to the fum cluster. Based on their annotations, the lac genes and the fum gene cluster are predicted to function in lactose and fucosyllactose/L-fucose metabolism, respectively.
Interestingly, when comparing the transcriptomes of a 2′-FL versus lactose-grown APCKJ1 culture, significant transcriptional upregulation was noted of gene BKKJ1_0429 (designated here as fumG), predicted to encode 1,2-PD oxidoreductase. 1,2-PD has previously been identified as a product of L-fucose metabolism in certain bifidobacteria33,39, and the presence of this gene is consistent with the results of the fermentation product analysis which suggests the utilisation of L-fucose derived from 2′-FL/3-FL by APCKJ1. Between the lacAS genes and the fum gene cluster we detected an apparently truncated LacI-type repressor-coding gene, lacR (Table 1).
Comparison of the transcriptomes of a 2′-FL versus sorbitol grown APCKJ1 culture revealed downregulation of three distinct gene clusters (with locus tags BKKJ1_0067-0069, BKKJ1_0206-0208 and BKKJ1_0336-0341), which, as expected, appear to be involved in sorbitol metabolism based on BLAST analysis (Table 1).
Similar results were obtained when we performed transcriptome analyses for B. kashiwanohense APCKJ1 grown on 3-FL (available in the GEO dataset provided here).
Biochemical characterisation of FumA1 and FumA2, and activity towards 2′-FL and 3-FL
In order to investigate the predicted fucosidase activities encoded by fumA1 (BKKJ1_2069) and fumA2 (BKKJ1_2070), the corresponding FumA1 and FumA2 proteins were purified as His-tagged versions (and designated here as FumA1His and FumA2His, respectively; see Materials and Methods). FumA1 is an 87.46 kDa protein belonging to the GH95 family fucosidases, and FumA2 is a 53.59 KDa protein belonging to the GH29 family fucosidases. Neither protein sequence contains a predicted signal peptide site, indicating their likely intracellular localisation. Assessment of substrate specificity was performed by incubating either FumA1His or FumA2His on their own or in combination with 2′-FL or 3-FL, and analysing the reaction products after varying times (20 minutes, 1 hour, 4 hours and 24 hours) by High-Performance Anion-Exchange Chromatography with Pulsed Amperometric Detection (HPAEC-PAD). Purified FumA1His was shown to fully liberate lactose and L-fucose from both 2′-FL (within 20 minutes) and 3-FL (within 4 hours) (Fig. 3), demonstrating hydrolytic activity towards both the Fuc-α1-2Gal linkage in 2′-FL and the Fuc-α1-3Glc connection in 3-FL, thus indicating a key role in the hydrolysis and utilisation of both sugars. Purified FumA2His was shown to fully cleave 3-FL (within 20 mins), but not 2′-FL, into lactose and L-fucose (Fig. 3), demonstrating hydrolytic activity solely towards the Fumα1-3Glc residue. These results indicate a high hydrolytic activity and specificity of FumA1His for 2′-FL, and similarly of the FumA2His enzyme for 3-FL, with FumA1His also capable of hydrolysing 3-FL, albeit at an apparently low rate.

Hydrolysis products of 2′-FL and 3-FL incubated with FumA1His and FumA2His. (A) HPAEC-PAD chromatogram profiles of (I) 2′-FL and (II) 3-FL, when incubated in MOPS buffer (pH7) with FumA1His at the time-points 0 minutes, 20 minutes, 1 hour, 4 hours and 24 hours. (B) HPAEC-PAD chromatogram profiles of (I) 2′-FL and (II) 3-FL, when incubated in MOPS buffer (pH7) with FumA2His at the time-points 0 minutes, 20 minutes, 1 hour, 4 hours and 24 hours. Figure adapted from thesis Figure 5.4; James, 201897.
According to our phylogenetic analysis the FumA1 protein is a as GH95 α-fucosidase based on the fact that it clusters with other known and characterized GH95 members (Fig. S2). Among these are Blon_2335 from Bifidobacterium longum subsp. infantis ATCC 1569744, AfcA from B. bifidum45 and Afc3 from Clostridium perfringens46. Of note, GH95 enzymes, also referred as 1,2-α-L-fucosidases (EC 3.2.1.63), specifically target α-1,2-fucosidic linkages44,46, thus corroborating our observation of FumA1 showing preference towards 2′-fucosyllactose. In contrast, FumA2 clusters with other previously characterized GH29 α-fucosidases, including BT_2970, BT_2192 and BT3798 from Bacteroides thetaiotaomicron VPI-548247,48,49, Blon_2336 from Bifidobacterium longum subsp. infantis ATCC 1569744, TM0306 from Thermotoga maritima50 and Afc2 from Clostridium perfrigens46. Previous studies have demostrated that the GH29 family is further divided into two sub-families with different substrate specificity24,48. Notably, FumA2 clusters with previously characterized representatives of GH29 subfamily B (e.g. BT2192 from B. thetaiotaomicron and Blon_2336 from B. longum subsp. infantis) (Fig. S2), indicating that FumA2 is a member of the B subfamily. GH29-B enzymes, also referred as 1,3-1,4-α-l-fucosidases (EC 3.2.1.111), have been shown to be specifically active towards α1,3/4-fucosidic linkages44,48, in line with our finding that FumA2 has preference towards 3-fucosyllactose.
Phenotypic analysis of B. breve UCC2003 strains harbouring B. kashiwanohense APCKJ1 genes implicated in 2′-FL/3-FL metabolism
It has previously been shown that L-fucose supports rather moderate growth of the infant-associated species B. breve18,51. While some strains of B. breve have been demonstrated to metabolize 2′-FL33,52, the majority of studied strains, including UCC2003, cannot use 2′-FL or 3-FL as a sole carbon source to any appreciable extent17,18,33. Thus, B. breve UCC2003 is capable of successfully metabolising the two individual components that make up fucosyllactose (i.e. lactose and L-fucose), yet not the 2′-FL or 3-FL compounds themselves, whereas B. kashiwanohense APCKJ1 is capable of growth on 2′-FL, 3-FL and lactose, but not on L-fucose. We therefore elected to heterologously express genes from the B. kashiwanohense APCKJ1 fum gene cluster, that are predicted to be required for fucosyllactose uptake and hydrolysis in an attempt to confer 2′-FL and/or 3-FL metabolism to B. breve UCC2003. The fumA1 gene was selected because of its ability to hydrolyse both 2′-FL and 3-FL (see above), while the fumS, fumT1 and fumT2 genes (corresponding to locus tags BKKJ1_2076–2078) were chosen as these genes are predicted to encode the (2′-FL and 3-FL) transporter components of the fum locus (Table 1). Accordingly, the fumA1 gene, and the fumS, fumT1 and fumT2-encompassing DNA fragment were cloned into two separate vectors, which were then introduced into B. breve UCC2003, generating strain B. breve UCC2003-fumA1-fumST1T2 (control strains were constructed containing the ‘empty’ cloning vectors, see Materials and Methods and below). B. breve UCC2003-fumA1-fumST1T2 was assessed for its ability to grow in mMRS supplemented with 2′-FL, 3-FL, L-fucose or a lactose control, as compared to the following strains: wild-type UCC2003, B. breve UCC2003-fumA1-pBC1.2, UCC2003-fumST1T2-pNZ44-strepR, and wild-type B. kashiwanohense APCKJ1 (Fig. 4). Strains B. breve UCC2003-fumA1-pBC1.2 and UCC2003-fumST1T2-pNZ44-strepR were generated and used as negative controls, as each of these strains expresses either of the two components predicted as required for 2′-FL/3-FL utilisation fumA1 and fumST1T2 (i.e. fucosidase activity and 2′-FL/3-FL transport, respectively), which are both expressed by B. breve UCC2003-fumA1-fumST1T2. Our results demonstrated that wild-type UCC2003, as well as the recombinant UCC2003 strains expressing either fumA1 or fumST1T2 on their own, were unable to grow on 2′-FL or 3-FL (final OD600nm < 0.5). In contrast, the recombinant UCC2003 strain expressing both fumA1 and fumST1T2 demonstrated good growth on either 2′-FL or 3-FL (final OD600nm > 2.0), which is comparable with that obtained by APCKJ1 grown on either of these substrates (Fig. 4). As expected all of the tested cultures grew to a high cell density on lactose (OD600nm > 3.0), and none of the strains demonstrated significant growth on L-fucose. Notably, this latter result contrasts with previous work which observed the ability of UCC2003 to grow on L-fucose, though at a modest level51. However, some L-fucose metabolism by UCC2003 was observed here, based not on growth ability but analysis of metabolic end products, as described below.

Growth assays of B. breve UCC2003 WT, B. kashiwanohense APCKJ1 WT and recombinant B. breve UCC2003-fumA1-fumST1T2 strains in lactose, L-fucose, 2′-FL or 3-FL. Final OD600nm values after 24 hours of growth of wild type B. breve UCC2003, wild type B. kashiwanohense APCKJ1 and recombinant B. breve UCC2003 strains in modified MRS containing 1% (wt/vol) lactose, L-fucose, 2′-FL or 3-FL as the sole carbon source. The results are the mean values obtained from three separate experiments. Error bars represent the standard deviation. Figure adapted from thesis Figure 5.5; James, 201897.
Thus, the transcriptome data, substrate hydrolysis profiles and growth results of various recombinant strains corroborate the notion that in B. kashiwanohense APCKJ1 the fucosidase encoded by fumA1 is specifically required for 2′-FL hydrolysis, and capable of hydrolysing 3-FL, while the solute binding protein and two permeases encoded by fumS, fumT1 and fumT2, respectively, are responsible for the uptake and internalisation of 2′-FL and 3-FL. These results also demonstrate the ability of a recombinant B. breve UCC2003-derivative to successfully internalise and utilise 2′-FL and 3-FL for growth, with the heterologous and concomitant expression of this transport system and fucosidase from B. kashiwanohense APCKJ1.
Analysis of metabolites generated by fucose and fucosyllactose utilisation
NMR spectroscopy of the supernatants generated during growth of B. kashiwanohense APCKJ1, B. breve UCC2003 or B. breve UCC2003-fumA1-fumST1T2 on lactose, L-fucose, 2′-FL or 3-FL, revealed the metabolic end-products of the fermentation of these sugars by these strains. A combination of standard 1D nuclear magnetic resonance spectroscopy (NMR) and 2D Homonuclear 1H–1H Total Correlation Spectroscopy (TOCSY) and 1H J-resolved (JRES) experiments confirmed the presence of a number of metabolites/carbohydrates, including: 1,2-PD, ethanol, lactate, acetate, methanol (contaminant of the 3-FL substrate, see below), formate, lactose, fucose, 2′-FL, 3-FL, α-glucose and β-glucose (Table 2).
These identified metabolites/carbohydrates were assigned to peaks in the resulting JRES spectra for each sample, using the TOCSY experiment to identify 1H-1H coupling in the molecules and allowed accurate identification and quantification of each individual compound within the analyzed samples. The millimolar concentrations of each of these compounds within a given sample, as well as in the medium of the uninoculated controls are represented in Table 2. For all three strains, growth on lactose resulted in a decrease in the amount of lactose present in the samples. This coincided with a substantial increase in the concentration of acetate, and to a lesser extent ethanol, lactate and formate, as compared to the uninoculated control containing 1% lactose. Interestingly, while none of the strains demonstrated significant growth on L-fucose as a substrate, the metabolite profiles of supernatants obtained following incubation of each of the three with this sugar did not match that of the uninoculated control containing 1% L-fucose. In particular, the supernatant of the wild type UCC2003 showed a marked decrease in the concentration of L-fucose present, with an increase in the levels of acetate and formate and 1,2-PD, as well as a small increase in ethanol concentration. The same trend was observed, but to a lesser extent, in the supernatants of UCC2003-fumA1-fumST1T2, and even less so, APCKJ1, when incubated in the presence of 1% L-fucose. The corresponding metabolite profiles of the supernatants containing 2′-FL and 3-FL were essentially identical, except for the difference between signals originating from the two sugars themselves. The supernatants of wild type UCC2003 incubated in the presence of 2′-FL or 3-FL showed no decrease in the concentration of either sugar, nor the accumulation of additional metabolites, as compared to the uninoculated controls. The supernatants of APCKJ1 grown on 2′-FL or 3-FL revealed a near-complete depletion of either sugar, with significant increases in the concentrations of 1,2-PD, formate, and in particular acetate. The same trend in metabolite profiles was observed for the supernatants of UCC2003-fumA1-fumST1T2 grown on either 2′-FL or 3-FL, with full depletion of these two sugars. These trends in metabolite profiles between the individual samples were further confirmed by PCA analysis, which showed distinct clustering of the B. kashiwanohense APCKJ1 WT and the UCC2003-fumA1-fumST1T2 strains with the exception of the isolates grown on fucose, which clustered independently regardless of bacterial strain (Fig. S3). The presence of a low level of methanol in all 3-FL-containing samples (including the uninoculated control) is presumed to be due to contamination of 3-FL used for these experiments with this alcohol.
Transcriptome analysis of recombinant B. breve UCC2003 grown on 2′-FL
The results of the metabolite analysis indicate that, while neither B. breve UCC2003 nor B. kashiwanohense APCKJ1 are capable of efficient growth using L-fucose as a sole carbohydrate source, they both still demonstrate the ability to consume a limited quantity of this sugar, and convert it into organic acids and 1,2-PD, in particular (WT) UCC2003. However, the genes involved in this metabolic process in B. breve UCC2003 cannot be identified by transcriptomic analysis during this metabolism of free L-fucose, as growth is not sufficient to allow this.
Thus, transcriptomic analyses were carried out on the recombinant B. breve strain generated in this study during growth on fucosyllactose, in order to identify the potential genes specifically involved in the metabolism of the L-fucose component of fucosyllactose. Global gene expression was determined by microarray analysis during growth of UCC2003-fumA1-fumST1T2 in mMRS supplemented with 2′-FL, as compared with gene expression during growth in mMRS supplemented with ribose. As lactose is the other constituent of 2′-FL, microarray analysis was also carried out to compare gene expression during growth of UCC2003-fumA1-fumST1T2 in mMRS supplemented with lactose (as compared to gene expression during growth in mMRS supplemented with ribose), in order to identify the lactose metabolism-specific genes expressed during the utilisation of fucosyllactose. Genes that were shown to be significantly upregulated in transcription above the designated cut-off (fold-change > 2.0, P < 0.001) are displayed in Table 3. A fold-change of 2.0 was chosen as the threshold for differential transcription, as this allowed a clear resolution of those genes that exhibited the highest level of increased transcription (from a presumed background of a high number of genes that elicit a lower level of increased transcription).
Among the genes that showed increased transcription when UCC2003-fumA1-fumST1T2 was grown on lactose or 2′-FL were genes from loci known to function in lactose metabolism, namely the lnt (Bbr_0526-0530)30 and lac loci (Bbr_1551-1553)53. Among the genes that were uniquely upregulated in transcription during growth on 2′-FL were those of the gene cluster Bbr_1739-1748 (containing genes, homologs of which are also present in the fum locus of APCKJ1) (Table 3 and Fig. S1). These results indicate that while expression of the non-native B. kashiwanohense APCKJ1 genes fumA1, fumS, fumT1 and fumT2 in B. breve UCC2003 allows the latter strain to take up 2′-FL and to hydrolyse it into lactose and L-fucose, expression of native UCC2003 genes of the lnt, lac and fum loci are presumed to enable strain UCC2003-fumA1-fumST1T2 to further metabolise the released carbohydrates.
Elucidation of the fucosyllactose/fucose metabolic pathway in B. kashiwanohense and B. breve
The results of the APCKJ1 transcriptome analyses indicate involvement of the fum locus and fumG in fucosyllactose metabolism, and the combined results of the in vitro hydrolytic assays and recombinant UCC2003 growth analysis implicate the APCKJ1 fucosidase-encoding genes fumA1 and fumA2, as well as the transporter-encoding genes fumS, fumT1 and fumT2 in the internalisation and hydrolysis of 2′-FL and 3-FL. However, the presence of further genes of the fum locus, as well as fumG, upregulated in transcription during growth on 2′-FL or 3-FL suggests the involvement of these genes in the metabolism of the liberated L-fucose, given that the genes for lactose metabolism appear to be constitutively expressed in strain APCKJ1. Likewise, transcriptional upregulation of genes in the UCC2003 fum locus during growth of the recombinant strain on 2′-FL implicate these genes in L-fucose metabolism in UCC2003. This, and the NMR data, suggests that both B. kashiwanohense APCKJ1 and B. breve UCC2003 possess functional pathways for the metabolism of L-fucose.
A pathway for the anaerobic catabolism of L-fucose leading to the formation of 1,2-PD (via non-phosphorylated intermediates) has recently been proposed for B. longum subsp. infantis ATCC1569733, based on sequence homology with enzymes from a fucose-dependent metabolic route previously identified in Xanthomonas campestris54. Genes involved in the proposed L-fucose utilisation pathway in B. longum subsp. infantis ATCC15697 were used in BLASTP searches for homologs in the genomes of B. kashiwanohense APCKJ1 and B. breve UCC2003. Homologs identified in the APCKJ1 and UCC2003 genomes, whose predicted annotations did not match the annotations of their ATCC15697 equivalents (based on their function in the fucose/fucosyllactose utilisation pathways33), were further checked for alternative annotations in the Pfam (http://pfam.xfam.org) and KEGG (http://www.genome.jp/kegg/) databases.
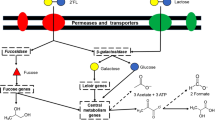
This search revealed the presence of homologs for all predicted fucose utilisation genes from ATCC15697 in APCKJ1 (Table S2), including seven of the genes of the fum locus, and the gene fumG, all of which were transcriptionally upregulated when APCKJ1 was grown on 2′-FL (see above). These results therefore suggest the presence of all genes necessary for L-fucose metabolism in B. kashiwanohense APCKJ1, via a pathway converting L-fucose into L-2-keto-3-deoxyfuconate (as proposed in B. breve, B. longum subsp. infantis, and B. longum subsp. suis33,39), but subsequently converted into L-lactaldehyde and pyruvate by means of a predicted L-2-keto-deoxy-fuconate aldolase (FumF) encoded by the fum locus (Fig. 5). The presence of an L-1,2-propanediol oxidoreductase (FumG) in the genome sequence of APCKJ1 allows the further reduction of L-lactaldehyde into L-1,2-PD (Fig. 5). These findings are supported by the observed accumulation of lactate, acetate, formate (all metabolites of pyruvate) and 1,2-PD in the 2′-FL and 3-FL fermentates analysed by HPLC and NMR, as mentioned above.

Fucose utilisation pathway in B. kashiwanohense and B. breve. Schematic representation of the pathway for the utilisation of fucosyllactose in B. kashiwanohense APCKJ1 (red arrows) and B. breve UCC2003 (grey arrows). Figure adapted from thesis Figure 5.6; James, 201897.
Homologs for the fum genes in APCKJ1 were identified in the UCC2003 genome (Table S2) and were shown to be transcriptionally upregulated when the recombinant B. breve UCC2003 was grown on 2′-FL (Table 3). Notably, a recent transcriptional study conducted on B. breve UCC2003 identified the FumG homologue (Bbr_1505) as constitutively expressed in this strain55.
The ATCC15697 genes lacking homologs in UCC2003 were the GH29 α-fucosidase-encoding gene and the L-fucose mutarotase-encoding gene, as well as those encoding the components the (three) components of the proposed fucosyllactose uptake system. While neither B. breve UCC2003 nor B. kashiwanohense APCKJ1 demonstrated significant growth on free L-fucose, our NMR data indicate active metabolism of this sugar in both species, generating the same metabolites (although at lower levels) as are observed following growth on 2′-FL or 3-FL. Thus, the apparent absence of an L-fucose mutarotase does not seem to prevent metabolism of L-fucose in UCC2003. While a GH95 fucosidase-encoding gene homologous to fumA1kw (Bbr_1288) was identified within an additional “HMO islet” in UCC2003 (Bbr_1288-91) (Fig. 6), the presence of 2′-FL or 3-FL do not seem to induce transcription of this region, as demonstrated in the recombinant UCC2003 fumA1-ST1T2 growth assays. It is possible that this locus containing fumA1br targets other fucosylated HMO structures, such as fucosyl-LN(n)T, as B. breve is known to efficiently consume free LN(n)T30. Alternatively, it may be that this is a non-functional remnant of a former FL-utilization gene cluster.

Fucose utilisation loci in infant-derived bifidobacterial strains. Comparison of the fucose/fucosyllactose metabolism genes of B. kashiwanohense APCKJ1 with corresponding homologous loci of B. longum subsp. infantis ATCC 15697, B. pseudocatenulatum DSM 20438 and B. breve UCC2003, as identified using the heatmap in Fig. 7. Each solid arrow represents an open reading frame. The length of each arrow is proportional to the size of the open reading frame, and the color code is indicative of the predicted gene function. The amino acid sequence identity of each predicted protein expressed as a percentage is also shown.
Distribution of fucose/fucosyllactose utilisation genes in the Bifidobacterium genus
The results of the microarray analysis implicated the protein products of fumG (BKKJ1_0429) as well as the genes of the fum locus (BKKJ1_2069-2079) in the utilisation of fucosyllactose by B. kashiwanohense APCKJ1. This is supported by the results of the phenotypic analysis of the B. breve UCC2003 strain expressing the fucosidase FumA1 and associated transporter proteins encoded by this locus. We selected four genes within the fum locus responsible for the intracellular metabolism of fucosyllactose and/or L-fucose identified in APCKJ1 in order to observe their distribution within the Bifidobacterium genus. The regulatory and transport-associated genes of the fum locus were excluded from comparative analysis, as carbohydrate transporter and transcriptional regulation-associated genes generally share a high degree of homology across bifidobacteria, thus lacking distinguishable sequence variability56,57.
This analysis revealed that the four genes included in the analysis are predominant across infant-associated bifidobacteria (e.g. B. kashiwanohense, B. breve, B. longum subsp. infantis and B. pseudocatenulatum), with the exception of B. bifidum. (Fig. 7). This corroborates the observation that members of the species B. bifidum employ extracellular fucosidases to liberate the lactose component of 2′-FL and 3-FL, which is subsequently metabolised, but are not capable of metabolising the released L-fucose58. The degree of similarity between the fucose utilisation genes from B. kashiwanohense APCKJ1 and their corresponding homologous loci in B. longum subsp. infantis ATCC15697 and B. breve UCC2003 are shown in Table S2.

Distribution of fum genes across the Bifidobacterium genus. Heatmap representing the distribution of homologs of four genes encoding proteins predicted to have a key role in fucose/fucosyllactose utilisation pathway from B. kashiwanohense APCKJ1 across the Bifidobacterium genus. Gene products from the representative type strain genomes of all online-available Bifidobacterium species with a significant homology of 50% iterative similarity over 50% of protein length are represented in the matrix. The colour code grading represents the degree of sequence similarity at protein level, with the species grouped by origin of isolation.
Discussion
B. kashiwanohense represents an example of an apparently infant-specific Bifidobacterium species, although just two publications have previously reported on the isolation of this species from infant faeces34,35. The bifidobacterial component of the infant gut microbiota is typically dominated by the species B. breve, B. longum subsp. longum and B. bifidum, with B. pseudocatenulatum, B. longum subsp. infantis and B. longum subsp. suis occurring at lower frequency13,59,60,61. However, the ability of members of B. kashiwanohense to utilise milk-derived substrates suggests that the infant gut may constitute its primary ecological niche. The ability of a B. kashiwanohense isolate to grow on the HMOs 2′-FL or 3-FL as its sole carbohydrate source has been reported33. HMO utilisation by certain members of the Bifidobacterium genus appears to be present in some infant-associated species8,18,19,29,33. As fucosylated sugars typically represent a substantial proportion of all HMOs2,36,37, the ability to utilise fucosyllactose by B. kashiwanohense represents a specific adaptation to the neonatal gut environment. Here, a novel B. kashiwanohense strain (APCKJ1) isolated from the faeces of a breastfed infant demonstrated the ability to consume 2′-FL or 3-FL as its sole carbohydrate source, which became the focus of this investigation.
Transcriptional analysis of APCKJ1 revealed an 18 kb gene cluster dedicated to the metabolism of fucosyllactose. This region contains the adjacent lac and fum loci, thus representing a potential ‘HMO island’ conferring the ability to utilize fucosyllated HMOs. Interestingly, the constitutive transcription of the lac locus (Table 1) is due to the presence of a truncated, non-functional LacI-type repressor (LacR) in APCKJ1 strain. Furthermore, homologs of lacR appear to be entirely absent in B. kashiwanohense DSM21854 and B. longum subsp. infantis ATCC1569733. A homolog of this ‘HMO island’ has been previously described in B. longum subsp. infantis26,62,63 and B. longum subsp. longum64, thus reinforcing the notion of specific adaptations to HMO utilisation by infant-associated bifidobacteria.
The combined results of in vitro hydrolysis assays, heterologous expression of FumA1 and FumA2 fucosidases in B. breve UCC2003, as well as growth results obtained with recombinant UCC2003 strains confirmed the predicted functions of some of the genes of the fum locus. Our findings are consistent with the notion that the three adjacent genes fumS, fumT1 and fumT2 encode transport components necessary for 2′-FL/3-FL uptake, while the fumA1 and fumA2 encoded fucosidases are responsible for the hydrolysis of 2′-FL and 3-FL, respectively. Of note, our findings corroborate what had previously been observed in a subset of FL-utilising bifidobacteria (members of B. breve, B. longum and B. pseudocatenulatum spp), where a solute binding protein of an ABC transporter system and an α-fucosidase (GH95 family) were shown to play an essential role in FL utilisation38. Interestingly, while FumA1 is capable of hydrolysing both 2′-FL and 3-FL, it possesses higher catalytic efficiency with the former; FumA2 instead can only hydrolyse 3-FL. Such apparent redundancy in the degradation of isomeric HMO structures is not unusual in bifidobacteria19,27,30. However, it is interesting that both FumA1 and FumA2 can hydrolyse 3-FL, as this HMO has been reported to be much less abundant in breastmilk compared to its isomer 2′-FL36. We can’t exclude that the true target substrate for FumA2 may be in fact another fucosylated HMO, such as fucosyl-LN(n)T, containing similar linkages.
While these results reveal the mechanisms of fucosyllactose uptake and intracellular hydrolysis in B. kashiwanohense APCKJ1, they do not reveal whether small fractions of L-fucose may be internalised and used by the cell. The combined results of the HPLC and NMR analyses of the various 2′-FL/3-FL and L-fucose fermentations, as well at the APCKJ1 microarrays, indicate the utilisation of the L-fucose mainly resulting by the hydrolysis of 2′-FL and 3-FL, with even some partial metabolism of free L-fucose. The comparative analysis performed between B. kashiwanohense APCKJ1 and B. longum subsp. infantis ATCC15697 revealed that all the presumed genetic components necessary for the utilisation of L-fucose are encoded by APCKJ1. This therefore represents the first evidence of a complete and functional fucose utilisation pathway in B. kashiwanohense, a species which has been previously suggested to extracellularly release the fucose component of fucosyllactose33,39. In addition to this we also identified an identical (predicted) pathway for L-fucose metabolism in B. breve UCC2003, as this species has recently been characterised for its utilisation of L-fucose39. The observed depletion of L-fucose and accumulation of its breakdown product 1,2-PD show that B. breve UCC2003 and B. kashiwanohense APCKJ1 possess the ability to at least partially metabolise free L-fucose, despite not growing to any significant degree in its sole presence. Interestingly, 1,2-PD excretion in human urine and faeces has been largely attributed to contamination since it is an excipient in several pharmaceutical preparations or food additives65. However, other research has shown that 1,2-PD is present in higher concentrations in faeces of breastfed babies66, which would be consistent with microbial influence.
Identification of homologs involved in fucose metabolism across the Bifidobacterium genus highlights the apparent importance of this pathway for colonisation of the infant gut. While the ability to utilise fucosyllactose undoubtedly provides an advantage for B. kashiwanohense establishment in the breastfed neonatal gut, this strategy is also employed by other infant-associated bifidobacteria, including B. bifidum, B. longum subsp. infantis, B. longum subsp. longum (and possibly B. pseudocatenulatum) as well as some species of B. breve, all of which are capable of utilising other HMO components, such as sialyllactose, LN(n)T, LNB, sialic acid and/or L-fucose8,19,20,21,22,23,24,25,26,27,28,29,30,31,32,64. Therefore, the narrow specialisation in HMO metabolism by B. kashiwanohense may in fact also be a hindrance in becoming one of the dominant species in the breastfed infant gut microbiota.
The findings of this study reinforce the importance of HMO metabolism for the successful proliferation of Bifidobacterium species in the breastfed infant gut. This allows the depiction of the metabolic pathway required for the utilisation of fucosyllactose by B. kashiwanohense APCKJ1 and a potential pathway for the metabolism of free L-fucose by B. breve UCC2003, and furthermore reveals the distribution of fucose-utilisation associated genes in Bifidobacterium species typically found in neonates. Our findings not only demonstrate the conservation of fucose metabolism in many infant-associated bifidobacteria, but also highlight the role of fucosylated HMOs in bifidobacterial establishment in the breastfed infant gut.
Thus, the outcome of our work significantly contributes to increasing our understanding of HMO metabolism by infant-associated bifidobacteria. In particular B. kashiwanohense, while previously shown to consume HMO33, remained until now uncharacterised for its manner of HMO utilisation. Our findings have generated insights into the pathway employed by a number of Bifidobacterium species for their metabolism of L-fucose freely available or liberated by the degradation of larger HMO structures. Ultimately, our gained understanding of HMO metabolism by a number of Bifidobacterium species can be used as a tool to identify infant-specific probiotic strains and conversely reinforces the potential of fucosylated HMOs as a prebiotic.
Materials and Methods
Strain isolation from infant faeces, and species identification
Pure cultures of Bifidobacterium strains, capable of utilising 2′-FL as their sole carbohydrate source, were isolated from the faeces of an exclusively breastfed neonate (aged 4 weeks). The faecal sample was obtained from the APC Microbiome Ireland human clinical sample collection (stored at −80 °C), originally collected for the InfantMet Cohort67. Mothers were approached for consent between February 2012 and May 2014 at the Cork University Maternity Hospital. All mothers provided informed consent for the use of the infant faeces in this study. Ethical approval provided by the Cork University Hospital Research Ethics Committee (ethical approval reference: ECM (w) 07/02/2012), with all methods performed in accordance with the relevant guidelines and regulations required by said committee. Bifidobacterium culture isolates were obtained using a method based on that employed in the InfantMet study67, though with the following modifications. All sample preparation and platings were carried out under anaerobic conditions in a modular atmosphere-controlled system (Davidson and Hardy, Belfast, Ireland) at 37 °C. One gram of frozen faecal sample was thawed anaerobically at 37 °C, and resuspended in 10 ml PBS (Sigma Aldrich, Ireland) supplemented with 0.05% L-cysteine hydrochloride (Sigma Aldrich, Ireland). Selection of bifidobacteria was performed by spread-plating 10 aliquots of 1 ml of the faecal resuspension on: modified de Man Rogosa and Sharpe (mMRS) agar68, supplemented with 1% (wt/vol) 2′-FL (Glycom A/S, Lyngby, Denmark), 0.05% L-cysteine-HCl, 100 μg/ml mupirocin (Oxoid, Fannin, Ireland) and 50 units nystatin suspension (Sigma Aldrich, Ireland). Agar plates were incubated anaerobically at 37 °C for 72 h. Emerging colonies from these plates were resuspended in 1 ml of PBS supplemented with 0.05% L-cysteine-HCl, after which the resuspensions from the 10 plates of a single sample were pooled and homogenised, and serial-diluted in the aforementioned PBS, and dilutions between 1 × 10−4 and 1 × 10−8 were spread-plated on 2′-FL-supplemented mMRS agar, as described above, and incubated anaerobically at 37 °C for 72 h. Single, isolated colonies were selected from the serial dilution plates, and re-streaked onto the same agar medium, and incubated anaerobically at 37 °C for 48 h. This sub-cultivation process was repeated twice, after which individual colonies were inoculated into modified mMRS medium, supplemented with 1% lactose and 0.05% L-cysteine-HCl. Pure cultures were stocked and stored at −80 °C. Isolates were confirmed as bifidobacteria based on the fructose-6-phosphate phosphoketolase activity test69, and species identity was determined by sequencing of their ITS region70.
Genome sequencing and annotation of novel isolate Bifidobacterium kashiwanohense APCKJ1
Genome sequencing of the B. kashiwanohense isolate was performed by GATC Biotech Ltd. (Germany) using Pacific Biosciences SMRT RSII technology. Raw sequencing reads were de novo assembled using the Hierarchical Genome Assembly Process (HGAP) protocol RS_Assembly.2 implemented in the SMRT Smart Analysis portal v.2.3 with default parameters (https://github.com/PacificBiosciences/SMRT-Analysis). Open Reading Frame (ORF) prediction and automatic annotation was performed using Prodigal v2.0 (http://prodigal.ornl.gov) for gene predictions, BLASTP v2.2.26 (cut-off e-value of 0.0001)71 for sequence alignments against a combined bifidobacterial genome-based database, and MySQL relational database to assign annotations. Predicted functional assignments were manually revised and edited using similarity searches against the non-redundant protein database curated by the National Centre for Biotechnology Information (ftp://ftp.ncbi.nih.gov/BLAST/db/) and PFAM database (http://pfam.sanger.ac.uk), allowing a more detailed, in silico characterization of hypothetical proteins. GenBank editing and manual inspection was performed using Artemis v18 (http://www.sanger.ac.uk/resources/soft-ware/artemis/). Transfer RNA genes were identified employing tRNAscan-SE v1.4 and ribosomal RNA genes were detected based on Rnammer v1.272 software supported by BLASTN v2.2.26.
Nucleotide sequence analysis and multiple sequence alignment
Sequence data were obtained from Artemis-mediated73 genome annotations of B. kashiwanohense strains APCKJ1, JCM1543974 and PV20-275, and B. breve UCC200376. Database searches were performed using non-redundant sequences accessible at the National Centre for Biotechnology Information (http://www.ncbi.nlm.nih.gov) using BLAST71. Sequences were verified and analysed using SeqMan and SeqBuilder programs of the DNAStar software (version 10.1.2; DNAStar, Madison, WI, USA). Multiple sequence alignments of amino acid sequences were created with Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalw/), using default settings77. Alignments were visualised with Genedoc v 2.7.0 (http://iubio.bio.indiana.edu/soft/molbio/ibmpc/), using physicochemical display mode (similar colouring based on common physical and chemical properties).
Bacterial strains, plasmids, culture conditions and Bifidobacterium growth assays
Bacterial strains and plasmids used in this study are listed in Table S3. B. breve and B. kashiwanohense cultures were incubated under anaerobic conditions in a modular atmosphere-controlled system (Davidson and Hardy, Belfast, Ireland) at 37 °C. B. breve UCC2003 was routinely cultured in either de Man Rogosa and Sharpe medium (MRS medium; Difco, BD, Le Pont de Claix, France) supplemented with 0.05% L-cysteine-HCl and 1% lactose, or reinforced clostridial medium (RCM; Oxoid Ltd., Basingstoke, England). B. kashiwanohense APCKJ1 was routinely cultured in modified mMRS supplemented with 0.05% L-cysteine-HCl and 1% lactose. Lactococcus lactis strains were cultivated in M17 broth (Oxoid Ltd., Basingstoke, England) containing 0.5% glucose78 at 30 °C. Escherichia coli strains were cultured in Luria-Bertani (LB) broth79 at 37 °C with agitation. Where appropriate, growth media contained chloramphenicol (Cm; 5 μg ml−1 for L. lactis and E. coli, 2.5 μg ml−1 for B. breve) or streptomycin (Strep; 400 μg ml−1).
Carbohydrate utilisation by bifidobacterial strains was examined in mMRS medium68, and excluding a carbohydrate source. Prior to inoculation, the mMRS medium was supplemented with cysteine-HCl (0.05%, wt/vol) and a particular carbohydrate source (1%, wt/vol). It has previously been shown that mMRS does not support growth of B. breve UCC2003 in the absence of an added carbohydrate68. Carbohydrates used were lactose, glucose, D-ribose, sorbitol, cellobiose, raffinose, melibiose (all purchased from Sigma Aldrich, Steinheim, Germany), and LNT, LNnT, lactosamine-hydrochloride (lactosamine HCl), N-acetylneuramic acid (sialic acid), L-fucose, 3-sialyllactose (3-SL), 6-sialyllactose (6-SL), 2′-FL and 3-FL (all obtained from Glycom, Lyngby, Denmark). A 1% wt/vol carbohydrate concentration was considered sufficient to analyse growth capabilities of a strain on a particular carbon source. To determine bacterial growth profiles and final optical densities, 5 ml of freshly prepared mMRS medium, including a particular carbohydrate (see above), was inoculated with 50 μl (1%) of a stationary phase culture of B. breve UCC2003 or B. kashiwanohense APCKJ1. Uninoculated mMRS medium was used as a negative control. Cultures were incubated anaerobically at 37 °C for 24 h, and the optical density at 600 nm (OD600nm) was determined manually. Growth assays were repeated in duplicate or triplicate, depending on carbohydrate availability.
HPLC analysis
Fermentation end-product profiles were assessed by HPLC following growth of selected bifidobacteria in mMRS medium containing 1% 2′-FL, 3-FL or lactose as the sole carbohydrate source. Collected culture samples were then prepared for HPLC analysis by centrifugation at 3270 × g for 5 min, after which the resulting supernatants were filter sterilized (0.45 μm filter, Costar Spin-X Column) and stored at −20 °C until further use. An Agilent 1200 HPLC system (Agilent Technologies, Santa Clara, CA) with a refractive index detector was used to detect generated metabolites. Metabolite peaks were identified based on retention times of known standards (2′-FL, 3-FL, L-fucose, lactose, lactic acid, acetic acid, formic acid, sodium pyruvate and 1,2-PD) at a concentration of 10 mM. Non-fermented mMRS medium containing carbohydrates (or the absence of any carbohydrate) served as controls. A REZEX 8 μm 8% H organic acid column (300 mm × 7.8 mm, Phenomenex, Torrance, CA, USA) was utilized and maintained at 65 °C. Elution was performed for 25 min using a 0.01 M H2SO4 solution at a constant flow rate of 0.6 mL/min.
Analysis of global gene expression using B. breve DNA microarrays
Global gene transcription was determined during log-phase growth of B. kashiwanohense APCKJ1 in mMRS supplemented with 2′-FL. The obtained transcriptome was compared to that determined for log-phase B. kashiwanohense APCKJ1 cells when grown in mMRS supplemented with sorbitol. The transcriptome for growth of APCKJ1 on 2′-FL, as compared to a lactose control, was also obtained by the same method. Likewise, global gene transcription was determined during log-phase growth of recombinant strain B. breve UCC2003-fumA1fumST1T2 in mMRS supplemented with either 2′-FL or lactose. The obtained transcriptome was compared to that determined for log-phase B. breve UCC2003-fumA1fumST1T2 cells when grown in mMRS supplemented with ribose.
Microarrays containing oligonucleotides representing each of the 1864 identified open reading frames (ORFs) on the genome of B. breve UCC2003, or each of the 2110 identified ORFs on the genome of B. kashiwanohense APCKJ1, were designed and obtained from Agilent Technologies (Palo Alto, Ca., USA). Methods for cell disruption, RNA isolation, RNA quality control, complementary DNA synthesis and labelling were performed as described previously80. Labelled cDNA was hybridized using the Agilent Gene Expression hybridization kit (part number 5188-5242) as described in the Agilent Two-Colour Microarray-Based Gene Expression Analysis v4.0 manual (publication number G4140-90050). Following hybridization, microarrays were washed in accordance with Agilent’s standard procedures and scanned using an Agilent DNA microarray scanner (model G2565A). Generated scans were converted to data files with Agilent’s Feature Extraction software (Version 9.5). DNA-microarray data were processed as previously described81,82,83. Differential expression tests were performed with the Cyber-T implementation of a variant of the student t-test84.
DNA Manipulations
Chromosomal DNA was isolated from B. breve UCC2003 and B. kashiwanohense APCKJ1 as previously described85. Plasmid DNA was isolated from E. coli, L. lactis and B. breve using the Roche High Pure Plasmid Isolation kit (Roche Diagnostics, Basel, Switzerland). An initial lysis step was performed using 30 mg ml−1 of lysozyme for 30 minutes at 37 °C prior to plasmid isolation from L. lactis, B. breve or B. kashiwanohense. DNA manipulations were essentially performed as described previously79. Restriction enzymes and T4 DNA ligase were used according to the supplier’s instructions (Roche Diagnostics, Basel, Switzerland). Synthetic single stranded oligonucleotide primers used in this study (Table S4) were synthesized by Eurofins (Ebersberg, Germany). Standard PCRs were performed using Extensor Hi-Fidelity PCR Master Mix (Thermo Scientific, Waltham, United States) or Q5 High-Fidelity 2X Mastermix (New England BioLabs, Herefordshire, United Kingdom) in a Life Technologies Pro-FLex PCR System (Thermo Scientific, Waltham, United States). PCR products were visualized by ethidium bromide staining following agarose gel electrophoresis (1% agarose). B. breve colony PCR reactions were performed as described previously86. PCR fragments were purified using the Roche high Pure PCR purification kit, while plasmid DNA was isolated using the Roche High Pure Plasmid Isolation kit (Roche Diagnostics, Basel, Switzerland). Plasmid DNA was introduced into E. coli by electroporation as described previously79. B. breve UCC200376 and L. lactis87 were transformed by electroporation according to published protocols. The correct fragment orientation and integrity of all plasmid constructs (see also below) were verified by DNA sequencing, performed at Eurofins (Ebersberg, Germany).
Construction of overexpression vectors, protein overproduction and purification
For the construction of plasmids pNZ-FucA1 and pNZ-FucA2, DNA fragments encompassing the predicted fucosidase-encoding genes fumA1 (corresponding to BKKJ1_2069) or fum2 (corresponding to BKKJ1_2070) were generated by PCR amplification from chromosomal DNA of B. kashiwanohense APCKJ1 using Q5 High-Fidelity DNA polymerase and the primer combinations 2069F and 2069R, or 2070F and 2070R, respectively (Table S4). An in-frame N-terminal His10-encoding sequence was incorporated into the forward primers 2069F and 2070F to facilitate downstream protein purification. The generated amplicons were digested with SmaI and XbaI, and ligated into the ScaI and XbaI-digested, nisin-inducible translational fusion plasmid pNZ815088. The ligation mixtures were introduced into L. lactis NZ9000 by electrotransformation and transformants were then selected based on chloramphenicol resistance. The plasmid content of a number of Cm-resistant transformants was screened by restriction analysis and the integrity of positively identified clones was verified by sequencing.
Nisin-inducible gene expression and protein overproduction was performed as described previously89,90,91. In brief, 400 ml of M17 broth supplemented with 0.5% (wt/vol) glucose was inoculated with a 2% inoculum of a particular L. lactis strain, followed by incubation at 30 °C until an OD600nm of 0.5 was reached, at which point protein expression was induced by addition of cell-free supernatant of a nisin-producing strain92, followed by continued incubation for a further 2 hours. Cells were harvested by centrifugation and protein purification achieved as described previously90. Protein samples were dialysed into 20 mM morpholinepropanesulfonic acid (MOPS) (pH 7.0) buffer using Merck Amicon Ultra-15 Centrifugal Filter Units and stored at 4 °C. Protein concentrations were determined using the Bradford method93.
Assay of individual and combined fucosidase activities
The individual or sequential hydrolytic activities specified by FumA1 and FumA2 were determined essentially as described previously89, using either 2′-FL or 3-FL as a substrate. Briefly, a 50 μl volume of each purified protein (protein concentration of 0.5 mg/ml) was added to 20 mM MOPS (pH 7.0) buffer and 1 mg ml−1 (wt/vol) of one of the above-mentioned sugars in a final volume of 1 ml, followed by incubation for 24 hours at 37 °C. In order to determine enzyme affinity, each enzyme was incubated individually or in combination, with 2′-FL or 3-FL, as described above, with 200 μl samples taken at 20 minutes, 1 hour, 4 hours and 24 hours. All samples were subject to a final enzyme denaturation step at 85 °C for 15 minutes, before storage at −20 °C.
HPAEC-PAD analysis
For HPAEC-PAD analysis, a Dionex (Sunnyvale, CA) ICS-3000 system was used. Carbohydrate fractions from the above-mentioned hydrolysis assays (25 μl aliquots) were separated on a CarboPac PA1 analytical-exchange column (dimensions, 250 mm by 4 mm) with a CarboPac PA1 guard column (dimensions, 50 mm by 4 mm) and a pulsed electrochemical detector (ED40) in PAD mode (Dionex). Elution was performed at a constant flow-rate of 1.0 ml/min at 30 °C using the following eluents for the analysis: eluent A, 200 mM NaOH; eluent B, 100 mM NaOH plus 550 mM Na acetate; eluent C, Milli-Q water. The following linear gradient of sodium acetate was used with 100 mM NaOH: from 0 to 50 min, 0 mM; from 50 to 51 min, 16 mM; from 51 to 56 min, 100 mM; from 56 to 61 min, 0 mM. Chromatographic profiles of standard carbohydrates were used for comparison of the results of their breakdown by FumA1 or FumA2 proteins. Chromeleon software (version 6.70; Dionex Corporation) was used for the integration and evaluation of the chromatograms obtained. A 1 mg/ml stock solution of each of the carbohydrates, as well as their putative breakdown products (where available) used as reference standards was prepared by dissolving the particular sugar in Milli-Q water. Chromatographic profiles of mMRS containing standard carbohydrates were used for comparison of the results of their fermentation by B. kashiwanohense APCKJ1. mMRS containing 1% of 2′-FL or 3-FL, as well as their putative breakdown products (where available) used as reference standards.
Generation of recombinant B. breve strains
DNA fragments encompassing fumA1 (corresponding to locus tag BKKJ1_2069); and fumS, fumT1 and fumT2 (corresponding to locus tags BKKJ1_2076-2078, including the presumed promoter-containing region located upstream of BKKJ1_2078; this fragment was designated fumST1T2) were generated by PCR amplification from B. kashiwanohense APCKJ1 using Q5 High-Fidelity Polymerase (New England BioLabs, Herefordshire, UK) and primer pairs 2069pNZ44F and 2069pNZ44R, and 2076-78pCB1.2F and 2076-78pBC1.2R, respectively (primers are listed in Table S4. The resulting fumA1-encompassing fragment was digested with KpnI and XbaI, and ligated to the similarly digested pNZ44-strR94. The ligation mixture was introduced into E. coli EC101 by electrotransformation and transformants were selected based on streptomycin resistance. Transformants were checked for plasmid content using colony PCR, restriction analysis of plasmid DNA, and verified by sequencing. Plasmids pNZ44-fumA1-strR and pNZ44-strR were introduced into E. coli EC101 harbouring pNZ-M.BbrII-M.BbrIII by electroporation, as previously described86, and transformants were selected based on Cm and Strep resistance. Methylation of the plasmid complement of transformants by the M.BbrIII (an isoschizomer of a methylase corresponding to the PstI recognition site) was confirmed by their observed resistance to PstI restriction86. Methylated pNZ44-fumA1-strR was introduced by electrotransformation into B. breve UCC2003 and transformants were selected based on streptomycin resistance, generating B. breve strain UCC2003-fumA1. The fucST1T2 fragment was digested with XmaI and XbaI, and ligated to similarly digested pBC1.2 to generate pBC1.2-fucST1T2. The ligation mixture was introduced into E. coli EC101 by electrotransformation and transformants selected based on chloramphenicol resistance. Transformants were checked for plasmid content using colony PCR, restriction analysis of plasmid DNA, and verified by sequencing. Plasmid pBC1.2-fucST1T2 was introduced into B. breve UCC2003-fumA1 by electrotransformation and transformants were selected based on streptomycin and chloramphenicol resistance, generating strain B. breve UCC2003-fumA1-fucST1T2. In addition, the ‘empty’ cloning vector pBC1.2 was introduced into strain B. breve UCC2003-fumA1 (to generate B. breve UCC2003-fumA1-pBC1.2), while ‘empty’ cloning vector pNZ44-strR was introduced into strain B. breve UCC2003-fucST1T2 (to generate B. breve UCC2003-fumST1T2-pNZ44-strR), in both cases to serve as negative controls. All plasmids and strains are listed in Table S3.
Sample preparation for NMR analysis
The culture media samples were thawed, vortexed and allowed to stand for 10 min at room temperature. To 540 μl of culture media, 60 μl of a 1.5 M KH2PO4 buffer (pH 7.4, 100% of deuterium oxide (D2O), 2 mM sodium azide and 1% of TSP (3-trimethylsilyl-[2,2,3,3,-2H4]-propionic acid sodium salt) was added. The mixture was centrifuged at 18,000 × g at 4 °C for 5 s. An aliquot of 580 μL of the supernatant was transferred into a 5 mm outer diameter NMR tube. The samples analysed were the cell-free supernatants of B. kashiwanohense APCKJ1, wild type B. breve UCC2003 or B. breve UCC2003-fumA1-fucST1T2 following 24 h growth in mMRS with 0.05% cysteine-HCL and 1% lactose, L-fucose, 2′-FL or 3-FL. Samples of uninoculated media, containing the respective sugars, as well as without any added substrate, were also analysed as controls.
NMR spectroscopy and data pre-processing
NMR analysis was performed at 300 K on a Bruker 800 MHz spectrometer equipped with a 5 mm CPTCI 1H-13C/15N/D Z-gradient cryoprobe, and automated tuning and matching. 1H NMR spectra were acquired using standard one-dimensional pulse sequence, with water presaturation (noesygppr1d sequence) during both the relaxation delay (RD = 4 s) and mixing time (τm = 10 ms). The receiver gain was set to 16, and acquisition time to 2.73 s for all experiments. Each culture media spectrum was acquired using 4 dummy scans, 32 scans and 64 K data-points with a spectral window set to 20 ppm. Prior to Fourier Transformation, each free induction decay (FID) was multiplied by an exponential function corresponding to a line broadening of 0.3 Hz. 1H NMR spectra were manually phased, baseline corrected and digitized over the range δ −0.5 to 11, and imported into MATLAB (2014a, MathWorks, Natick, U.S.). FIDs were referenced to TSP at δ 0.0 ppm. Spectral regions containing residual water (δ 4.69 to 5.18), TSP (δ −0.50 to 0.77), and noise (δ 10.26 to 11.00) were removed prior to probabilistic quotient normalization95. Principal component analysis (PCA) was applied to the processed Pareto-scaled NMR data using SIMCA software (v. 14.1; Umetrics, Sweden). This model was validated by a 7-fold cross-validation.
Identification of metabolites
Metabolites have signals at specific positions in the 1D 1H NMR frequency domain spectrum and can have multiple peaks providing a fingerprint for each molecule. The multiplicities, chemical shifts and peak area integration of resonance signals were used for structural identification of metabolites. 2D Homonuclear 1H–1H Total Correlation SpectroscopY (TOCSY) and 1H J-resolved (JRES) experiments were acquired for a representative sample, to confirm the presence of metabolites. TOCSY spectra of selected samples were acquired using the MLEV17 spin-lock scheme and a spectral width of 12 KHz in both dimensions and a mixing time of 60 ms. 32 transients per increment (16 dummy scans) were collected into 452 increments with a relaxation delay of 2 s and acquisition time of 0.34 s. The spectrum was processed into a resolution of 16k (F2) and 1k (F1) using a QSINE function. A JRES spectrum was acquired for the same samples using 8 scans (16 dummy scans) per increment and 80 increments with an acquisition time of 0.41 s, and a relaxation delay of 2 s. JRES spectra were processed using a SINE window function in both dimensions with SSB = 0 and a resolution of 16k (F2) and 256 (F1). The peaks were subsequently tilted by 45° and symmetrised. Databases such as the Human Metabolome Data Base (HMDB; http://hmdb.ca/) or the Biological Magnetic Resonance Data Bank (BMRB; http://www.bmrb.wisc.edu) were used for confirmation of assignments. Metabolite concentrations were calculated relative to the CH3 signal of alanine which was consistent for all samples and remained unmodified by any of the bacterial strains.
Bioinformatic analysis
Sequences of proteins identified as upregulated when B. kashiwanohense APCKJ1 was grown on 2′-FL as the sole carbon source were used to search for homologs across type strains of all available Bifidobacterium species, using BLASTP71 at default settings. On-line available genomic data sets of bifidobacteria were first retrieved from the NCBI website (http://www.ncbi.nlm.nih.gov) and aligned using an all-vs-all BLASTP approach96, using cut-off values of 50% of similarity across 50% of protein length and a 0.0001 e-value as a significance for the identification of homologous proteins across all available Bifidobacterium species. The resulting alignment was subsequently clustered in Markov Cluster Algorithm (MCL) families of orthologous genes using the mclblastline algorithm96. The resulting output was used to first build a presence/absence binary matrix, and then the genes of interest were selected and represented in a heatmap employing a code colour grading that represents the degree of sequence similarity, with species ordered by origin of isolation. The products of the genes with locus tags BKKJ1_2072 (designated here as fumF and encoding a putative L2-keto-deoxy-fuconate aldolase), BKKJ1_2073 (fumD encoding a predicted L-fuconolactone hydrolase), BKKJ1_2074 (fumC encoding a putative L-fucose dehydrogenase) and BKKJ1_2075 (fumE predicted to specify a L-fuconate dehydratase) were selected as key enzymes of the fucose utilisation pathway in B. kashiwanohense APCKJ1. A fucose utilisation cluster was established as present across bifidobacterial genomes when all the four genes fumF, fumD, fumC and fumE were found located within the same genomic region with an average similarity of 70% of the four genes at protein level. Automatic protein annotations of the identified clusters were manually checked using the information retrieved from alternative databases such as Pfam (http://pfam.xfam.org) and KEGG (http://www.genome.jp/kegg/). The sequence similarity of these genes and their homologs in bifidobacterial genomes were represented in a heatmap. The identified fucose utilisation clusters identified in B. longum subsp. infantis ATCC 15697, B. pseudocatenulatum DSM 20438 and B. breve UCC2003 were directly compared and represented in a locus homology map.
Protein sequences of eighty three GH29 and GH95 α-fucosidases were retrieved from the Cazy (http://www.cazy.org/Glycoside-Hydrolases.html) and GenBank (https://www.ncbi.nlm.nih.gov/genbank) databases. Phylogenetic inference was performed on the eighty three α-fucosidases, with the E. coli K-12 DnaA protein (b3702) as an outgroup, using the MEGA7 suite (http://www.megasoftware.net/). Protein sequence alignments were performed using the Muscle module available within MEGA7, and the resulting phylogenetic tree was built using the neighbour joining method based on a statistical assessment of 1000 bootstrap replicates. Finally, the iTOL viewer (https://itol.embl.de) was employed in order to visualize and produce the final tree.
Ethics approval and consent to participate
The infant faeces used in this study were collected as part of the INFANTMET study cohort. Mothers were approached for consent between February 2012 and May 2014 in Cork University Maternity Hospital, with ethical approval provided by the Cork University Hospital Research Ethics Committee. Ethical approval reference: ECM (w) 07/02/2012.
Data availability
The genome sequence of B. kashiwanohense APCKJ1 was deposited at the GenBank under Accession No. CP026729. The microarray data obtained in this study have been deposited in NCBI’s Gene Expression Omnibus database and are accessible through GEO Series Accession Number GSE107439.
Change history
09 October 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Engfer, M. B., Stahl, B., Finke, B., Sawatzki, G. & Daniel, H. Human milk oligosaccharides are resistant to enzymatic hydrolysis in the upper gastrointestinal tract. Am J Clin Nutr 71, 1589–1596 (2000).
Kunz, C., Rudloff, S., Baier, W., Klein, N. & Strobel, S. Oligosaccharides in human milk: structural, functional, and metabolic aspects. Annual review of nutrition 20, 699–722, https://doi.org/10.1146/annurev.nutr.20.1.699 (2000).
Brussow, H. Human microbiota: ‘the philosophers have only interpreted the world in various ways. The point, however, is to change it’. Microbial biotechnology 8, 11–12, https://doi.org/10.1111/1751-7915.12259 (2015).
Bezirtzoglou, E., Tsiotsias, A. & Welling, G. W. Microbiota profile in feces of breast- and formula-fed newborns by using fluorescence in situ hybridization (FISH). Anaerobe 17, 478–482, https://doi.org/10.1016/j.anaerobe.2011.03.009 (2011).
Charbonneau, M. R. et al. Sialylated Milk Oligosaccharides Promote Microbiota-Dependent Growth in Models of Infant Undernutrition. Cell 164, 859–871, https://doi.org/10.1016/j.cell.2016.01.024 (2016).
Wang, M. et al. Fecal microbiota composition of breast-fed infants is correlated with human milk oligosaccharides consumed. Journal of pediatric gastroenterology and nutrition 60, 825–833, https://doi.org/10.1097/MPG.0000000000000752 (2015).
Asakuma, S. et al. Physiology of consumption of human milk oligosaccharides by infant gut-associated bifidobacteria. The Journal of biological chemistry 286, 34583–34592, https://doi.org/10.1074/jbc.M111.248138 (2011).
Sela, D. A. & Mills, D. A. Nursing our microbiota: molecular linkages between bifidobacteria and milk oligosaccharides. Trends in microbiology 18, 298–307, https://doi.org/10.1016/j.tim.2010.03.008 (2010).
Sela, D. A. Bifidobacterial utilization of human milk oligosaccharides. International journal of food microbiology 149, 58–64, https://doi.org/10.1016/j.ijfoodmicro.2011.01.025 (2011).
Gibson, G. R. & Roberfroid, M. B. Dietary modulation of the human colonic microbiota: introducing the concept of prebiotics. The Journal of nutrition 125, 1401–1412 (1995).
Bode, L. Human Milk Oligosaccharides at the Interface of Maternal-Infant Health. Breastfeeding medicine: the official journal of the Academy of Breastfeeding Medicine 13, S7–s8, https://doi.org/10.1089/bfm.2018.29073.ljb (2018).
Bottacini, F., van Sinderen, D. & Ventura, M. Omics of bifidobacteria: research and insights into their health-promoting activities. The Biochemical journal 474, 4137–4152, https://doi.org/10.1042/BCJ20160756 (2017).
Turroni, F. et al. Diversity of bifidobacteria within the infant gut microbiota. PloS one 7, e36957, https://doi.org/10.1371/journal.pone.0036957 (2012).
Simone, M. et al. The probiotic Bifidobacterium breve B632 inhibited the growth of Enterobacteriaceae within colicky infant microbiota cultures. BioMed research international 2014, 301053, https://doi.org/10.1155/2014/301053 (2014).
Di Gioia, D., Aloisio, I., Mazzola, G. & Biavati, B. Bifidobacteria: their impact on gut microbiota composition and their applications as probiotics in infants. Applied microbiology and biotechnology 98, 563–577, https://doi.org/10.1007/s00253-013-5405-9 (2014).
Milani, C. et al. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol Mol Biol Rev 81, https://doi.org/10.1128/MMBR.00036-17 (2017).
LoCascio, R. G. et al. Glycoprofiling of bifidobacterial consumption of human milk oligosaccharides demonstrates strain specific, preferential consumption of small chain glycans secreted in early human lactation. Journal of agricultural and food chemistry 55, 8914–8919, https://doi.org/10.1021/jf0710480 (2007).
Ward, R. E., Ninonuevo, M., Mills, D. A., Lebrilla, C. B. & German, J. B. In vitro fermentability of human milk oligosaccharides by several strains of bifidobacteria. Mol Nutr Food Res 51, 1398–1405, https://doi.org/10.1002/mnfr.200700150 (2007).
Kitaoka, M. Bifidobacterial enzymes involved in the metabolism of human milk oligosaccharides. Advances in nutrition (Bethesda, Md.) 3, 422s–429s, https://doi.org/10.3945/an.111.001420 (2012).
Wada, J. et al. Bifidobacterium bifidum lacto-N-biosidase, a critical enzyme for the degradation of human milk oligosaccharides with a type 1 structure. Applied and environmental microbiology 74, 3996–4004, https://doi.org/10.1128/aem.00149-08 (2008).
Moller, P. L., Jorgensen, F., Hansen, O. C., Madsen, S. M. & Stougaard, P. Intra- and extracellular beta-galactosidases from Bifidobacterium bifidum and B. infantis: molecular cloning, heterologous expression, and comparative characterization. Applied and environmental microbiology 67, 2276–2283, https://doi.org/10.1128/aem.67.5.2276-2283.2001 (2001).
Sakurama, H. et al. Lacto-N-biosidase encoded by a novel gene of Bifidobacterium longum subspecies longum shows unique substrate specificity and requires a designated chaperone for its active expression. The Journal of biological chemistry 288, 25194–25206, https://doi.org/10.1074/jbc.M113.484733 (2013).
Miwa, M. et al. Cooperation of beta-galactosidase and beta-N-acetylhexosaminidase from bifidobacteria in assimilation of human milk oligosaccharides with type 2 structure. Glycobiology 20, 1402–1409, https://doi.org/10.1093/glycob/cwq101 (2010).
Ashida, H. et al. Two distinct alpha-L-fucosidases from Bifidobacterium bifidum are essential for the utilization of fucosylated milk oligosaccharides and glycoconjugates. Glycobiology 19, 1010–1017, https://doi.org/10.1093/glycob/cwp082 (2009).
Kiyohara, M. et al. An exo-alpha-sialidase from bifidobacteria involved in the degradation of sialyloligosaccharides in human milk and intestinal glycoconjugates. Glycobiology 21, 437–447, https://doi.org/10.1093/glycob/cwq175 (2011).
Sela, D. A. et al. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc Natl Acad Sci USA 105, 18964–18969, https://doi.org/10.1073/pnas.0809584105 (2008).
Yoshida, E. et al. Bifidobacterium longum subsp. infantis uses two different beta-galactosidases for selectively degrading type-1 and type-2 human milk oligosaccharides. Glycobiology 22, 361–368, https://doi.org/10.1093/glycob/cwr116 (2012).
Garrido, D., Ruiz-Moyano, S. & Mills, D. A. Release and utilization of N-acetyl-D-glucosamine from human milk oligosaccharides by Bifidobacterium longum subsp. infantis. Anaerobe 18, 430–435, https://doi.org/10.1016/j.anaerobe.2012.04.012 (2012).
Xiao, J. Z. et al. Distribution of in vitro fermentation ability of lacto-N-biose I, a major building block of human milk oligosaccharides, in bifidobacterial strains. Applied and environmental microbiology 76, 54–59, https://doi.org/10.1128/aem.01683-09 (2010).
James, K., Motherway, M. O., Bottacini, F. & van Sinderen, D. Bifidobacterium breve UCC2003 metabolises the human milk oligosaccharides lacto-N-tetraose and lacto-N-neo-tetraose through overlapping, yet distinct pathways. Scientific reports 6, 38560, https://doi.org/10.1038/srep38560 (2016).
Sela, D. A. et al. An infant-associated bacterial commensal utilizes breast milk sialyloligosaccharides. The Journal of biological chemistry 286, 11909–11918, https://doi.org/10.1074/jbc.M110.193359 (2011).
Sela, D. A. et al. Bifidobacterium longum subsp. infantis ATCC 15697 alpha-fucosidases are active on fucosylated human milk oligosaccharides. Applied and environmental microbiology 78, 795–803, https://doi.org/10.1128/AEM.06762-11 (2012).
Bunesova, V., Lacroix, C. & Schwab, C. Fucosyllactose and L-fucose utilization of infant Bifidobacterium longum and Bifidobacterium kashiwanohense. BMC microbiology 16, 248, https://doi.org/10.1186/s12866-016-0867-4 (2016).
Vazquez-Gutierrez, P. et al. Bifidobacteria strains isolated from stools of iron deficient infants can efficiently sequester iron. BMC microbiology 15, 3, https://doi.org/10.1186/s12866-014-0334-z (2015).
Morita, H. et al. Bifidobacterium kashiwanohense sp. nov., isolated from healthy infant faeces. International journal of systematic and evolutionary microbiology 61, 2610–2615, https://doi.org/10.1099/ijs.0.024521-0 (2011).
McGuire, M. K. et al. What’s normal? Oligosaccharide concentrations and profiles in milk produced by healthy women vary geographically. The American journal of clinical nutrition 105, 1086–1100, https://doi.org/10.3945/ajcn.116.139980 (2017).
Bode, L. Human milk oligosaccharides: every baby needs a sugar mama. Glycobiology 22, 1147–1162, https://doi.org/10.1093/glycob/cws074 (2012).
Matsuki, T. et al. A key genetic factor for fucosyllactose utilization affects infant gut microbiota development. Nature communications 7, 11939, https://doi.org/10.1038/ncomms11939 (2016).
Schwab, C. et al. Trophic Interactions of Infant Bifidobacteria and Eubacterium hallii during L-Fucose and Fucosyllactose Degradation. Front Microbiol 8, 95, https://doi.org/10.3389/fmicb.2017.00095 (2017).
Engels, C., Ruscheweyh, H. J., Beerenwinkel, N., Lacroix, C. & Schwab, C. The Common Gut Microbe Eubacterium hallii also Contributes to Intestinal Propionate Formation. Front Microbiol 7, 713, https://doi.org/10.3389/fmicb.2016.00713 (2016).
Rada, V. Detection of Bifidobacterium species by enzymatic methods and antimicrobial susceptibility testing. Biotechnology Techniques 11, 909–912 (1997).
Simpson, P. J., Fitzgerald, G. F., Stanton, C. & Ross, R. P. The evaluation of a mupirocin-based selective medium for the enumeration of bifidobacteria from probiotic animal feed. Journal of microbiological methods 57, 9–16, https://doi.org/10.1016/j.mimet.2003.11.010 (2004).
Pokusaeva, K., Fitzgerald, G. F. & van Sinderen, D. Carbohydrate metabolism in Bifidobacteria. Genes &. nutrition 6, 285–306, https://doi.org/10.1007/s12263-010-0206-6 (2011).
Sela, D. A. et al. Bifidobacterium longum subsp. infantis ATCC 15697 α-Fucosidases Are Active on Fucosylated Human Milk Oligosaccharides. Applied and environmental microbiology 78, 795–803, https://doi.org/10.1128/AEM.06762-11 (2012).
Katayama, T. et al. Molecular cloning and characterization of Bifidobacterium bifidum 1,2-alpha-L-fucosidase (AfcA), a novel inverting glycosidase (glycoside hydrolase family 95). Journal of bacteriology 186, 4885–4893, https://doi.org/10.1128/jb.186.15.4885-4893.2004 (2004).
Fan, S. et al. Cloning, characterization, and production of three alpha-L-fucosidases from Clostridium perfringens ATCC 13124. Journal of basic microbiology 56, 347–357, https://doi.org/10.1002/jobm.201500582 (2016).
Lammerts van Bueren, A., Popat, S. D., Lin, C. H. & Davies, G. J. Structural and thermodynamic analyses of alpha-L-fucosidase inhibitors. Chembiochem: a European journal of chemical biology 11, 1971–1974, https://doi.org/10.1002/cbic.201000339 (2010).
Sakurama, H. et al. Differences in the substrate specificities and active-site structures of two alpha-L-fucosidases (glycoside hydrolase family 29) from Bacteroides thetaiotaomicron. Bioscience, biotechnology, and biochemistry 76, 1022–1024, https://doi.org/10.1271/bbb.111004 (2012).
Guillotin, L., Lafite, P. & Daniellou, R. Unraveling the substrate recognition mechanism and specificity of the unusual glycosyl hydrolase family 29 BT2192 from Bacteroides thetaiotaomicron. Biochemistry 53, 1447–1455, https://doi.org/10.1021/bi400951q (2014).
Sulzenbacher, G. et al. Crystal structure of Thermotoga maritima alpha-L-fucosidase. Insights into the catalytic mechanism and the molecular basis for fucosidosis. The Journal of biological chemistry 279, 13119–13128, https://doi.org/10.1074/jbc.M313783200 (2004).
Egan, M. et al. Cross-feeding by Bifidobacterium breve UCC2003 during co-cultivation with Bifidobacterium bifidum PRL2010 in a mucin-based medium. BMC microbiology 14, 282, https://doi.org/10.1186/s12866-014-0282-7 (2014).
Ruiz-Moyano, S. et al. Variation in consumption of human milk oligosaccharides by infant gut-associated strains of Bifidobacterium breve. Applied and environmental microbiology 79, 6040–6049, https://doi.org/10.1128/AEM.01843-13 (2013).
O’Connell Motherway, M., Kinsella, M., Fitzgerald, G. F. & van Sinderen, D. Transcriptional and functional characterization of genetic elements involved in galacto-oligosaccharide utilization by Bifidobacterium breve UCC2003. Microb Biotechnol 6, 67–79, https://doi.org/10.1111/1751-7915.12011 (2013).
Yew, W. S. et al. Evolution of enzymatic activities in the enolase superfamily: L-fuconate dehydratase from Xanthomonas campestris. Biochemistry 45, 14582–14597, https://doi.org/10.1021/bi061687o (2006).
Bottacini, F. et al. Global transcriptional landscape and promoter mapping of the gut commensal Bifidobacterium breve UCC2003. BMC Genomics 18, 991, https://doi.org/10.1186/s12864-017-4387-x (2017).
Turroni, F. et al. Analysis of predicted carbohydrate transport systems encoded by Bifidobacterium bifidum PRL2010. Applied and environmental microbiology 78, 5002–5012, https://doi.org/10.1128/AEM.00629-12 (2012).
Khoroshkin, M. S., Leyn, S. A., Van Sinderen, D. & Rodionov, D. A. Transcriptional Regulation of Carbohydrate Utilization Pathways in the Bifidobacterium Genus. Front Microbiol 7, 120, https://doi.org/10.3389/fmicb.2016.00120 (2016).
Turroni, F. et al. Bifidobacterium bifidum as an example of a specialized human gut commensal. Front Microbiol 5, 437, https://doi.org/10.3389/fmicb.2014.00437 (2014).
Benno, Y., Sawada, K. & Mitsuoka, T. The intestinal microflora of infants: composition of fecal flora in breast-fed and bottle-fed infants. Microbiology and immunology 28, 975–986 (1984).
Guaraldi, F. & Salvatori, G. Effect of breast and formula feeding on gut microbiota shaping in newborns. Frontiers in cellular and infection microbiology 2, 94, https://doi.org/10.3389/fcimb.2012.00094 (2012).
Solis, G., Reyes-Gavilan, deL., Fernandez, C. G., Margolles, N. & Gueimonde, A. M. Establishment and development of lactic acid bacteria and bifidobacteria microbiota in breast-milk and the infant gut. Anaerobe 16, 307–310, https://doi.org/10.1016/j.anaerobe.2010.02.004 (2010).
LoCascio, R. G., Desai, P., Sela, D. A., Weimer, B. & Mills, D. A. Broad conservation of milk utilization genes in Bifidobacterium longum subsp. infantis as revealed by comparative genomic hybridization. Applied and environmental microbiology 76, 7373–7381, https://doi.org/10.1128/aem.00675-10 (2010).
Zivkovic, A. M., German, J. B., Lebrilla, C. B. & Mills, D. A. Human milk glycobiome and its impact on the infant gastrointestinal microbiota. Proc Natl Acad Sci USA 108(Suppl 1), 4653–4658, https://doi.org/10.1073/pnas.1000083107 (2011).
Garrido, D. et al. A novel gene cluster allows preferential utilization of fucosylated milk oligosaccharides in Bifidobacterium longum subsp. longum SC596. Scientific reports 6, 35045, https://doi.org/10.1038/srep35045 (2016).
Casazza, J. P., Song, B. J. & Veech, R. L. Short chain diol metabolism in human disease states. Trends in biochemical sciences 15, 26–30 (1990).
Jackson, F. Metabolic phenotyping and metagenomic analysis of developing infants. Doctor of Philosophy (PhD) thesis, Imperial College London (2016).
Hill, C. J. et al. Evolution of gut microbiota composition from birth to 24 weeks in the INFANTMET Cohort. Microbiome 5, 4, https://doi.org/10.1186/s40168-016-0213-y (2017).
Watson, D. et al. Selective carbohydrate utilization by lactobacilli and bifidobacteria. Journal of applied microbiology 114, 1132–1146, https://doi.org/10.1111/jam.12105 (2013).
Scardovi, V. Genus Bifidobacterium. 1418–1434 (Williams and Wilkins: Baltimore, Md., 1986).
Turroni, F. et al. Exploring the diversity of the bifidobacterial population in the human intestinal tract. Applied and environmental microbiology 75, 1534–1545, https://doi.org/10.1128/AEM.02216-08 (2009).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J Mol Biol 215, 403–410, https://doi.org/10.1016/s0022-2836(05)80360-2 (1990).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic acids research 35, 3100–3108, https://doi.org/10.1093/nar/gkm160 (2007).
Rutherford, K. et al. Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945 (2000).
Morita, H. et al. Complete Genome Sequence of Bifidobacterium kashiwanohense JCM 15439 T, Isolated from Feces from a Healthy Japanese Infant. Genome announcements 3, https://doi.org/10.1128/genomeA.00255-15 (2015).
Vazquez-Gutierrez, P. et al. Complete and Assembled Genome Sequence of Bifidobacterium kashiwanohense PV20-2, Isolated from the Feces of an Anemic Kenyan Infant. Genome announcements 3, https://doi.org/10.1128/genomeA.01467-14 (2015).
O’Connell Motherway, M. et al. Functional genome analysis of Bifidobacterium breve UCC2003 reveals type IVb tight adherence (Tad) pili as an essential and conserved host-colonization facto. r. Proceedings of the National Academy of Sciences of the United States of America 108, 11217–11222, https://doi.org/10.1073/pnas.1105380108 (2011).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular systems biology 7, 539, https://doi.org/10.1038/msb.2011.75 (2011).
Terzaghi, B. E. & Sandine, W. E. Improved medium for lactic streptococci and their bacteriophages. Appl Microbiol 29, 807–813 (1975).
Sambrook, J. F. E. & Maniatis, T. Molecular cloning: a laboratory manual, 2nd ed., (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY 1989).
Zomer, A. et al. An interactive regulatory network controls stress response in Bifidobacterium breve UCC2003. Journal of bacteriology 191, 7039–7049, https://doi.org/10.1128/jb.00897-09 (2009).
Garcia de la Nava, J. et al. Engene: the processing and exploratory analysis of gene expression data. Bioinformatics 19, 657–658 (2003).
van Hijum, S. A., García de la Nava, J., Trelles, O., Kok, J. & Kuipers, O. P. MicroPreP: a cDNA microarray data pre-processing framework. Applied Bioinformatics 2, 241–244 (2003).
van Hijum, S. A. et al. A generally applicable validation scheme for the assessment of factors involved in reproducibility and quality of DNA-microarray data. BMC genomics 6, 77, https://doi.org/10.1186/1471-2164-6-77 (2005).
Long, A. D. et al. Improved statistical inference from DNA microarray data using analysis of variance and a Bayesian statistical framework. Analysis of global gene expression in Escherichia coli K12. The Journal of biological chemistry 276, 19937–19944, https://doi.org/10.1074/jbc.M010192200 (2001).
O’Riordan, K. & Fitzgerald, G. F. Evaluation of bifidobacteria for the production of antimicrobial compounds and assessment of performance in cottage cheese at refrigeration temperatur. e. Journal of applied microbiology 85, 103–114 (1998).
O’Connell Motherway, M., O’Driscoll, J., Fitzgerald, G. F. & Van Sinderen, D. Overcoming the restriction barrier to plasmid transformation and targeted mutagenesis in Bifidobacterium breve UCC2003. Microb Biotechnol 2, 321–332, https://doi.org/10.1111/j.1751-7915.2008.00071.x (2009).
Law, J. et al. A system to generate chromosomal mutations in Lactococcus lactis which allows fast analysis of targeted genes. Journal of bacteriology 177, 7011–7018 (1995).
Mierau, I. & Kleerebezem, M. 10 years of the nisin-controlled gene expression system (NICE) in Lactococcus lactis. Applied microbiology and biotechnology 68, 705–717, https://doi.org/10.1007/s00253-005-0107-6 (2005).
O’Connell, K. J. et al. Metabolism of four alpha-glycosidic linkage-containing oligosaccharides by Bifidobacterium breve UCC2003. Applied and environmental microbiology 79, 6280–6292, https://doi.org/10.1128/aem.01775-13 (2013).
O’Connell Motherway, M., Fitzgerald, G. F. & van Sinderen, D. Metabolism of a plant derived galactose-containing polysaccharide by Bifidobacterium breve UCC2003. Microbial biotechnology 4, 403–416, https://doi.org/10.1111/j.1751-7915.2010.00218.x (2011).
Pokusaeva, K. et al. Cellodextrin utilization by bifidobacterium breve UCC2003. Applied and environmental microbiology 77, 1681–1690, https://doi.org/10.1128/aem.01786-10 (2011).
de Ruyter, P. G., Kuipers, O. P. & de Vos, W. M. Controlled gene expression systems for Lactococcus lactis with the food-grade inducer nisin. Applied and environmental microbiology 62, 3662–3667 (1996).
Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical biochemistry 72, 248–254 (1976).
Bottacini, F. et al. Comparative genomics and genotype-phenotype associations in Bifidobacterium breve. Scientific reports 8, 10633, https://doi.org/10.1038/s41598-018-28919-4 (2018).
Dieterle, F., Ross, A., Schlotterbeck, G. & Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1 H NMR metabonomics. Analytical chemistry 78, 4281–4290, https://doi.org/10.1021/ac051632c (2006).
Enright, A. J., Van Dongen, S. & Ouzounis, C. A. An efficient algorithm for large-scale detection of protein families. Nucleic acids research 30, 1575–1584 (2002).
James, K. Metabolism of human milk oligosaccharides by infant-associated bifidobacteria. Doctorate of Philosophy (PhD) thesis, University College Cork (2018).
Acknowledgements
The authors would like to sincerely thank Glycom A/S (Hoersholm, Denmark) for the provision of purified HMO samples used in this study under their donation program. The authors gratefully acknowledge the financial support of this work by the Mizutani Foundation for Glycoscience (ref no. 190049). This study was funded in part by the Irish Research Council, under the Postgraduate Research Project Award; Project ID GOIPG/2013/651. In addition, the authors are supported by Science Foundation Ireland (SFI) (Grant No. SFI/12/RC/2273) and Mary O’Connell Motherway is a recipient of a HRB postdoctoral fellowship (Grant No. PDTM/20011/9). Francesca Bottacini is a recipient of a FEMS Research Grant (FEMS-RG-2016-0103) and FEMS/ESCMID Award. Elaine Holmes and Jose Ivan Serrano Contreras are supported by the National Institute for Health Research (NIHR) Imperial Biomedical Research Centre (BRC) within the Gut Health Theme.
Author information
Authors and Affiliations
Contributions
D.v.S., K.J., E.H. and M.O.C.M. conceived the experiments. K.J. conducted the experiments. K.J. and M.V. carried out the faecal screening. M.E. assisted with microarray design. J.I.S.C. and E.H. carried out the metabolite analysis experiments. F.B. conducted the bioinformatic analysis. K.J., D.v.S. and F.B. wrote the manuscript. All authors analysed the results and contributed at some stage to the writing of the manuscript. Part of the work in this publication has been previously published as a part of the thesis entitled “Metabolism of human milk oligosaccharides by infant-associated bifidobacteria”97. This includes Figure(s) 1, 2, 3, 4 and 5, and Supplementary Figure S1, which have been adapted from the thesis figures.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
James, K., Bottacini, F., Contreras, J.I.S. et al. Metabolism of the predominant human milk oligosaccharide fucosyllactose by an infant gut commensal. Sci Rep 9, 15427 (2019). https://doi.org/10.1038/s41598-019-51901-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-51901-7
This article is cited by
-
Fungal β-glucan-facilitated cross-feeding activities between Bacteroides and Bifidobacterium species
Communications Biology (2023)
-
Priority effects shape the structure of infant-type Bifidobacterium communities on human milk oligosaccharides
The ISME Journal (2022)
-
Human milk oligosaccharide-sharing by a consortium of infant derived Bifidobacterium species
Scientific Reports (2022)
-
Key bacterial taxa and metabolic pathways affecting gut short-chain fatty acid profiles in early life
The ISME Journal (2021)
-
Strain-specific strategies of 2′-fucosyllactose, 3-fucosyllactose, and difucosyllactose assimilation by Bifidobacterium longum subsp. infantis Bi-26 and ATCC 15697
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
