
Abstract
Tuberculosis remains the world’s leading cause of death from an infectious agent, and is a serious health problem in Papua New Guinea (PNG) with an estimated 36,000 new cases each year. This study describes the genetic diversity of Mycobacterium tuberculosis among tuberculosis patients in the Balimo/Bamu region in the Middle Fly District of Western Province in PNG, and investigates rifampicin resistance-associated mutations. Archived Ziehl-Neelsen-stained sputum smears were used to conduct microbead-based spoligotyping and assess genotypic resistance. Among the 162 samples included, 80 (49.4%) generated spoligotyping patterns (n = 23), belonging predominantly to the L2 Lineage (44%) and the L4 Lineage (30%). This is consistent with what has been found in other PNG regions geographically distant from Middle Fly District of Western Province, but is different from neighbouring South-East Asian countries. Rifampicin resistance was identified in 7.8% of the successfully sequenced samples, with all resistant samples belonging to the L2/Beijing Lineage. A high prevalence of mixed L2/L4 profiles was suggestive of polyclonal infection in the region, although this would need to be confirmed. The method described here could be a game-changer in resource-limited countries where large numbers of archived smear slides could be used for retrospective (and prospective) studies of M. tuberculosis genetic epidemiology.
Similar content being viewed by others


Introduction
Papua New Guinea (PNG) is one of the ten high tuberculosis (TB) burden countries worldwide with a TB notification rate of 333 per 100,000 people (n = 28,244) in 20161. The World Health Organization (WHO)-estimated incidence rate was 432 cases per 100,000 population (95% CI 352 to 521) in 2017, accounting for 36,000 new cases and 4,300 deaths2. The TB burden can be even higher in rural areas of PNG, with estimated incidence rates of 1,290 cases per 100,000 population in Gulf Province and 550 cases per 100,000 in Western Province (WP)3,4. Considering that HIV (a major driver of TB in sub-Saharan Africa) accounts for only about 5–10% of TB cases in PNG, these estimated incidence rates are remarkably high4. PNG is also considered to have a high burden of rifampicin-resistant TB (RR-TB) and multidrug-resistant TB (MDR-TB), defined as resistance to at least rifampicin (RIF) and isoniazid (INH)2. In 2017, the proportion of RR-TB and MDR-TB (referred to as MDR/RR-TB) was estimated to be 3.4% of new TB cases and 26% of retreatment cases2.
Comparing the genetic diversity of Mycobacterium tuberculosis (Mtb) within and between different populations provides insights into TB dynamics at different levels, and may help improve control strategies developed by national TB control programs. Seven lineages of human adapted M. tuberculosis complex (MTBC) have been identified5. The so-called “modern lineages” (Lineages 2, 3 and 4) are widespread, while the “ancient” (Lineages 1, 5 and 6) show spatial heterogeneity6. Modern lineages have been suggested to have increased virulence or transmissibility, which may have evolved as a response to co-evolutionary interactions with particular human populations, or to mass BCG vaccination and use of antituberculosis drugs7. In PNG, molecular epidemiology studies based on MTBC clinical isolates are scarce. Two studies, published in 20128 and in 20149 investigated the diversity of Mtb in different PNG provinces with cultured samples using spoligotyping, a method using the CRISPR locus (clustered regularly interspaced short palindromic repeats) as a target for Mtb subtyping10. Mtb isolates originating from adult TB patients from Madang and surrounds belonged predominantly to Lineage 4 and Lineage 2, and 44% of isolates were molecularly clustered, suggesting active Mtb transmission in the community8. Spoligotyping of Mtb isolates from Goroka (Eastern Highlands), Madang (Madang Province) and Alotau (Milne Bay) revealed L4, L2, and L1 Lineages as the main Mtb lineages at all three sites9. More recent studies also used variable number of tandem repeats (VNTR) and whole genome sequencing (WGS) to understand the genetic diversity of circulating strains, and revealed drug resistance-associated mutations on Daru Island, in the South Fly District WP, PNG11,12. In Daru, a cluster of MDR/RR-TB (n = 95) was identified as belonging to a single L2/Beijing sub-lineage 2.2.1.112. Nevertheless, the diversity of Mtb in PNG remains largely unexplored, and no information is available from the Middle Fly District WP.
Overall, facilities for culturing clinical isolates and testing them for drug susceptibility are lacking in PNG. In the Middle Fly District WP, the catchment area for Balimo District Hospital (BDH) primarily includes the population of the ‘Balimo Urban’ and ‘Gogodala Rural’ local level government (LLG) areas, and to a lesser degree the ‘Bamu Rural’ LLG. This area is hereafter referred to as the “Balimo/Bamu region”. BDH does not have facilities for performing Mtb culture and is not equipped with a GeneXpert (Cepheid) instrument, so TB diagnosis relies primarily on clinical presentation and direct sputum smear microscopy, based on the PNG National Tuberculosis Management Protocol13,14,15. DNA extracted from Ziehl-Neelsen (ZN) stained sputum smears has already proven useful in molecularly confirming the microscopy diagnosis of TB in this region of PNG14, and spoligotyping has been undertaken successfully on DNA extracted from cultures10,16, from decontaminated sputa17, as well as from ZN-stained sputum smears18,19,20,21. Direct sputum smear microscopy is routinely performed in diagnostic laboratories worldwide, including in developing countries, and stained slides can be stored and transported at room temperature, which makes them a prime target for spoligotyping of Mtb samples collected in remote settings21. Spoligotyping has been historically performed by reverse hybridization on a nylon membrane22 but microbead-based systems improve throughput and sensitivity10,23. We performed microbead-based spoligotyping and molecular analysis of ZN-stained sputum smears collected in the Balimo/Bamu region in the Middle Fly District WP, PNG, in order to (i) assess the efficacy of the spoligotyping method when using archived samples collected under routine programmatic conditions in a remote location, (ii) analyse the genetic diversity and distribution of Mtb, and (iii) identify RIF-resistant TB among newly diagnosed TB patients.
Results
Clinical sample set
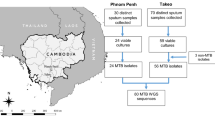
A total of 345 smeared sputa were collected from June 2012 to March 2014, of which 206 showed evidence of mycobacterial DNA based on combined TaqMan qPCR assays (see14 for details). Out of those 206 qPCR-positive samples, the spoligotyping and resistance analyses focused on the ones with the lowest Cq values after qPCR, with a cutoff of 36 chosen from preliminary spoligotyping results. Samples included in the analyses (n = 162) are summarised in Table 1, with their detailed microscopy results. The excluded samples (n = 44; Cq > 36) were mostly negative in microscopy (36 negative, 3 scanty, 5 unknown).
Genotypic diversity of the clinical isolates
Spoligotyping of Mtb was successfully conducted in 49.4% (80/162) samples (Table 1). Interpretable spoligotyping results were obtained for 92% (57/62) of the sputum smears showing ‘3+’ and ‘2+’ microscopy results; the spoligotyping success dropped to 41% (12/29) for the sputum smears with ‘1+’ and ‘scanty’ microscopy results. Three of seven worldwide reported Mtb lineages were detected in the Balimo/Bamu region (Table 1). Among the 80 independent samples with successful genotyping, modern Lineages L2 and L4 were most prevalent, representing 44% (35/80) and 30% (24/80) of the samples respectively. In contrast, ancient L1 Lineage was very rare (1%; 1/80). Nine spoligotype profiles were atypical (only one reported in SITVIT2, one reported in Madang, PNG, and all giving contradictory labellings with TBminer), and careful exploration of raw signals showed intermediate values for several spacers, especially sp35 and sp36 that are absent in L4 Lineage and present in L2 Lineage. These mixed patterns likely corresponded to mixed infections of L2 and L4 strains as reported in other studies (see Discussion) and represented 25% (20/80) of the samples (Table 1). When excluding ‘mixed’ samples, the prevalences per lineage were 2% (1/60) for L1, 58% (35/60) for L2, and 40% (24/60) for L4.
Twenty-three different spoligotyping patterns belonged to 13 different families and five clusters with two or more samples (Fig. 1). The largest clusters were L2/Beijing SIT1 (n = 34), one new L4 pattern (likely L4.2/Ural1 according to TBminer) (n = 9), and one pattern labelled as ‘mixed’ (n = 11). Of the 23 patterns identified, eight patterns (n = 44 samples) have already been described in PNG, including four patterns described in the two previous PNG studies8,9 as well as ours: L2/Beijing SIT1, L4/H3 SIT50, L4/T1 SIT53 and L4/T1 SIT393 (Fig. 1). The other 15 patterns (n = 36 samples) were “new” in PNG. Of note, eight patterns were detected in the two previous PNG studies8,9 but were not identified in the Balimo/Bamu region, including L4/X1 SIT119 and L4/LAM9 SIT42 that each accounted for 6% of spoligotypes in Madang8.

Spoligotype patterns and lineages detected in our study and in previous studies from different provinces in PNG. The present study focuses on patients from the Balimo/Bamu region (Middle Fly District of Western Province), all diagnosed at the Balimo District hospital. The Baillif et al. 2012 study8 focuses on patients from Madang Province, East Sepik Province and “other”, all diagnosed at the main hospital of Madang Province (Modilon General Hospital). The Ley et al. 2014 study9 focuses on patients from Madang (Madang Province), Goroka (Eastern Highlands Province) and Alotau (Milne Bay Province).
Spatial analysis of Mtb lineages in PNG
Figure 2 shows the distribution of Mtb diversity according to previous investigations in PNG (Fig. 2B) and the present study (Fig. 2B,C). L2 and L4 Lineages were present in all PNG settings, while L1 Lineage was detected in samples from three locations out of the five regions investigated, including the Balimo/Bamu region where it was detected in one sample (Fig. 2C). However, the relative percentage of each lineage varied between locations at the national level (Fig. 2B) as well as at the regional level (Fig. 2C). In the present study, the biggest spatial cluster (n = 8) corresponds to the town of Balimo (Fig. 2C).

Spatial distribution of Mtb spoligotyping-defined lineages in Papua New Guinea. (A) Localization of previous study settings, as well as the present study in the Balimo/Bamu region, Middle Fly District of Western Province (red square). (B) Distribution of Mtb lineages at the regional level in Goroka, Alotau, Madang9, Madang Province and East Sepik Province8 and the Balimo/Bamu region (this study). (C) Distribution of Mtb lineages at the subregional level in the Balimo/Bamu region. Size of circle is proportional to the number of TB cases (n). Local level government (LLG) areas are delimited in grey lines, with TB cases in (C) located in the Gogodala Rural (southwest, including the Balimo Urban LLG) and Bamu Rural (northeast) LLGs. Undet: undetermined (including intermediate L2/L4 patterns; see main text for further explanation).
RIF-resistance associated mutations
RIF-resistance was assessed by sequencing a 147-bp region of the rpoB gene (including RRDR). Out of the 162 qPCR-positive sputum smears included in the analyses, 116 (71.6%) were successfully sequenced (Table 2). Mutations were identified in 7.8% (95% CI 2.9–12.6%) of the successfully sequenced samples, and all corresponded to the rpoB S450L codon mutation (Table 2). Of note, one sample that tested positive for mycobacterial DNA based on qPCR resulted in a rpoB sequence originating from Pseudomonas aeruginosa. Considering that this sample showed a late positive qPCR (Cq 33.3 and 35.5), a negative microscopy result, but a ‘1 +’ microscopy result on another slide from the same patient sampled the previous month, we believe this patient to be a true but low MTBC positive and a mixed sample containing MTBC and Pseudomonas.
Among the 79 sputum smears with both rpoB and spoligotyping results, 72 were rpoB wild type and seven were S450L mutants, with all the RIF-resistant samples belonging to the L2/Beijing Lineage (Fig. S1). Drug resistance was thus linked with Lineage 2 (Fisher exact test p = 0.003), as already observed in a study conducted at Modilon General Hospital, Madang Province8.
Discussion
The spoligotyping method is known to have limited discriminatory capacity due to the fact that it targets only a single genetic locus, covering less than 0.1% of the Mtb genome, and when used alone, is not sufficient for epidemiological linking studies24. Nevertheless, spoligotyping allows identification of most if not all genotypes of significant, clinical, and epidemiological relevance, such as the “Beijing” genotype24. In this study, we used spoligotyping to describe the genetic diversity of Mtb from patients presenting at BDH, originating from the Middle Fly District WP, PNG. We show a wide diversity of spoligotype patterns in the Balimo/Bamu region, and confirm the presence of the modern L2 and L4 Lineages, with a single ancient L1 Lineage case, originating from the Bamu LLG. We also analysed drug-resistance associated mutations in the rpoB gene, and identified 7.8% of the successfully sequenced samples showing RIF resistance, all being L2/Beijing Lineage isolates.
Populations in the Highlands are the oldest settlers of PNG, as shown by human genetic studies25, and have been isolated from the outside world for much longer than the populations in coastal sites. For this reason it was expected that evolutionary ancient lineages of Mtb (e.g. L1 Lineage) would be found in the low density populations of the Highlands, as opposed to modern lineages of Mtb (L2 and L4 Lineages) that would be found in the high density populations of the coastal regions9. However, a recent human genetic study determined that the genetic differentiation between populations from highland and lowland PNG does not seem to be as significant as previously thought26, and no statistically significant difference in the prevalence of L1 Lineage could be found in Goroka, Eastern Highlands, compared to coastal regions9. In the end, the pattern of dominance of modern lineages over ancient L1 Lineage seems consistent across PNG, with the 2% of L1 Lineage found in the Balimo/Bamu region not dissimilar to the 0.6% (1/173) to 8% (3/38) observed in the North Coast (East Sepik, Madang), Goroka or Alotau8,9. This pattern is strikingly different from what has been described in neighbouring South-East Asian countries. In Western New Guinea (WNG), i.e. the Indonesian part of the island, the same three lineages have been reported, but with a higher proportion of L1 Lineage (33.7%)27. This is also true in other countries of the region, with L1 Lineage representing 30.2% of samples in Makassar, Indonesia28, 56.4% in Malaysia29, and 36.3% in southern Vietnam30. This might be due to different importation histories of TB strains in PNG compared to the rest of South-East Asia. For example, it has been suggested that ancient TB lineages might have been replaced early on by modern and more successful lineages with the arrival of Europeans in PNG in the late 19th century31.
In L4 Lineage, Latin-American and Mediterreanean (LAM) families (Lineage 4.3) were the most prevalent in WNG, as compared to PNG where other L4 families were found in high proportion in most of the locations. Interestingly, L4/SIT393 is reported in the three independent studies performed so far in PNG. L4/SIT393 is characterized by the absence of spacer sp14, a character that has not yet been shown to be monophyletic. A total of 60 cases have now been reported in PNG (Fig. 1), whereas until now, only 36 cases were described worldwide in SITVIT232. The largest and best collection of L4 Lineage characterized so far in PNG has been described by Stucki and colleagues33 that report, within the L4 Lineage, up to 40% of L4.10/PGG3 (Principal Genetic Group 3) strains, characterized by the katG463-CGG (Arg) plus gyrA95-AGC (Ser) combination and the Rv1501 C- > A allelic change34,35. We suggest that L4/SIT393 isolated in PNG could belong to the L4.10/PGG3 lineage. Future work should focus on SNP characterization of these isolates in PNG, and related analyses should shed interesting light into diversity at the sublineage level.
An interesting result from our analysis was the likely identification of many mixed (or polyclonal) infections, which are infections with several different genotypes/strains of Mtb in a single patient36. In our study, many spoligotype patterns could be assigned as MANU2 in SITVIT2, but have been assigned as “mixed” patterns in our analysis, with strains harboring superimpositions of L2 and L4 patterns (Table 1), and seven orphans showing similar patterns. It has been suggested that mixed infections in clinical specimens can challenge TB spoligotyping and sequencing techniques, and might have been the cause of false spoligotypes previously reported37,38. MANU2 spoligotype has been suggested to be one of the five genotypes derived from mixed infections in clinical specimens39. Studies showing the prevalence of MANU2 and discussing a link between MANU2 and mixed infection have been conducted in Iran37, Mozambique40, and Sudan41. The study in Mozambique concluded that some of the MANU2 genotypes could be derived from mixed infections of L2/Beijing and L4/T1, or L2/Beijing and L4/T2. Further studies on samples showing “mixed” patterns are required to more fully establish whether polyclonal infections (from either coinfection or superinfection) are present in the Balimo/Bamu region of PNG. This information may be epidemiologically relevant as it may indicate high contact rates of individuals with diverse Mtb strains.
Typical L2/Beijing strains are reported at high frequency worldwide and have been associated with drug resistance and increased transmissibility42,43. Sequencing of a 147-bp rpoB region identified seven samples as RIF-resistant, all presenting the rpoB S450L codon mutation. This is in agreement with previous findings in the Balimo/Bamu region WP44 and in South Fly District WP12 and other provinces in PNG8,9. All RIF-resistant samples belonged to the L2/Beijing Lineage, as found in Vietnam30,45. Molecular epidemiological studies have suggested that certain Mtb genotypes are more successful than others, including some L2/Beijing genotypes that have a higher fitness and have evolved unique pathogenic characteristics42,46,47. Phylogenetic analysis has provided evidence that the more recently diversified strains (so-called modern/typical Beijing L2.3) are adapted to spread and cause disease, compared to other sublineages (so-called ancient/atypical Beijing L2.2 or proto-Beijing, L2.1)48. On Daru Island in the South Fly District WP, a drug-resistant TB outbreak was recently characterized using WGS and was found to be driven by a Beijing L2.2.1.1 strain12, corresponding to a modern Beijing L2.3 in the latest classification from Liu and colleagues48. Unfortunately, our study did not provide any information at the sublineage level regarding the L2/Beijing strains. To complement our spoligotyping, further genomic studies of this sublineage in the Balimo/Bamu region of PNG will be needed to assess the presence of super-spreaders, association with drug resistance, or any other genetic signature of epidemiological interest.
Our study confirms the limitation that clean spoligotyping results can be obtained mostly from AFB 3+ and 2+ slides. Nevertheless, the collected information about genetic diversity is invaluable for better understanding the epidemiology of TB, and archived smear slides are of sufficient quality to collect such data, as seen in this study. It is likely that prior targeted enrichment techniques may have improved our direct genotyping results from sputum smears49,50. However, such methods are not yet appropriate to local processing of samples. Before more sensitive methods can be introduced in countries such as PNG, huge numbers of archived smear slides could be used for retrospective and prospective studies of Mtb diversity.
In summary we demonstrate that, in countries where conservation of samples might be an issue, and/or hospital laboratories do not have the capacity for culture or Xpert MTB/RIF-based diagnosis of TB, uncultured biological material could be used to obtain rich genomic information using archived sputum smears. Increasing local in vitro diagnostic capacities using devices such as the GeneXpert (Cepheid, USA) is important to increase awareness and improve public health. Alternatively, research on efficiency of new lab-on-chip devices, that should be as simple and inexpensive as possible to be used in level 1 laboratories and run as close as possible to the bedside of the patient, could be another way to improve TB control in rural regions of PNG.
Methods
Study setting and sample collection
BDH is located in the town of Balimo in the Middle Fly District WP, PNG. Passive case detection for TB is conducted at BDH among individuals self-presenting for health care. Presumptive TB cases presenting at BDH were from the Balimo Urban, Gogodala Rural, and Bamu Rural LLG areas, i.e. three of the five LLGs belonging to the Middle Fly District WP. Sputum samples are collected routinely for diagnostic purposes from patients with clinical signs of TB, and are ZN-stained and examined for AFB (acid-fast bacilli) by light microscopy, with slides then archived and stored. Each sputum sample was smeared on several slides, with up to five slides per sputum sample (see14 for details). Archived slides were transferred to James Cook University, in Townsville, Australia, for DNA extraction from the sputum smears.
DNA extraction and detection of Mycobacterium species
DNA extracts were obtained using a modified version of a previously described Chelex DNA extraction method in biosafety level 2 laboratories21; the modified protocol has been published elswhere14. The presence of MTBC in the extracted DNA was analyzed using a TaqMan real-time PCR51 and the molecular diagnosis results have been published elsewhere14. Regarding duplicate sputum smears (i.e. originating from a single patient), only the smear showing the lowest Cq value was selected for further analysis. DNA aliquots were sent to the Institute for Integrative Cell Biology (Gif-sur-Yvette, France) for high-throughput spoligotyping.
Microbead-based spoligotyping
Microbead-based hybridization was performed as previously described10,23 using the ‘TB-SPOL’ kits purchased from Beamedex (Beamedex, Orsay, France; www.beamedex.com). Detection was performed using a Luminex® 200 platform (Luminex Corp, Austin, TX, USA) and XPONENT software for LX100/LX200 (version 3.1.871.0). Interpretation of raw data was undertaken jointly by two experts (CS, GR) who idependently assessed spoligotyping patterns as previously described52. When no clear-cut interpretation was possible, typing was considered as failed; in other cases, intermediate quantitative signals for some spacers only allowed categorization of the samples as “mixed patterns” (see Discussion). Individual spoligotyping patterns were further compared with the International Spoligotyping Database (SITVIT2) of Pasteur Institute of Guadeloupe (http://www.pasteur-guadeloupe.fr:8081/SITVIT2/indexfr.jsp), the latest updated release of SITVITWEB database32. Shared International Types (SIT) and spoligotype lineages and families were assigned according to signatures provided in SITVIT2 and to the latest genome-based lineage designations, i.e. L1-L7 Lineage labels and sublineage labels when possible, according to53 and34. Spoligotyping patterns absent in SITVIT2 were reported as “orphans”, as well as the patterns already described as “orphans” in the SITVIT2 database with no assigned SIT. Additional exploration of the family to which strains belonged was performed using TBminer (https://info-demo.lirmm.fr/tbminer/)54. This tool provides assignation according to different classifications that largely overlap. Contradictory assignations between the different classifications are indicative of either newly described subfamilies or of artefacts in the genotyping data (unpublished results). Spoligotyping from smear slides has previously shown better results with AFB-positive sputa quantified as '2+' and '3+' because of higher DNA loads55. However, instead of using microscopy results, we took into account Cq values obtained from qPCR targeting MTBC to select samples showing high DNA loads (see14 for detailed qPCR results). The Cq cutoff to be used was chosen from preliminary spoligotyping results.
rpoB mutations analysis
MDR/RR-TB was inferred from drug resistance-associated gene mutations of Mtb, focusing on the well-described rpoB gene. A qPCR was used to identify mutations in the rpoB gene using the DNA extracted from the TB clinical samples as a template. The rpoB primer set generating a 147-bp product was designed in-house and synthesized by Macrogen Inc. (Republic of Korea): Mtb-rpoB-147-F (5′-ATC AAG GAG TTC TTC GGC ACC AG-3′) and Mtb-rpoB-147-R (5′-CAC GTC GCG GAC CTC CAG-3′). Each qPCR reaction (20 μL) contained 1X GoTaq qPCR Master Mix (Promega, Madison, USA), 0.8 μM each of forward and reverse primer, and 2 μL of DNA template. The positive control used MTB H37Rv (GenBank: AL123456.3). Thermal cycling was as follows: hold at 95 °C for 2 min, followed by 45 cycles of 95 °C for 5 s and 60 °C for 15 s. Melting curve analysis was performed at a linear temperature transition rate of 0.1 °C/s from 70 to 95 °C. The qPCR assays were optimized and undertaken on a Rotor-Gene qPCR machine (Rotor-Gene software version 2.0.2.4). Samples that were reactive (Cq ≤ 38) were sequenced in both directions (Macrogen Inc., Republic of Korea). Sequence chromatograms were analysed using Geneious R10 (Biomatters Limited, Auckland, New Zealand), with the consensus sequences of each sample aligned with and compared to the MTB H37Rv reference sequence (GenBank: AL123456.3) using the “Map to Reference” Geneious tool. The consensus numbering system used for the RIF-resistance associated rpoB gene mutations is according to that described previously for MTB H37Rv56.
Mapping the data
Patient data was obtained from the BDH TB patient and laboratory registers, with patient locations identified as the first residential address recorded. Each patient’s location was matched to a census unit, based primarily on PNG census data obtained from the PNG National Statistical Office, or alternatively from the 2012 and 2017 PNG government election polling schedules57,58. Latitude and longitude coordinates were obtained from the PNG National Statistical Office and census data, based on census unit-level data for villages, and averaged coordinates of multiple census units for Balimo and larger villages where the precise census unit was unknown. Instances of alternate local names were checked and confirmed locally. Out of the 162 included samples, the residential address was not able to be determined for 14 (address absent from the register for 12 patients; unknown villages ‘Sarau’ and ‘Owa’ for two patients). Maps were created using a Mac OSX Version of QGIS v2.18 Las Palmas de Gran Canaria59 (available from: http://www.kyngchaos.com/software/qgis). A licence-free PNG map with administrative areas (level 1) was downloaded as a shapefile from http://www.diva-gis.org/gdata.
Ethics statement
Ethics approval was obtained from the PNG Medical Research Advisory Council and registered under the reference MRAC No. 17.02. The information sheet and written informed consent forms were available where required. However, the need for written informed consent was waived by the Ethics Committee due to generally low levels of literacy, and verbal consent was obtained in the majority of the cases. The study was conducted with the permission and support of the Middle Fly District Health Service and the Evangelical Church of PNG Health Service, and at all times permission was obtained prior to sampling activities.
Data availability
All data generated or analysed during this study are included in the published article.
References
Aia, P. et al. Epidemiology of tuberculosis in Papua New Guinea: analysis of case notification and treatment-outcome data, 2008–2016. Western Pac Surveill Response J 9, 9–19, https://doi.org/10.5365/wpsar.2018.9.1.006 (2018).
World Health Organization. Global Tuberculosis Report 2018. (World Health Organization, Geneva, Switzerland, 2018).
Cross, G. B. et al. TB incidence and characteristics in the remote gulf province of Papua New Guinea: a prospective study. BMC Infect Dis 14, 93, https://doi.org/10.1186/1471-2334-14-93 (2014).
McBryde, E. S. Evaluation of Risks of Tuberculosis in Western Province Papua New Guinea. 56 (Burnet Institute, Australia, 2012).
Comas, I. et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet 45, 1176–1182, https://doi.org/10.1038/ng.2744 (2013).
Coscolla, M. & Gagneux, S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin Immunol 26, 431–444, https://doi.org/10.1016/j.smim.2014.09.012 (2014).
Brites, D. & Gagneux, S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol Rev 264, 6–24, https://doi.org/10.1111/imr.12264 (2015).
Ballif, M. et al. Genetic diversity of Mycobacterium tuberculosis in Madang, Papua New Guinea. Int J Tuberc Lung Dis 16, 1100–1107, https://doi.org/10.5588/ijtld.11.0779 (2012).
Ley, S. D. et al. Diversity of Mycobacterium tuberculosis and drug resistance in different provinces of Papua New Guinea. BMC Microbiol 14, 307, https://doi.org/10.1186/s12866-014-0307-2 (2014).
Zhang, J. et al. Mycobacterium tuberculosis complex CRISPR genotyping: improving efficiency, throughput and discriminative power of ‘spoligotyping’ with new spacers and a microbead-based hybridization assay. J Med Microbiol 59, 285–294, https://doi.org/10.1099/jmm.0.016949-0 (2010).
Bainomugisa, A. et al. A complete high-quality MinION nanopore assembly of an extensively drug-resistant Mycobacterium tuberculosis Beijing lineage strain identifies novel variation in repetitive PE/PPE gene regions. Microb Genom 4, https://doi.org/10.1099/mgen.0.000188 (2018).
Bainomugisa, A. et al. Multi-clonal evolution of multi-drug-resistant/extensively drug-resistant Mycobacterium tuberculosis in a high-prevalence setting of Papua New Guinea for over three decades. Microb Genom 4, https://doi.org/10.1099/mgen.0.000147 (2018).
Diefenbach-Elstob, T. et al. The epidemiology of tuberculosis in the rural Balimo region of Papua New Guinea. Trop Med Int Health 23, 1022–1032, https://doi.org/10.1111/tmi.13118 (2018).
Guernier, V. et al. Molecular diagnosis of suspected tuberculosis from archived smear slides from the Balimo region, Papua New Guinea. Int J Infect Dis 67, 75–81, https://doi.org/10.1016/j.ijid.2017.12.004 (2018).
Papua New Guinea Department of Health. Papua New Guinea: National tuberculosis management protocol. (Department of Health, Port Moresby, Papua New Guinea, 2011).
Cowan, L. S., Diem, L., Brake, M. C. & Crawford, J. T. Transfer of a Mycobacterium tuberculosis genotyping method, Spoligotyping, from a reverse line-blot hybridization, membrane-based assay to the Luminex multianalyte profiling system. J Clin Microbiol 42, 474–477 (2004).
Heyderman, R. S. et al. Pulmonary tuberculosis in Harare, Zimbabwe: analysis by spoligotyping. Thorax 53, 346–350 (1998).
Gomgnimbou, M. K. et al. Spoligotyping of Mycobacterium africanum, Burkina Faso. Emerg Infect Dis 18, 117–119, https://doi.org/10.3201/eid1801.110275 (2012).
Molina-Moya, B. et al. Microbead-based spoligotyping of Mycobacterium tuberculosis from Ziehl-Neelsen-stained microscopy preparations in Ethiopia. Sci Rep 8, 3987, https://doi.org/10.1038/s41598-018-22071-9 (2018).
Suresh, N., Arora, J., Pant, H., Rana, T. & Singh, U. B. Spoligotyping of Mycobacterium tuberculosis DNA from Archival Ziehl-Neelsen-stained sputum smears. J Microbiol Methods 68, 291–295, https://doi.org/10.1016/j.mimet.2006.09.001 (2007).
Van Der Zanden, A. G., Te Koppele-Vije, E. M., Vijaya Bhanu, N., Van Soolingen, D. & Schouls, L. M. Use of DNA extracts from Ziehl-Neelsen-stained slides for molecular detection of rifampin resistance and spoligotyping of Mycobacterium tuberculosis. J Clin Microbiol 41, 1101–1108 (2003).
Kamerbeek, J. et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol 35, 907–914 (1997).
Molina-Moya, B. et al. Molecular Characterization of Mycobacterium tuberculosis Strains with TB-SPRINT. Am J Trop Med Hyg 97, 806–809, https://doi.org/10.4269/ajtmh.16-0782 (2017).
Jagielski, T. et al. Current methods in the molecular typing of Mycobacterium tuberculosis and other mycobacteria. Biomed Res Int 2014, 645802, https://doi.org/10.1155/2014/645802 (2014).
Stoneking, M., Jorde, L. B., Bhatia, K. & Wilson, A. C. Geographic variation in human mitochondrial DNA from Papua New Guinea. Genetics 124, 717–733 (1990).
Lee, E. J., Koki, G. & Merriwether, D. A. Characterization of population structure from the mitochondrial DNA vis-a-vis language and geography in Papua New Guinea. Am J Phys Anthropol 142, 613–624, https://doi.org/10.1002/ajpa.21284 (2010).
Chaidir, L. et al. Predominance of modern Mycobacterium tuberculosis strains and active transmission of Beijing sublineage in Jayapura, Indonesia Papua. Infect Genet Evol 39, 187–193, https://doi.org/10.1016/j.meegid.2016.01.019 (2016).
Sasmono, R. T. et al. Heterogeneity of Mycobacterium tuberculosis strains in Makassar, Indonesia. Int J Tuberc Lung Dis 16, 1441–1448, https://doi.org/10.5588/ijtld.12.0055 (2012).
Ismail, F. et al. Study of Mycobacterium tuberculosis complex genotypic diversity in Malaysia reveals a predominance of ancestral East-African-Indian lineage with a Malaysia-specific signature. PLoS One 9, e114832, https://doi.org/10.1371/journal.pone.0114832 (2014).
Buu, T. N. et al. Increased transmission of Mycobacterium tuberculosis Beijing genotype strains associated with resistance to streptomycin: a population-based study. PLoS One 7, e42323, https://doi.org/10.1371/journal.pone.0042323 (2012).
Ley, S. D., Riley, I. & Beck, H. P. Tuberculosis in Papua New Guinea: from yesterday until today. Microbes Infect 16, 607–614, https://doi.org/10.1016/j.micinf.2014.06.012 (2014).
Couvin, D., David, A., Zozio, T. & Rastogi, N. Macro-geographical specificities of the prevailing tuberculosis epidemic as seen through SITVIT2, an updated version of the Mycobacterium tuberculosis genotyping database. Infect Genet Evol pii: S1567-1348, 30969–30969, https://doi.org/10.1016/j.meegid.2018.12.030 (2018).
Stucki, D. et al. Standard genotyping overestimates transmission of Mycobacterium tuberculosis among immigrants in a low incidence country. J Clin Microbiol, https://doi.org/10.1128/JCM.00126-16 (2016).
Stucki, D. et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat Genet 48, 1535–1543, https://doi.org/10.1038/ng.3704 (2016).
Sreevatsan, S. et al. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci USA 94, 9869–9874 (1997).
Balmer, O. & Tanner, M. Prevalence and implications of multiple-strain infections. Lancet Infect Dis 11, 868–878, https://doi.org/10.1016/S1473-3099(11)70241-9 (2011).
Kargarpour Kamakoli, M. et al. Challenge in direct Spoligotyping of Mycobacterium tuberculosis: a problematic issue in the region with high prevalence of polyclonal infections. BMC Res Notes 11, 486, https://doi.org/10.1186/s13104-018-3579-z (2018).
Sanoussi, C. N., Affolabi, D., Rigouts, L., Anagonou, S. & de Jong, B. Genotypic characterization directly applied to sputum improves the detection of Mycobacterium africanum West African 1, under-represented in positive cultures. PLoS Negl Trop Dis 11, e0005900, https://doi.org/10.1371/journal.pntd.0005900 (2017).
Lazzarini, L. C. et al. Mycobacterium tuberculosis spoligotypes that may derive from mixed strain infections are revealed by a novel computational approach. Infect Genet Evol 12, 798–806, https://doi.org/10.1016/j.meegid.2011.08.028 (2012).
Viegas, S. O. et al. Molecular diversity of Mycobacterium tuberculosis isolates from patients with pulmonary tuberculosis in Mozambique. BMC Microbiol 10, 195, https://doi.org/10.1186/1471-2180-10-195 (2010).
Khalid, F. A. et al. Molecular identification of Mycobacterium tuberculosis causing Pulmonary Tuberculosis in Sudan. Eur Acad Res 4, 7842–7855 (2016).
Hanekom, M. et al. A recently evolved sublineage of the Mycobacterium tuberculosis Beijing strain family is associated with an increased ability to spread and cause disease. J Clin Microbiol 45, 1483–1490, https://doi.org/10.1128/JCM.02191-06 (2007).
Ribeiro, S. C. et al. Mycobacterium tuberculosis strains of the modern sublineage of the Beijing family are more likely to display increased virulence than strains of the ancient sublineage. J Clin Microbiol 52, 2615–2624, https://doi.org/10.1128/JCM.00498-14 (2014).
Diefenbach-Elstob, T. et al. Molecular Evidence of Drug-Resistant Tuberculosis in the Balimo Region of Papua New Guinea. Trop Med Infect Dis 4, https://doi.org/10.3390/tropicalmed4010033 (2019).
Huyen, M. N. et al. Tuberculosis relapse in Vietnam is significantly associated with Mycobacterium tuberculosis Beijing genotype infections. J Infect Dis 207, 1516–1524, https://doi.org/10.1093/infdis/jit048 (2013).
European Concerted Action on New Generation Genetic Markers and Techniques for the Epidemiology and Control of Tuberculosis. Beijing/W genotype Mycobacterium tuberculosis and drug resistance. Emerg Infect Dis 12, 736–743, https://doi.org/10.3201/eid1205.050400 (2006).
Glynn, J. R., Whiteley, J., Bifani, P. J., Kremer, K. & van Soolingen, D. Worldwide occurrence of Beijing/W strains of Mycobacterium tuberculosis: a systematic review. Emerg Infect Dis 8, 843–849, https://doi.org/10.3201/eid0805.020002 (2002).
Liu, Q. Y. et al. China’s tuberculosis epidemic stems from historical expansion of four strains of Mycobacterium tuberculosis. Nat Ecol Evol 2, 1982–1992, https://doi.org/10.1038/s41559-018-0680-6 (2018).
Avanzi, C. et al. Transmission of Drug-Resistant Leprosy in Guinea-Conakry Detected Using Molecular Epidemiological Approaches. Clin Infect Dis 63, 1482–1484, https://doi.org/10.1093/cid/ciw572 (2016).
Bos, K. I. et al. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature 514, 494–497, https://doi.org/10.1038/nature13591 (2014).
Broccolo, F. et al. Rapid diagnosis of mycobacterial infections and quantitation of Mycobacterium tuberculosis load by two real-time calibrated PCR assays. J Clin Microbiol 41, 4565–4572 (2003).
Gomgnimbou, M. K. et al. Spoligoriftyping, a dual-priming-oligonucleotide-based direct-hybridization assay for tuberculosis control with a multianalyte microbead-based hybridization system. J Clin Microbiol 50, 3172–3179, https://doi.org/10.1128/JCM.00976-12 (2012).
Coll, F. et al. PolyTB: a genomic variation map for Mycobacterium tuberculosis. Tuberculosis (Edinb) 94, 346–354, https://doi.org/10.1016/j.tube.2014.02.005 (2014).
Aze, J. et al. Genomics and Machine Learning for Taxonomy Consensus: The Mycobacterium tuberculosis Complex Paradigm. PLoS One 10, e0130912, https://doi.org/10.1371/journal.pone.0130912 (2015).
Molina-Moya, B. et al. Mycobacterium tuberculosis complex genotypes circulating in Nigeria based on spoligotyping obtained from Ziehl-Neelsen stained slides extracted DNA. PLoS Negl Trop Dis 12, e0006242, https://doi.org/10.1371/journal.pntd.0006242 (2018).
Andre, E. et al. Consensus numbering system for the rifampicin resistance-associated rpoB gene mutations in pathogenic mycobacteria. Clin Microbiol Infect 23, 167–172, https://doi.org/10.1016/j.cmi.2016.09.006 (2017).
Election commencing 23 June 2012: polling schedule for Middle Fly electorate. (ed Papua New Guinea Electoral Commission) (Port Moresby, Papua New Guinea, 2012).
Election commencing 20th April 2017: polling schedule for Middle Fly Open electorate. (ed Papua New Guinea Electoral Commission) (Port Moresby, Papua New Guinea, 2017).
QGIS Geographic Information System (Open Source Geospatial Foundation Project, 2019).
Acknowledgements
We are grateful to the continued support from the Balimo District Hospital staff, the Middle Fly and ECPNG health services, the local community and the PNG Medical Research Advisory Committee (MRAC 17.02). Research undertaken by Tanya Diefenbach-Elstob was supported by an Australian Government Research Training Program (RTP) Scholarship. Research undertaken by Bernice J. Klotoe was supported by the Beamedex SAS® company (Orsay, France) with support from French CNRS (Centre National de la Recherche Scientifique) and ANRT (Agence Nationale de la Recherche et la Technologie). This study was funded by two unrestricted grants from the Queensland Government Department of Science, Information, Technology and Innovation (DSITI) through the Australian Institute of Tropical Health and Medicine (AITHM). The grants recipients were (1) Emma McBryde and Jeffrey Warner; and (2) Catherine Rush and Jeffrey Warner.
Author information
Authors and Affiliations
Contributions
Conception and design of the study: V.G.-C., E.S.M., C.R., C.S. and J.W. Experimental design and laboratory procedures: V.G.-C. T.D.-E., G.B., D.P., R.D., B.G. and J.W. Acquisition of data: V.G.-C., T.D.-E., B.J.K., D.P. and J.W. Analysis and interpretation of data: V.G.-C., T.D.-E., G.R., C.S. and J.W. Drafting the article: V.G.-C., G.R., C.S. and J.W. Revising it critically for important intellectual content and final approval of the version to be published: all authors.
Corresponding author
Ethics declarations
Competing interests
C. Sola is among the founders of the Beamedex SAS company that produces and commercializes the spoligotyping (TB-SPOL) kit as a “Research Use Only” assay, to be run on Luminex 200 and on MagPix devices (Luminex Corp., Austin, TX). C. Sola does not hold any share in Beamedex and does not hold any share in Luminex Corporation nor was financed by Luminex. The other investigators have no financial interest or financial conflict with the subject matter or materials discussed in this report.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guernier-Cambert, V., Diefenbach-Elstob, T., Klotoe, B.J. et al. Diversity of Mycobacterium tuberculosis in the Middle Fly District of Western Province, Papua New Guinea: microbead-based spoligotyping using DNA from Ziehl-Neelsen-stained microscopy preparations. Sci Rep 9, 15549 (2019). https://doi.org/10.1038/s41598-019-51892-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-51892-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
