
Abstract
Although numerous attempts have been made to alter the sex ratio of the progeny of mammals, the limitations of current technologies have prevented their widespread use in farm animals. The presence or absence of a Y chromosome determines whether a mammalian embryo develops as a male or female, and non-invasive genetic reporters such as fluorescence protein markers have been intensively applied in a variety of fields of research. To develop a non-invasive and instantaneous method for advance determination of the sex of embryos, we developed a Y chromosome-linked eGFP mouse line that stably expresses green fluorescent protein under the control of the CAG promoter. The development of the CRISPR/Cas9 system has made it easy to deliver an exogenous gene to a specific locus of a genome, and linking a tracer to the Y chromosome has simplified the process of predicting the sex of embryos collected by mating a Y-Chr-eGFP transgenic male with a wild-type female. XY embryos appeared green, under a fluorescence microscope, and XX embryos did not. Y chromosome-linked genes were amplified by nested PCR to further confirm the accuracy of this method, and the simultaneous transplantation of green and non-green embryos into foster mothers indicated that 100% accuracy was achieved by this method. Thus, the Y-Chr-eGFP mouse line provides an expeditious and accurate approach for sexing pre-implantation embryos and can be efficiently used for the pre-selection of sex.
Similar content being viewed by others

Introduction
The ability to produce offspring of a desired sex is significant for livestock production, and several approaches have been developed to determine the sex of embryos prior to their transfer in mammals. Although Veerhuis et al.1 reported improved accuracy of sexing bovine embryos by using a cell surface histocompatibility-Y (H-Y) antigen, this methodology was not repeatable2. Subsequent research has used embryo biopsy3 and polymerase chain reaction (PCR)-based assays of sex chromosome-linked genes4,5, but these methods are generally labour intensive and error prone and may even damage normal embryonic development2,6.
A non-invasive genetic marker has also been reported to allow pre-selection of the sex of progeny7: a D4/XEGFP transgenic mouse line harbouring an X-linked GFP was generated by embryonic stem (ES) cell-mediated transgenesis in this study, but the incorporation of the GFP gene was random. Furthermore, X chromosome inactivation in mammals during embryologic development8,9,10,11 could terminate the expression of the X-linked transgene7. As one type of non-invasive genetic reporter, fluorescence proteins have been applied in many fields of research, such as identifying the genotype of transgenic animals12,13,14, tracing a specific cell lineage15,16, and studying the expression status of genes in time and space7,17,18. Therefore, non-invasive genetic reporters inspired us to consider how to use a non-invasive label to sex mammalian embryos.
Since the Y chromosome determines the male sex19, only male progeny can inherit Y chromosome-linked genetic markers. A ubiquitously expressed sex chromosome-linked reporter gene could be used to indicate the presence of an X or Y chromosome in the mouse embryo. Therefore, the expression of this reporter gene would reflect the sex of the progeny. Ultimately, a Y-linked transgenic mouse line could more stably transmit a genetic marker to single-sex progeny, and the presence of a labelled marker in the Y chromosome enables visual detection of the sex of embryos and animals.
The site-specific integration of exogenous genes can be readily achieved using CRISPR (clustered regularly interspaced short palindromic repeats)/Cas920,21,22, and the type II bacterial CRISPR/Cas9 system has recently been engineered into an efficient genome-editing tool consisting of the Cas9 nuclease and a single guide RNA (sgRNA). The CRISPR/Cas9 system causes a double-strand break (DSB) at a specific gene locus to inactivate genes and has been used in many organisms23,24,25. Precise gene editing can be achieved by the homology-directed repair (HDR) mechanism25,26,27 when a donor DNA template is provided.
Based on the approach described above, we now demonstrate for the first time that the use of the XYeGFP mouse line can help to distinguish between males and females. The ubiquitous expression of enhanced green fluorescence protein in males can be directly linked to sex and is suitable for routine embryo sex identification in mammals.
Materials and Methods
Mice
C57BL/6J and ICR mice were raised under specific pathogen-free (SPF) conditions. All animal experimental protocols were performed in accordance with the relevant ethical guidelines and regulations. All animal procedures used in this study were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (8th edition, released by the National Research Council, USA) and were approved by the Institutional Animal Care and Use Committee (IACUC) of Guangxi University.
Construction of plasmids
The donor vector contained a 0.75-kb left homologous arm (HA), a CAG-eGFP-bGH-PolyA cassette, and a 0.78-kb right HA. The backbone of the donor was pMD-19T (Takara, Kusatsu, Japan), and bGH-PolyA was amplified by PCR from px330 (Addgene, Watertown, MA, USA) and then inserted into the EcoRI/HindIII-digested pMD-19T vector using the ClonExpress II One Step Cloning Kit (Vazyme, Nanjing, China). To add a number of cloning sites in the vector, oligos were synthesized and purified through polyacrylamide gel electrophoresis (PAGE) (GENEWIZ, SuZhou, China) and subcloned into pMD-19T-bGH at the EcoRV restriction sites. The CAG-EGFP cassette was produced by digesting the plasmid CAG-EGFP-IRES-CRE (Addgene, Watertown, MA, USA) using SalI and NotI. Then, the CAG-EGFP cassette was inserted into the plasmid of pMD-19T-bGH-Oligos linearized by SalI and NotI to produce the vector pMD-19T-CAG-EGFP-bGH. The sequences of LHR and RHR were amplified by PCR from the genome and were subcloned into pMD-19T-CAG-EGFP-bGH at the 5′ KpnI/ClaI and 3′ BbsI/EcoRI restriction sites to complete the donor vector. The primer sequences used for PCR of bGH-PolyA, the 5′ and 3′ HAs, and the oligos are shown in Supplementary Table S2. All of the restriction enzymes were purchased from New England Biolabs (Ipswich, MA, USA). All of the enzymes used to amplify DNA in the construction of the donor were in 2 × Phanta Max Master Mix (Vazyme, Nanjing, China). PCR was performed with an initial denaturation at 95 °C for 3 min; 35 cycles of 95 °C for 15 s, 60 °C for 15 s and 72 °C for 40 s; and a final extension at 72 °C for 5 min.
Production of Cas9 mRNA and sgRNA
The T7 promoter was added to Cas9 and sgRNA by PCR amplification of px260 (Addgene, Watertown, MA, USA) and px330 (Addgene, Watertown, MA, USA) using the primers listed in Table 1. The T7-Cas9 PCR product was purified and used as the template for in vitro transcription (IVT) using the mMESSAGE mMACHINE T7 Ultra Kit (Ambion, Austin, Texas, USA), and the T7-sgRNA PCR product was purified and used as the template for IVT using the MEGAshortscriptTM High Transcription Kit (Ambion, Austin, Texas, USA) according to the manufacturer’s instructions. Both Cas9 mRNA and sgRNA were purified using the MEGA Clear Kit (Ambion, Austin, Texas, USA) according to the manufacturer’s instructions and eluted in RNase-free water.
Zygote microinjection, embryo culture and transplantation
Zygote microinjection was performed as previously described28. Briefly, mouse zygotes were obtained by mating super-ovulated female C57BL/6J mice (4 weeks old) with C57BL/6J male mice, and fertilized eggs were collected from their oviducts. Cas9 mRNA (100 ng/µl), sgRNA (50 ng/µl) and the donor vector (100 ng/µl) were mixed and injected into the cytoplasm of zygotes with well-defined pronuclei in a droplet of HEPES-CZB (Millipore, Billerica, MA, USA) medium containing 5 µg/ml cytochalasin B (Sigma, ST. Louis, MO, USA) using a FemtoJet micro-injector (Eppendorf, Hamburg, Germany) with constant flow settings. The injected zygotes were cultured in KSOM medium with amino acids (Millipore, Billerica, MA, USA) at 37 °C under 5% CO2 in air until the blastocyst stage at 3.5 days. Then, 15–25 blastocysts were transferred into the uterus of pseudo-pregnant ICR females at 2.5 dpc as described previously24.
Blastocyst genotyping analysis
The collection and transfer of single blastocysts were performed using a glass capillary under a dissection microscope as described in Zuo et al.29. A single blastocyst was picked based on its fluorescence and directly transferred into a PCR tube containing 2 µl of lysis buffer (0.1% Tween-20, 0.1% TritonX-100 and 4 µg/ml proteinase K). The samples were incubated for 20 min at 56 °C, and proteinase K was inactivated by heating at 95 °C for 10 min. PCR amplification was performed using nested primer sets (Table 2). Ex-Taq (Takara, Kusatsu, Japan) was activated at 95 °C for 3 min, and primary PCR was performed for 30 cycles at 95 °C for 30 s, 60 °C for 30 s and 72 °C for 1 min, with a final extension at 72 °C for 5 min. Secondary PCR was performed using 1 µl of the primary PCR product and a nested inner primer. PCR was carried out in the same reaction mixture, and the product was then gel purified and sequenced.
Detection of insertion mutations in mice and off-target analyses
Mouse tail DNA was purified using a TIANamp Genomic DNA Kit (TIANGEN, China) according to the manufacturer’s instructions. The integration site of the CAG-EGFP cassette was detected by primers located separately in the mouse genome and at the CAG-eGFP-bGH cassette in 5′ and 3′ junctions. The fragment length of the 5′ junction was amplified by placing the forward primer in the mouse genome and the reverse primer in the CAG promoter. The 3′ junction was detected by placing the forward primer in the polyA signal and the reverse primer in the mouse genome. The primer target in the genome flanks the HA to exclude interference from the original plasmid. The primer sequences used for PCR are listed in Table 3. The enzymes used for PCR were from Vazyme (Nanjing, China), and PCR was performed as follows: initial denaturation at 95 °C for 3 min; 35 cycles of 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 40 s; and a final extension at 72 °C for 5 min. The products were stored at 4 °C for evaluation via agarose gel electrophoresis.
As previously reported30, potential off-target sites of sgRNA were predicted using online software (http://www.rgenome.net/cas-offinder/). We collected all the off-target sites that had no more than 3 mismatches for the sgRNA and then broadened the search regions to 250 base pairs up- and downstream of the potential off-target sites. For amplification of the loci, PCR amplifications were performed as follows: initial denaturation at 95 °C for 3 min; 35 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min; and a final extension at 72 °C for 5 min. The products were then stored at 4 °C, and the primers used are listed in Table 4.
Statistical analysis
Data are presented as the mean ± SEM. Statistical analysis was performed using Prism 7 (GraphPad, San Diego, CA, USA), and significant differences were determined using Student’s unpaired t test. P > 0.05 was considered not significantly different.
Results
Strategy for the generation of Y-Chr-eGFP mice with the CAG promoter
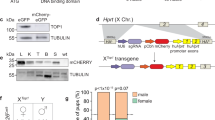
A specified locus for the integration of eGFP was selected in the intergenic region of the two contiguous genes Ddx3y and Uty of the Y chromosome short arm (Fig. 1A). Our experimental design is based on that described by Chu et al.31 and involves the co-injection of Cas9 mRNA, sgRNA, and a donor plasmid. The donor plasmid contains an 800 base pair homology sequence upstream and downstream of the insertion cassette. Cas9 is guided by the sgRNA and results in the incorporation of the donor plasmid via homology-dependent repair (Fig. 1B). The reason for employing a cytomegalovirus early enhancer element and chicken β-actin (CAG) is that it is a strong promoter and can enhance the expression level of the transgene32. Fertilized eggs were collected from oviducts by crossing super-ovulated female C57BL/6J mice with C57BL/6J males, and Y-Chr-eGFP mice were produced from zygotes by microinjection and embryo transfer (Fig. 1C).

Strategy for the generation of Y-Chr-eGFP mice with a CAG promoter. (A) Targeted locus in the Y chromosome: intergenic region sequence of the Ddx3y and Uty genes, which are both located on the short arm of the Y chromosome. (B) Schematic overview of the homologous independent DSB repair pathway at the target locus. (C) Flowchart for the generation of the gene-edited mice.
Generation of Y-Chr-eGFP transgenic mice
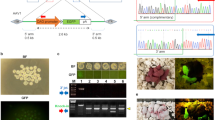
We first generated Y-Chr-eGFP transgenic mice through CRISPR/Cas9-mediated homology-dependent repair. Whole fluorescent mice could be identified when the GFP-positive founder was exposed to ultraviolet light, as shown in Fig. 2A. We produced germline-chimeric mice, and then Y-Chr-eGFP mice were produced through test-cross analysis with wild-type (WT) female mice. To verify the accuracy of the integration site of the GFP gene in the F1 generation of Y-Chr-eGFP mice, genotyping analysis was performed by PCR. The amplified bands (Fig. 2B) of the 5′ and 3′ junctions had sizes that were consistent with expectations, and their exact sizes were determined by sequencing (Fig. 2C). The results showed that the length of the fragment was 1172 base pairs for the 5′ junction and 1016 base pairs for the 3′ junction. Furthermore, these results indicate that the GFP gene was successfully inserted into the specific sites.

Generation of Y-Chr-eGFP mice. (A) Photograph of Y-Chr-eGFP mouse #1 and the control male mouse. (B) Genotyping analysis of the Y-Chr-eGFP mice: PCR products amplified from the 5′ and 3′ junction sites of DNA samples from Y-Chr-eGFP mouse #1. NC, negative control from WT male mice. M, DNA marker. (C) Sequencing results of the integration sites in Y-Chr-eGFP mice. DNA sequencing of the PCR products amplified from the 5′ and 3′ junction sites of DNA samples from Y-Chr-eGFP mouse #1.
Off-target analysis of Y-Chr-eGFP mice
To assess the off-target effects of the CRISPR/Cas9 system in Y-Chr-eGFP mice, we examined 9 off-target sites for the sgRNA used in Y-Chr-eGFP mice (Fig. 3A). DNA sequencing of the PCR products that covered the potential off-target loci showed that no mutations occurred (Fig. 3B).

Off-target analyses in the Y-Chr-eGFP mice. (A) Off-target sites resulting from sgRNA targeting were predicted using online software. Mismatches for up to 10 potential off-target sites were selected for analysis. Red indicates a mismatch with the targeted sequence. (B) Sequencing results of the off-target sites in Y-Chr-eGFP mice. DNA sequencing of the PCR products amplified from these genomic sites were TA cloned and sequenced.
Sex identification of embryos from (Y-Chr-eGFP♂ x WT♀)
We produced germline-chimeric mice, and then Y-Chr-eGFP mice were produced through test-cross analysis with WT female mice. The expression of green fluorescent protein can occur only in male offspring of the F1 generation (Fig. S4). Thus, the GFP-positive founder is a germline chimaera, and the eGFP gene of the Y-Chr-eGFP founder can be stably transmitted to the male offspring by germline transmission. Then, we collected the zygotes by mating Y-Chr-eGFP transgenic male mice (F1) to WT females. Green fluorescence was observed in half of the blastocysts that developed from the zygotes (Fig. 4A); the expression of green fluorescent protein in the offspring showed the stable germline transmission of the eGFP gene. Green blastocysts and non-green blastocysts were easily observed and separated under a fluorescence microscope when there was high expression of the eGFP gene. To evaluate the concordance between the reporter gene and PCR-based assays for sexing, 21 blastocysts were subjected to PCR analysis. Tyrosinase (Tyr) (Gene ID: 22173) is a key enzyme in melanin biosynthesis that is located on chromosome 7 and can serve as a positive control; lysine (K)-specific demethylase 5D (Kdm5d) (Gene ID: 20592) is located on the Y chromosome and can be used to identify males. As shown in Fig. 4B, a positive band for Tyr was observed at the 788 base pair position in all 21 embryos, but a positive band for Kdm5d was observed at the 457 base pair position only in the 11 green embryos (Fig. 4B). Furthermore, when the GFP-positive and GFP-negative blastocysts were implanted into foster mothers, the offspring formed from GFP-positive embryos were all males, and those formed from the other embryos were all females. All of these results reflect that the Y-Chr-eGFP founder is a germline chimaera, and the eGFP gene of the Y-Chr-eGFP founder can be stably transmitted to the male offspring by germline transmission. To test whether carrying the GFP gene in the Y chromosome impacted the development ability of XYeGFP embryos, we separately mated Y-Chr-eGFP mice and WT male mice with super-ovulated female mice. Then, zygotes were collected and cultured in vitro, and 3.5 days later, all embryos were subjected to PCR to identify their sex. The results showed no difference in the sex ratio of males compared with WT (Fig. 4C).

Sex identification of embryos. (A) Representative fluorescence images of embryos. Mixed zygotes cultured for 3.5 days were collected by crossing a Y-Chr-eGFP male mouse with a WT female mouse. Female, GFP-negative embryos under a fluorescence microscope; male, GFP-positive embryos under a fluorescence microscope. (B) Electrophoresis results of PCR products for sexing GFP+ and GFP− embryos. The Tyr gene is located on the autosome and serves as a control. The Kdm5d gene is located on the Y chromosome and serves as a male-specific control. (C) Sex ratio of embryos obtained from mating superovulation females with Y-Chr-eGFP male mice and WT male mice. There was no significant difference between Y-Chr-eGFP mice and WT male mice. “n” is the sample size of numbered embryos.
Discussion
The ability to produce offspring of the desired sex to take advantage of the different values of males and females is a significant benefit for livestock production, and several techniques have been developed to determine the sex of pre-implantation embryos in previous research. These techniques include embryo karyotyping33, fluorescence in situ hybridization (FISH) based on a sex chromosome-specific DNA probe34, and PCR-based assays35,36,37, but the sexing efficiencies of these techniques are closely related to the size of the embryo biopsy and may result in decreased pregnancy rates following the transfer of desmi-embryos38,39.
The CRISPR/Cas9 system has been used to produce gene-edited animals by the microinjection of Cas9 mRNA, sgRNA, and a donor template into the cytoplasm of zygotes, which greatly shortens the amount of time required for producing transgenic animals. In our current study, the XYeGFP mouse line was produced to enable alteration of the sex ratio in pregnancy. As shown in Hadjantonakis et al.7, labelled X chromosomes can also be used to sex pre-implantation embryos; however, it is difficult to define the sex after X chromosome inactivation during embryologic development. Furthermore, X-linked GFP male mice can pass their labelled X chromosome only to their female offspring. The XGFPY mutant male mouse line is difficult to maintain by only crossing with XGFPX or XGFPXGFP female mice in accordance with Mendelian inheritance. Therefore, XGFPX or XGFPXGFP female mice are needed to breed X-Chr-eGFP male mice. However, Y-linked GFP can exclusively be inherited by male offspring, and all of the resulting male genotypes are XYeGFP. The first report on Y chromosome tracing by a labelled gene was by Yamamoto et al., who demonstrated that a marked Y chromosome was helpful for monitoring the presence of the Y chromosome in Tg mouse-derived ES cell lines40. Their research is a great inspiration for us. However, there are many differences between our study and that of Yamamota et al. First, the purpose of our study is different. The main purpose of Yamamota et al. was to monitor the presence of the Y chromosome in Tg mouse-derived ES cell lines, while our purpose was to control the sex ratio of mammals by sexing pre-implantation embryos. Second, we used a different method to produce gene-modified mice. The CRISPR-mediated integration of exogenous DNA is more efficient than traditional approaches. We summarize the knock-in efficiency in offspring in Supplementary Table S1. Only one positive founder was identified, and the efficiency of the HR donor could reach 2.4%. As shown in Yao et al., the knock-in efficiency was no greater than 5% in blastocysts at different target sites25. As reported by Wang et al., the efficiency of CRISPR/HDR is in the range of 0.5–20%41. The HDR-mediated knock-in efficiency is still very low efficiency in animal embryos and tissues in vivo42,43, which is closely related to the length of the HA, the insertion fragment, and the target sites. At present, many studies have been conducted to improve the knock-in efficiency, such as using an inhibitor of the NHEJ pathway43, and optimizing different strategies based on targeted integration22,25, but it is still relatively low. The factors affecting knock-in efficiency remain to be further investigated to facilitate the widespread use of gene editing tools. However, it is still far higher than that of the traditional method (10−6~10−9)44. Third, the choice of target locus was different. In our study, the eGFP gene was inserted into a pre-determined site in the intergenic region of the Ddx3y and Uty genes in the Y chromosome, which overcomes the defect of random integration, thus guaranteeing the accuracy and reliability of the method. The integration locus is approximately 5.5 kb from the Uty gene and 9.3 kb from the Ddx3y gene, as proposed by Zuo et al.28,29. All male mice with deletions of these two genes and their F1 male offspring were fertile and had normal reproductive ability. The mRNA expression levels of the Ddx3y and Uty genes in the MEFs derived from Y-Chr-eGFP and WT mice were identified by Q-PCR, and no significant differences were observed, as shown in supplementary Fig. S1. In addition, our method has no effect on the development of XY embryos in vitro compared with the results in WT embryos, and there was no skewing of the sex ratio, which indicates that our method can be widely used for sex selection in agricultural production.
Although low transgene expression levels may be unsuited for simple visual examination, as suggested by Cornett et al.45, this work shows that GFP works well in distinguishing labelled and non-labelled mouse embryos when under the control of a strong CAG promoter. As indicated in previous research14, no adverse effect was detected in living cells as a result of the expression of eGFP. The onset of transgene expression in embryos is dependent on its particular promoter32, and based on the research performed by Hadjantonakis et al.7, expression of an exogenous gene could be detected at the morula stage. As shown by the experimental results, Y-linked eGFP can be used for visual detection of the sex of pre-implantation embryos with 100% accuracy.
Brief irradiation is not detrimental to the viability of embryos12,46, and as shown in our research, early blastocysts exposed to brief irradiation under a fluorescence microscope successfully developed into pups after transfer to the uterus of pseudo-pregnant foster mothers. Therefore, green male and non-green female embryos were separated at the early blastocyst stage for specific embryo transfer to control the sex bias of the offspring. No off-target effects were detected in XYeGFP mice produced by CRISPR/Cas9, which was consistent with previous studies that showed that off-target mutations are rare in gene-edited animals produced by the CRISPR/Cas9 technique47.
The procedures reported here demonstrate for the first time that a Y chromosome tracer can be used to sex mammalian pre-implantation embryos produced by the CRISPR/Cas9 system. This study also provides a rapid and non-invasive approach for identifying the sex of embryos without compromising the accuracy, feasibility and practicality of the method, and it will have to be modified for similar work in other species.
References
Veerhuis, R. et al. The production of anti-H-Y monoclonal antibodies: their potential use in a sex test for bovine embryos. Vet Immunol Immunopathol 42, 317–330 (1994).
Bredbacka, P. Recent developments in embryo sexing and its field application. Reproduction, nutrition, development 38, 605–613 (1998).
Harper, J. C. et al. Identification of the sex of human preimplantation embryos in two hours using an improved spreading method and fluorescent in-situ hybridization (FISH) using directly labelled probes. Hum Reprod 9, 721–724 (1994).
Fu, Q. et al. Cloning the swamp buffalo SRY gene for embryo sexing with multiplex-nested PCR. Theriogenology 68, 1211–1218 (2007).
Zhu, H. et al. Study of a Simple and Rapid PCR Sex Identification of Bovine Embryo. J Anim Vet Adv 11, 1847–1852 (2012).
van Vliet, R. A., Verrinder Gibbins, A. M. & Walton, J. S. Livestock embryo sexing: A review of current methods, with emphasis on Y-specific DNA probes. Theriogenology 32, 421–438 (1989).
Hadjantonakis, A. K., Gertsenstein, M., Ikawa, M., Okabe, M. & Nagy, A. Non-invasive sexing of preimplantation stage mammalian embryos. Nat Genet 19, 220–222 (1998).
van den Berg, I. M. et al. X chromosome inactivation is initiated in human preimplantation embryos. Am J Hum Genet 84, 771–779 (2009).
Matsui, J., Goto, Y. & Takagi, N. Control of Xist expression for imprinted and random X chromosome inactivation in mice. Hum Mol Genet 10, 1393–1401 (2001).
Monk, M. & Kathuria, H. Dosage compensation for an X-linked gene in pre-implantation mouse embryos. Nature 270, 599–601 (1977).
Betteridge, K. J. & Fléchon, J. E. The anatomy and physiology of pre-attachment bovine embryos. Theriogenology 29, 155–187 (1988).
Fink, D., Yau, T. Y., Kolbe, T. & Rulicke, T. Non-invasive instant genotyping of fluorescently labelled transgenic mice. ALTEX 32, 222–227 (2015).
Garrels, W., Cleve, N., Niemann, H. & Kues, W. A. Rapid non-invasive genotyping of reporter transgenic mammals. Biotechniques 52 (2012).
Ikawa, M. et al. A rapid and non-invasive selection of transgenic embryos before implantation using green fluorescent protein (GFP). FEBS Lett 375, 125–128 (1995).
Sato, M. et al. New approach to cell lineage analysis in mammals using the Cre-loxP system. Mol Reprod Dev 56, 34–44 (2000).
Xia, J. et al. Targeting of the enhanced green fluorescent protein reporter to adrenergic cells in mice. Mol Biotechnol 54, 350–360 (2013).
de Kloet, A. D. et al. Reporter mouse strain provides a novel look at angiotensin type-2 receptor distribution in the central nervous system. Brain Struct Funct 221, 891–912 (2016).
Kawamoto, S. et al. A novel reporter mouse strain that expresses enhanced green fluorescent protein upon Cre-mediated recombination. FEBS Lett 470, 263–268 (2000).
Page, D. C. et al. The sex-determining region of the human Y chromosome encodes a finger protein. Cell 51, 1091–1104 (1987).
Auer, T. O., Duroure, K., De Cian, A., Concordet, J. P. & Del Bene, F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res 24, 142–153 (2014).
Suzuki, K. et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540, 144–149 (2016).
Yao, X. et al. CRISPR/Cas9-mediated Targeted Integration In Vivo Using a Homology-mediated End Joining-based Strategy. Journal of visualized experiments: JoVE (2018).
Hwang, W. Y. et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature biotechnology 31, 227–229 (2013).
Wang, H. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 (2013).
Yao, X. et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Res 27, 801–814 (2017).
Chen, F. et al. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat Methods 8, 753–755 (2011).
Li, X. et al. Efficient SSA-mediated precise genome editing using CRISPR/Cas9. FEBS J 285, 3362–3375 (2018).
Zuo, E. et al. One-step generation of complete gene knockout mice and monkeys by CRISPR/Cas9-mediated gene editing with multiple sgRNAs. Cell Res 27, 933–945 (2017).
Zuo, E. et al. CRISPR/Cas9-mediated targeted chromosome elimination. Genome Biol 18, 224 (2017).
Chiang, C. et al. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods 12, 966–968 (2015).
Chu, V. T. et al. Efficient generation of Rosa26 knock-in mice using CRISPR/Cas9 in C57BL/6 zygotes. BMC Biotechnol 16, 4 (2016).
Niwa, H., Yamamura, K. & Miyazaki, J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–199 (1991).
Handyside, A. H. et al. Biopsy of human preimplantation embryos and sexing by DNA amplification. Lancet 1, 347–349 (1989).
Lee, J. H., Park, J. H., Lee, S. H., Park, C. S. & Jin, D. I. Sexing using single blastomere derived from IVF bovine embryos by fluorescence in situ hybridization (FISH). Theriogenology 62, 1452–1458 (2004).
Blanes, M. S., Tsoi, S. C. & Dyck, M. K. Accurate and Phenol Free DNA Sexing of Day 30 Porcine Embryos by PCR. Journal of visualized experiments: JoVE, 53301 (2016).
Kontogianni, E. H., Griffin, D. K. & Handyside, A. H. Identifying the sex of human preimplantation embryos in X-linked disease: amplification efficiency of a Y-specific alphoid repeat from single blastomeres with two lysis protocols. J Assist Reprod Genet 13, 125–132 (1996).
Martinhago, C. et al. Development of a real-time PCR method for rapid sexing of human preimplantation embryos. Reproductive biomedicine online 20, 75–82 (2010).
Kippax, I. S., Christie, W. B. & Rowan, T. Effects of method of splitting, stage of development and presence or absence of zone pellucida on foetal survival in commercial bovine embryo transfer of bisected embryos, Vol 35 (1991).
Wood, C. Embryo splitting: a role in infertility? Reprod Fertil Dev 13, 91–93 (2001).
Yamamoto, S. et al. Rapid selection of XO embryonic stem cells using Y chromosome-linked GFP transgenic mice. Transgenic Res 23, 757–765 (2014).
Wang, B. et al. Highly efficient CRISPR/HDR-mediated knock-in for mouse embryonic stem cells and zygotes. Biotechniques, 59, 201–202, 204, 206–208 (2015).
Cox, D. B., Platt, R. J. & Zhang, F. Therapeutic genome editing: prospects and challenges. Nat Med 21, 121–131 (2015).
Maruyama, T. et al. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nature biotechnology 33, 538–542 (2015).
Capecchi, M. R. Altering the genome by homologous recombination. Science 244, 1288–1292 (1989).
Cornett, J. C., Landrette, S. F. & Xu, T. Characterization of fluorescent eye markers for mammalian transgenic studies. PLoS One 6, e29486 (2011).
Dewhirst, M. W. et al. Intravital fluorescence facilitates measurement of multiple physiologic functions and gene expression in tumors of live animals. Dis Markers 18, 293–311 (2002).
Iyer, V. et al. Off-target mutations are rare in Cas9-modified mice. Nat Methods 12, 479 (2015).
Acknowledgements
This study was supported by the Natural Science Foundation of Guangxi (2015GXNSFAA139062). We thank Bernard Goodman for his linguistic assistance during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
M.Z., Y.L. and E.Z. initiated and designed the study. X.Z. performed the majority of the experiments in the study and wrote the manuscript. W.W., H.P., D.C. and J.N. performed the microinjection experiments. P.Z., F.C. and Q.F. performed the embryo culture experiments.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, X., Wei, W., Pan, H. et al. Identification of the Sex of Pre-implantation Mouse Embryos Using a Marked Y Chromosome and CRISPR/Cas9. Sci Rep 9, 14315 (2019). https://doi.org/10.1038/s41598-019-50731-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-50731-x
This article is cited by
-
Exploiting a Y chromosome-linked Cas9 for sex selection and gene drive
Nature Communications (2021)
-
Y chromosome in health and diseases
Cell & Bioscience (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
